
Abstract
After neuronal injury or death glial cells become reactive, exhibiting dramatic changes in morphology and patterns of gene expression and ultimately engulfing neuronal debris. Rapid clearance of degenerating neuronal material is thought to be crucial for suppression of inflammation and promotion of functional recovery. Here we demonstrate that Drosophila c-Jun N-terminal kinase (dJNK) signaling is a critical in vivo mediator of glial engulfment activity. In response to axotomy, we find glial dJNK signals through a cascade involving the upstream mitogen-activated protein kinase kinase kinases Slipper and Tak1, the mitogen-activated protein kinase kinase MKK4, and ultimately the Drosophila activator protein 1 (AP-1) transcriptional complex composed of Jra and Kayak to initiate glial phagocytosis of degenerating axons. Interestingly, loss of dJNK also blocked injury-induced upregulation of Draper levels in glia, and glial-specific overexpression of Draper was sufficient to rescue engulfment defects associated with loss of dJNK signaling. This work identifies that the dJNK pathway is a novel mediator of glial engulfment activity and a primary role for the glial Slipper/Tak1→MKK4→dJNK→dAP-1 signaling cascade appears to be activation of draper expression after axon injury.
Similar content being viewed by others


Main
Cell death occurs throughout the life of most metazoans. The presence of cell corpses and cellular debris triggers rapid responses from neighboring professional or non-professional phagocytes, ultimately promoting corpse engulfment.1, 2, 3 In the central nervous system (CNS) glial cells are the primary immune cell type, constantly surveying the neural environment for signs of cell death or injury. Neural injury results in rapid changes in glial cellular phenotypes – glia become ‘reactive’ whereby they exhibit dramatic changes in morphology and patterns of gene expression and they ultimately phagocytose degenerating neuronal debris.4, 5, 6, 7 Reactive glial responses are an early feature of most neurodegenerative diseases8, 9 and occur after any brain injury,10, 11 but remarkably little is known about how glial cells sense neuronal injury, become phagocytic, and eventually engulf neuronal debris.
A key mediator of glial responses to neuronal death or injury in Drosophila is the engulfment receptor Draper,12, 13 which encodes the Drosophila ortholog of the Caenorhabditis elegans cell corpse engulfment receptor CED-1.14 In the Drosophila embryo, glial Draper is required for efficient clearance of neuronal cell corpses from the developing CNS.12 In the adult brain, Draper is essential for activation of glial responses after axotomy and promotion of glial phagocytosis of degenerating axonal debris. For instance, after olfactory receptor neuron (ORN) axotomy, glial cells dramatically increase levels of Draper, extend membranes to degenerating axons, engulf axonal debris, and then terminate their responses and return to a resting state.13, 15 In drapernull mutants, glial cells fail to show any morphological or molecular responses to axonal injury and unengulfed axonal debris lingers in the adult brain for the lifespan of the fly.
During glial engulfment of degenerating axons, the Draper receptor appears to signal in a manner similar to the mammalian Fc, B-, and T-cell immunoreceptors with activation of Draper resulting in downstream engulfment signaling through an Src family kinase signaling cascade. Src42a phosphorylates an intracellular immunoreceptor tyrosine-based activation motif (ITAM) domain on Draper, the non-receptor tyrosine kinase Shark then binds to the Draper ITAM, and engulfment is activated.16 The phosphotyrosine-binding domain-containing protein dCed-6 also appears to signal downstream of the Draper receptor,17 but additional signaling molecules that promote engulfment activity in glia remain poorly defined. Here we show that the Drosophila c-Jun amino-terminal kinase (dJNK) plays a critical role in glial responses to degenerating axonal debris. Loss of glial dJNK signaling potently blocks axon injury-induced upregulation of Draper, activation of a glial phagocytic phenotype, and glial clearance of axonal debris. Our work identifies the dJNK pathway as a novel molecular cascade required for glial engulfment signaling and we propose that a primary role for dJNK is transcriptional activation of the draper gene after axotomy.
Results
Basket signaling is required in adult brain glia for engulfment of axonal debris
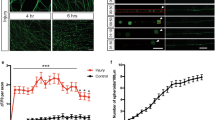
In an RNAi-based screen for novel genes required for glial responses to axonal injury, we identified basket (bsk), encoding dJNK, as a key regulator of glial engulfment of axonal debris. For glial-specific dJNK knockdown, we drove the expression of upstream-activating sequences (UAS)-bskRNAi-104569 with the pan-glial driver repo-Gal4 in a background where a subset of maxillary ORNs were labeled with membrane-tethered green fluorescent protein (mCD8::GFP). We then severed ORN axons by maxillary palp ablation and assayed clearance of axonal debris 5 days after axotomy. Whereas control animals cleared the vast majority of axonal debris within 5 days of axotomy, we found that this glial engulfment activity was potently suppressed by glial bskRNAi (Figures 1a). To confirm this phenotype was specific to bsk, we performed glial-specific RNAi knockdown using two additional RNAi lines targeting different regions of the bsk transcript (bskRNAi-34138 and bskRNAi-TRiP) and found similar results (Supplementary Figure 1). BskRNAi suppression of axonal clearance was robust (near 100%) and persistent – we observed axonal debris lingering for as many as 30 days after axotomy in glial bskRNAi animals, which is longer than the median lifespan of adult Drosophila.

dJNK is required for clearance of axonal debris after nerve injury. (a) Control animals (OR85e-mCD8::GFP/+; repo-Gal4/+) 0, 5, and 15 days after axotomy. (b) Glial knockdown of dJNK using repo-Gal4 to express UAS-dJNK RNAi (OR85e::mCD8::GFP/UAS-bskRNAi104569; repo-Gal4/+). (c) Glial overexpression of UAS-puckered using repo-Gal4 (OR85e::mCD8::GFP/+; repo-Gal4/UAS-puckered). (d) Quantification of GFP intensity in glomeruli for (a)–(c). Error bars depict ±S.E.M. ***P<0.001. (e) Quantification of maxillary nerves with GFP+ debris for (a)–(c)
We next overexpressed puckered (puc), a phosphatase that negatively regulates dJNK activity.18 Glial-specific expression of Puc phenocopied glial bskRNAi with nearly all axonal debris lingering in the CNS for 30 days after axotomy (Figures 1c). We note that axonal fragmentation appears to occur normally and on schedule (i.e. within 1 day) in these backgrounds, indicating that glial Bsk function is not required for axonal degradation.
dJNK signaling is known to be important for the development of a number of cell types. We examined glial morphology, numbers, position, and expression of the engulfment genes draper and dCed-6 in the CNS of glial bskRNAi and UAS-puc animals, but found no noticeable defects in glial cell development or engulfment gene regulation before injury (Supplementary Figure 2). Nevertheless, to exclude the possibility that engulfment phenotypes might arise from requirements for bsk in glial development, we conditionally expressed bskRNAi in adult brain glia using the temperature-sensitive Gal80ts repressor. At 18 °C, Gal80ts represses activation of the Gal4/UAS binary system; however, at 29 °C Gal80ts is inactivated, thereby derepressing Gal4/UAS. We crossed Gal80ts into control and glial bskRNAi backgrounds, raised animals at 18 °C, shifted them to 29 °C after eclosion for 7 days, and then performed axotomies. Consistent with a role for dJNK signaling in mature glia after axotomy, we found that adult-specific knockdown of bsk was sufficient to block glial engulfment of degenerating axons (Figures 2a and b).

The requirement for dJNK in glial engulfment of axonal debris is adult- and ensheathing-glia-specific. (a) Control animals (OR85e::mCD8::GFP, tub-Gal80ts/+; repo-Gal4/+) and bskRNAi animals (OR85e::mCD8::GFP, tub-Gal80ts/UAS-bskRNAi104569; repo-Gal4/+) were raised at 18 °C. After eclosion, adults were shifted to 29 °C for at least 7 days, maxillary palps were ablated, and flies were kept at 29 °C for 5 days before assaying axon clearance. (b) Quantification for (a). Error bars depict mean±S.E.M. ***P<0.001. (c) Control animals (OR85e::mCD8::GFP/+; mz0709-Gal4/+) or animals with ensheathing glia-specific knockdown of dJNK (OR85e::mCD8::GFP/UAS-bskRNAi104569; mz0709-Gal4/+) were axotomized and assayed for axon clearance 5 days after axotomy. (d) Quantification for (c). Error bars depict mean±S.E.M. ****P<0.0001
Ensheathing glia are the primary cell type in the Drosophila neuropil that express molecular components of the engulfment machinery, including the Draper receptor and dCed-6 adaptor molecule, and they phagocytose axonal debris after axotomy.19 To determine whether Bsk functions in ensheathing glia, we drove the expression of bskRNAi or UAS-puc with the ensheathing glia-specific driver mz0709-Gal4 and assayed glial clearance of degenerating axons. We found that suppression of dJNK signaling in ensheathing glia was sufficient to block the glial engulfment function (Figures 2c and d). Notably, suppressing dJNK signaling in astrocytes, the only other subtype of glial cells present in the antennal lobe neuropil, had no effect on glial clearance of axonal debris (data not shown).
These data identify dJNK signaling as a novel mediator of glial responses to axonal injury. Our data further showed that dJNK signaling is required in mature ensheathing glia – the same glial subtype in which Draper signals to promote phagocytosis of axonal debris.
Glial dJNK signaling involves a mitogen-activated protein kinase cascade that signals to the nucleus
dJNK has been shown to function downstream of a wide range of receptor types and to execute signaling events with a diversity of kinases. Precisely which signaling molecules dJNK interacts with appears to be context specific.20 To identify additional molecules involved in dJNK signaling during glial phagocytic clearance of degenerating axons, we assayed the roles of known components of JNK signaling pathways in Drosophila (Figure 3a). We found that glial-specific knockdown of the mitogen-activated protein kinase kinase kinases Slipper and Tak1, the mitogen-activated protein kinase kinase MKK4, and the transcriptional factors Jun-related antigen (Jra, Drosophila c-Jun) and Kayak (Drosophila c-Fos) significantly suppressed glial clearance of degenerating axonal debris 5 days after axotomy (Figure 3a and Supplementary Figure 3). We confirmed the specificity of these RNAi transgenes by using additional RNAi lines that target non-overlapping regions of each transcript.

Glial c-Jun kinase signaling to the nucleus after axotomy involves Slpr, Tak, MKK4, Jra, and Kay. (a) UAS-RNAi constructs for the indicated genes were driven in glia using the repo-Gal4 driver, axons were severed in adults, and axonal debris was scored 5 days after axotomy as above. See Supplementary Figure 3 for quantifications. (b) Slpr, tak double mutants (slprBS06, tak1/Y; OR85e::mCD8::GFP/+) were assayed for defects in axon clearance. (c) Model of dJNK signaling to the nucleus to induce engulfment gene expression. (d) Expression of the dAP-1 transcriptional reporter TRE-eGFP in uninjured animals (top) and animals where third antennal segments were ablated 1 day prior (bottom)
We next sought to provide genetic support for a role for this pathway in glial engulfment signaling and to explore the effects of complete loss of function of this JNK signaling pathway in the clearance of axonal debris. We therefore performed axotomies in slipper and tak1 mutants and examined their effects on the clearance of GFP-labeled degenerating axons. Clearance of axonal debris was largely normal in slipper- (slipperBS506) and tak1- (tak12) null mutant backgrounds (not shown); however, neuronal debris persisted at significant levels after axotomy in slipperBS06, tak12 double mutants (Figure 3b and Supplementary Figure 2). These data argue that the Slipper and Tak1 kinases likely function in a redundant manner in the glial cells responding to axonal injury.
Positive dJNK signaling often leads to changes in gene expression through the activator protein 1 (AP-1) complex. Drosophila AP-1 (dAP-1) is composed of a heterodimer of Jra and Kayak. The significant phenotypes in Jra and kayak RNAi backgrounds argue for a role for dAP-1 in glial transcriptional responses to axonal injury. To confirm this, we assayed activation of a Drosophila transcriptional reporter for dAP-1 activity, TRE-eGFP, which contains dAP-1 binding sites upstream of the eGFP gene.21 In uninjured brains, we detected only low levels of eGFP in the TRE-eGFP background, suggesting weak basal dAP-1 signaling. In contrast, 1 day after antennal ablation, we found robust upregulation of the TRE-eGFP reporter in ensheathing glia surrounding the antennal lobe as well as the local cortex glia (Figure 3d and Supplementary Figure 4).
Taken together, these data identify a novel role for a dJNK signaling pathway involving a Slipper/Tak1→MKK4→Bsk→dAP-1 cascade (Figure 3c) in promoting glial engulfment of degenerating axons after axonal injury. As loss of multiple components of this pathway (i.e. Bsk, Tak1, Slpr, Jra and Kay) block the clearance of a significant amount of axonal debris, positive dJNK signaling appears to be a central regulator of glial responses to axonal injury. On the basis of the requirements for dAP-1 and its activation after axotomy (based on the TRE-eGFP reporter), we propose that dJNK signaling acts to regulate glial engulfment behavior through dAP-1-dependent transcriptional changes.
JNK signaling is required for axotomy-induced increases in glial Draper levels
To explore the potential roles of dJNK signaling in regulating the expression of glial genes, we examined the expression of the engulfment receptor Draper in bskRNAi and UAS-puc backgrounds. Before injury, Draper levels were indistinguishable from control animals when we drove bskRNAi or UAS-puc in adult brain glia as judged by immunofluorescence (Figure 4a and Supplementary Figures 2A and 5A) and western blot (Supplementary Figure 2B). Therefore, loss of JNK signaling does not appear to affect basal levels of Draper expression.

dJNK is required for Draper upregulation after axotomy and activation of glial phagocytic function. (a) Immunostains for Draper in control or glial dJNKRNAi animals before or after antennal ablation. Arrows highlight upregulation of Draper in ensheathing glia (control) or lack thereof (bskRNAi). (b) Draper (red) localization along the maxillary nerve (arrows) before or 1 day after maxillary palp ablation. (c) Draper immunostains (red) in a single antennal lobe before or after axotomy. A subset of maxillary palp ORN axons are shown (green). Note the lack of Draper staining within the glomerulus of glial dJNKRNAi animals compared with controls (arrows). (d) Lysotracker Red stains for lysosomal activity one day after maxillary palp ablation. Note the punctate accumulation of Lysotracker Red in the OR85e+ glomerulus (green, white dotted circle) in controls but not in glial dJNKRNAi animals
Antennal ablation severs ∼88% of all ORN axons innervating the antennal lobe, resulting in widespread axonal degeneration throughout this brain region and in turn a robust increase in Draper levels in glia surrounding the site of injury in wild-type (Figure 4a).13, 19 Interestingly, we found a complete absence of this axotomy-induced increase in Draper levels when we drove glial-specific expression of bskRNAi (Figure 4a) or UAS-puc (Supplementary Figure 5A). Thus, loss of JNK signaling specifically blocks axotomy-induced changes in glial Draper levels after antennal ablation. These data are consistent with the notion that dAP-1 might regulate draper expression.
Glia with depleted dJNK signaling recruit Draper to local axonal debris
The above data raised the possibility that glia lacking dJNK signaling are incapable of responding to axonal injury – specifically, glia might be unable to extend membranes to degenerating axons and activate a phagocytic phenotype. To determine whether glial cells lacking dJNK signaling were capable of recognizing an injury and recruiting Draper specifically to severed axons, we assayed axon clearance and Draper localization after maxillary palp ablation. Surgical removal of maxillary palps severs only ∼12% of all ORN axons which innervate six uniquely identifiable antennal lobe glomeruli. This modest injury does not lead to detectable increases in Draper levels in glia surrounding the antennal lobe, but one can examine Draper recruitment to (1) degenerating axons within identifiable antennal lobe glomeruli or (2) ORNs housed within the maxillary nerve as it projects to the antennal lobe (Figures 4b and c).13 Assaying antennal lobe glomerular localization of Draper is especially useful for determining whether Draper or other components of the engulfment machinery can be recruited to axonal debris, as the engulfing cells must extend membranes into glomeruli to reach engulfment targets (see below). In contrast, ensheathing glia found along the maxillary nerve are already immediately adjacent to their engulfment targets (i.e. they do not have to migrate), so the maxillary nerve is useful for determining whether glia exhibit any changes in Draper localization after axotomy even in backgrounds where glia are incapable of migratory behavior.13
One day after maxillary palp ablation in control animals, we found a strong increase in Draper staining along the maxillary nerve (Figure 4b) and Draper levels returned to baseline levels within 7 days after axotomy. Surprisingly, we found that glial expression of either bskRNAi or UAS-puc did not suppress increases in Draper around the maxillary nerve: in both bskRNAi and UAS-puc backgrounds Draper accumulated on severed maxillary palp ORN axons, although at levels slightly lower than controls (Figure 4b and Supplementary Figures 5B, E, and 6B). Thus, glial cells with depleted dJNK signaling are capable of detecting axonal injury and responding by recruiting the Draper receptor to severed axons. This is notably different from what is observed when the Draper signaling pathway is inactivated – elimination of the non-receptor tyrosine kinase Shark or Src family kinase Src42a from glia completely suppresses Draper recruitment to severed axons along the maxillary nerve. This indicates that dJNK signaling acts downstream of initial recruitment of Draper to axonal debris.
Interestingly, while we observe initial recruitment of Draper to degenerating maxillary ORN axons is normal in animals deficient in glial dJNK signaling, glial cells appear incapable of terminating their responses to axonal injury. Even 30 days after axotomy, Draper levels along the maxillary nerve in glial bskRNAi or UAS-puc backgrounds remained elevated (data not shown). We interpret these data to mean that in addition to its role in engulfment of axonal debris, dJNK signaling is also critical termination of glial responses to axonal injury.
Loss of dJNK signaling suppresses extension of glial membranes to degenerating axons and activation of phagocytic function
We next asked whether loss of dJNK signaling blocked the extension of glial membranes to degenerating axons. We therefore assayed glial recruitment of Draper to degenerating maxillary ORNs in the antennal lobe, where glia must extend membranes into glomeruli to reach their engulfment targets. In control animals, Draper immunoreactivity was detectable one day after maxillary palp ablation throughout the glomeruli housing degenerating axonal debris (Figure 4c and Supplementary Figures 5 and 6). In contrast, while we observed an increase in Draper immunoreactivity around glomeruli in glial bskRNAi or UAS-puc animals, Draper immunoreactivity failed to accumulate in central regions of these structures (Figure 4c and Supplementary Figures 5 and 6). Thus, dJNK signaling is essential for glial membrane extension to engulfment targets.
Once glia have inserted membranes into glomeruli, they activate a phagocytic program – glia become Lysotracker positive (a marker for lysosomal activity) and internalize axonal fragments.22 When we examined Lysotracker activity 1 day after maxillary palp ablations, we observed strong punctate Lysotracker staining within glomeruli containing degenerating axons in control animals. However, Lysotracker staining was absent from these glomeruli in glial bskRNAi animals (Figure 4d). Taken together, these data indicate that dJNK signaling is required for glial membrane extension into glomeruli for engulfment of degenerating axonal material and for activation of lysosomal activity during glia phagocytic function.
Re-expression of draper in glia is sufficient to bypass requirements for dJNK signaling
dJNK is known to function upstream of the transcription factor (dAP-1). We have shown activation of dAP-1 reporters in glia respond to axonal injury and that loss of dAP-1 components suppresses glial engulfment of degenerating axons. In addition, we observed that glia responding to axonal injury in bskRNAi or UAS-puc animals failed to upregulate Draper in response to antennal ORN axotomy. Taken together, these observations raised the intriguing possibility that glial dJNK signaling might promote glial responses to axon injury through activation of draper expression. To explore this possibility, we drove the expression of Draper-I in control, glial bskRNAi, and UAS-puc animals, and assayed clearance of degenerating axonal debris. Draper-I, the longest isoform of Draper (there are three in total) and the only isoform capable of promoting glial engulfment of axonal debris, was made by cloning the full-length cDNA GH24127 into the pUAST vector.15 Expression of Draper-I in control animals did not affect axon clearance as axonal debris was cleared within 5 days (Figure 5). As described above, axonal debris remained 5 days after axotomy in glial bskRNAi or UAS-puc animals. However, when we expressed Draper-I in glial bskRNAi or UAS-puc animals, we found that engulfment defects were completely rescued (Figure 5b). Thus, increased expression of Draper-I is sufficient to overcome engulfment defects in animals with depleted glial dJNK signaling. These data argue that a primary role for dJNK signaling is increasing levels of Draper expression to promote axon clearance after axotomy.

Overexpression of Draper rescues engulfment defects in dJNK knockdown animals. (a) Control, glial dJNKRNAi, or glial expressed Puc animals before and 5 days after axotomy. (b) Control, glial dJNKRNAi, or glial expressed Puc animals 5 days after axotomy with UAS-Draper-I. (c) Quantification from (a) and (b). Error bars depict mean±S.E.M. ***P<0.001
Discussion
This study reveals that axonal injury in the Drosophila brain activates a glial signaling pathway composed of Slipper and Tak, MKK4, dJNK and the dAP-1 complex, which ultimately leads to increased levels of Draper in glia and activation of phagocytic function. Interestingly, we have recently found that draper gene expression is transcriptionally upregulated after axonal injury;15 however, the signaling mechanisms that transduce injury signals from Draper to the nucleus have remained unclear. dAP-1 is, to our knowledge, the first transcriptional regulator shown to modulate glial phagocytic activation and clearance of degenerating axons, and, based on our rescue experiments, it likely does so through the regulation of Draper levels.
Loss of glial dJNK, or suppression of dJNK signaling by overexpression of the phosphatase Puc, resulted in axonal debris lingering in the brain for at least 30 days after axotomy (i.e. longer than the mean lifespan of Drosophila). This is among the strongest glial engulfment phenotypes observed in Drosophila; hence, dJNK is a critical in vivo regulator of glial engulfment activity. We suspect that activation of dJNK is downstream of Draper activation. Loss of Draper or its immediate downstream signaling molecules Src42A, Shark and Rac116, 22 also block injury-induced upregulation of Draper, and Rac123, 24 and Src molecules25 have been shown to be capable of activating the Slipper/Tak complex upstream of dJNK. In addition, we note that loss of JNK signaling closely phenocopies draper-null mutants. In draper null animals, glia fail to respond morphologically to axonal injury, do not activate phagolysosome maturation, and axons are not cleared from the brain.13, 22 However, it remains an open possibility that dJNK signaling is activated by a yet-to-be-identified additional engulfment receptor that is stimulated by axonal injury.
In animals with depleted glial dJNK, glia successfully recruit the small amount of Draper that is present in these cells before injury (referred to as basal Draper) to severed axons in the maxillary nerve, indicating that dJNK is not required for glial cells to receive the injury signal or for Draper accumulation on degenerating axons. However dJNK-deficient glia fail to increase Draper levels in response to injury, to activate phagocytic pathways, and to engulf axonal debris. We interpret these data to mean that dJNK-dependent increases in levels of Draper after axotomy are critical for axon clearance. Multiple lines of evidence support this notion. First, we show activation of a dAP-1 reporter in glia after axotomy and a requirement for the dAP-1 transcriptional complex in glial engulfment of axonal debris. Second, we have recently demonstrated that draper is transcriptionally upregulated after axonal injury.15 Finally, we show that overexpression of Draper is sufficient to rescue dJNKRNAi or Puc misexpression phenotypes. We propose that basal Draper (i.e. that before injury) is present in glia in the healthy brain to sense injury; upon activation, dJNK signaling promotes dramatic increases in Draper through the cascade described above and increased Draper after injury allows efficient clearance of axonal debris.
JNK signaling (as measured with P-JNK antibodies) is activated in response to a broad range of neural injuries in mammals including sciatic nerve lesion,26 accumulation of glial fibrillary acidic protein in models for Alexanders’ disease,27 or focal demyelination.28 To date, the primary roles for JNK in mammalian glia appear to include modulating astrocyte proliferation or proinflammatory responses after neural injury. For example, JNK is activated in astrocytes in response to focal demyelination, and in primary astrocyte cultures, JNK appears to act upstream of injury-induced glial proliferation.28 In addition, lipopolysaccharide treatment of astrocytes in vitro leads to JNK-dependent increases in CPEB1 phosphorylation and activation of reactive oxygen species, which promote inflammation.29 Intriguingly, peripheral nerve injury also leads to activation of JNK signaling in mammalian Schwann cells through the p38 mitogen-activated protein kinase cascade, apparently promoting Schwann cell de-differentiation as a part of nerve repair.30 Moreover, expression of mammalian Draper, MEGF10, is dramatically upregulated in Schwann cells after sciatic nerve lesion.31 Thus, activation of glial JNK signaling and ultimately expression of Draper/MEGF10 appears to be a conserved glial response to axonal injury. Our data would argue that a primary role for dJNK activation in Drosophila glia after axonal injury is increasing levels of Draper and promoting phagocytic activity of engulfing glia. Whether the same is true in mammalian astrocytes is unclear, but it is an exciting possibility awaiting exploration.
Materials and Methods
Drosophila stocks
The following Drosophila strains were used: repo-GAL4,32 OR85e-mCD8::GFP (gift from B Dickson, Research Institute of Molecular Pathology, Vienna, Austria), mz0709-GAL,33 pUAST-mCD8::GFP,34 OR22a-GAL4,35 OR85e-GAL4 (kindly provided by J Carlson, Yale University, New Haven, CT, USA), slprBS06, tak12527/FM7 (gift from B Stronach, University of Pittsburgh, Pittsburgh, PA, USA), and pUAST-Draper-I.15 The following RNAi stocks were obtained from VDRC36: 104569 (bsk), 34138 (bsk), 34898 (tak), 33516 (slpr), 33518 (slpr), 106449 (slpr), 26928 (MKK4), 26929 (MKK4), 108561 (MKK4), 10835 (Jra), 107997 (Jra), and 6212 (kay). The following stocks were obtained from the Bloomington Stock Center (Indiana University, Bloomington, IN, USA): y-w-::UAS-puckered (pucScer\UAS.cMa), and y[ 1 ] sc[*] v[ 1 ]; P{y[+t7.7] v[+t1.8]=TRiP.HMS00777}attP2.
Immunolabeling and confocal microscopy
Standard methods were used for dissection, fixation, and antibody labeling of the adult Drosophila brain.13, 37 Primary antibodies were used at the following concentrations: mouse anti-GFP, 1 : 250 (Life Technologies, Grand Island, NY, USA); rabbit anti-Draper, 1 : 500;12 FITC anti-mouse IgG, 1 : 200 (Jackson ImmunoResearch, West Grove, PA, USA); and Cy3 anti-rabbit IgG, 1 : 200 (Jackson ImmunoResearch).
Lysotracker staining was performed as described in Ziegenfuss et al.22 Briefly, brains were dissected in PBS, Lysotracker Red was added (1 : 5000), and samples were rocked in the dark at room temperature for 15 min. Brains were washed with PBS five times quickly, and then rocked in PBS for 15 min at room temperature in the dark. Brains were then fixed with 4% formaldehyde in PBS/Triton for 30 min, rocking at room temperature in the dark. The standard antibody staining protocol was then performed as described above. Vectashield was applied to the brains and they were stored at 4 °C for 1 h before imaging on an Intelligent Imaging Innovations Everest spinning disk confocal microscope.
Images were quantified as previously described in MacDonald et al.13 Statistics were performed using GraphPad Prism (San Diego, CA, USA). In all figures except for Figure 1, Figure 5, and Supplementary Figure 1, data were analyzed using a two-tailed Student’s t-test. In Figure 1, Figure 5, and Supplementary Figure 1, a one-way ANOVA and Tukey’s multiple comparison test was used.
Western blot
Drosophila brains of the indicated genotype were dissected in PBS and homogenized in SDS loading buffer (60 mM Tris (pH 6.8), 10% glycerol, 2% SDS, 1% mercaptoethanol, 0.01% bromophenol blue). For western analysis, sample containing approximately four brains were loaded onto 10% SDS-PAGE gels (Bio-Rad, Hercules, CA, USA), transferred to nitrocellulose membranes (Bio-Rad), and probed with rabbit anti-Draper12 or rabbit anti-dCed-638 antibody at 1 : 1000 diluted in PBS/0.01% Tween-20/5% dry milk. Blots were incubated overnight at 4°C, washed several times in PBS/0.01% Tween-20, and probed with the appropriate HRP-conjugated secondary antibody for 2 h at room temperature. Additional washes were performed and the blot was developed using chemiluminescence (Amersham ECL Plus, GE Healthcare, Pittsburgh, PA, USA), and detected with a Fujifilm Luminescent Imager (Minato-ku, Tokyo, Japan). The protein blot was stripped with mild stripping buffer (0.2 M glycine, 0.1% sodium dodecyl sulfate, 1% Tween, pH 2.2) at room temperature, followed by washes in 1 × PBS and 1 × PBS+0.01% Tween-20, and then re-probed with mouse anti-tubulin (Sigma-Aldrich, St. Louis, MO, USA), 1 : 1000.
Abbreviations
- dJNK:
-
Drosophila c-Jun N-terminal kinase
- CNS:
-
central nervous system
- ITAM:
-
immunoreceptor tyrosine-based activation motif
- ORN:
-
olfactory receptor neuron
- AP-1:
-
activator protein 1
- UAS:
-
upstream activating sequences
- GFP:
-
green fluorescent protein
References
Gumienny TL, Hengartner MO . How the worm removes corpses: the nematode C. elegans as a model system to study engulfment. Cell Death Differ 2001; 8: 564–568.
Giorgi F, Deri P . Cell death in ovarian chambers of Drosophila melanogaster. J Embryol Exp Morphol 1976; 35: 521–533.
Sears HC, Kennedy CJ, Garrity PA . Macrophage-mediated corpse engulfment is required for normal Drosophila CNS morphogenesis. Development 2003; 130: 3557–3565.
Aldskogius H, Kozlova EN . Central neuron-glial and glial–glial interactions following axon injury. Prog Neurobiol 1998; 55: 1–26.
Sofroniew MV . Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 2009; 32: 638–647.
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG et al. Genomic analysis of reactive astrogliosis. J Neurosci 2012; 32: 6391–6410.
Murray M, Wang SD, Goldberger ME, Levitt P . Modification of astrocytes in the spinal cord following dorsal root or peripheral nerve lesions. Exp Neurol 1990; 110: 248–357.
Gerber YN, Sabourin JC, Rabano M, Vivanco MD, Perrin FE . Early functional deficit and microglial disturbances in a mouse model of amyotrophiclateral sclerosis. PLoS One 2012; 7: e36000.
Perry VH, Nicoll JA, Holmes CM . Microglia in neurodegenerative disease. Nat Rev Neurol 2010; 6: 193–201.
Loane DJ, Byrnes KR . Role of microglia in neurotrauma. Neurotherapeutics 2010; 7: 366–377.
Nedergaard M, Dirnagl U . Role of glial cells in cerebral ischemia. Glia 2005; 50: 281–286.
Freeman MR, Delrow J, Kim J, Johnson E, Doe CQ . Unwrapping glial biology: Gcm target genes regulating glial development, diversification, and function. Neuron 2003; 38: 567–580.
MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR . The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 2006; 50: 869–881.
Zhou Z, Hartwieg E, Horvitz HR . CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 2001; 104: 43–56.
Logan MA, Hackett R, Doherty J, Sheehan A, Speese SD, Freeman MR . Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat Neurosci 2012; 15: 722–730.
Ziegenfuss JS, Biswas R, Avery MA, Hong K, Sheehan AE, Yeung YG et al. Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature 2008; 453: 935–939.
Liu QA, Hengartner MO . Candidate adaptor protein CED-6 promotes the engulfment of apoptotic cells in C. elegans. Cell 1998; 93: 961–972.
Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM et al. Puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 1998; 12: 557–570.
Doherty J, Logan MA, Taşdemir OE, Freeman MR . Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci 2009; 29: 4768–4781.
Stronach B . Dissecting JNK signaling, one KKKinase at a time. Dev Dyn 2005; 232: 575–584.
Chatterjee N, Bohmann DA . Versatile ΦC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. PLoS One 2012; 7: e34063.
Ziegenfuss JS, Doherty J, Freeman MR . Distinct molecular pathways mediate glial activation and engulfment of axonal debris after axotomy. Nat Neurosci 2012; 15: 979–987.
Nagata K, Lamarche N, Hall A . A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-κB signalling pathways. EMBO J 1998; 17: 1395–1404.
Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS . Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol Chem 1996; 271: 27225–27228.
Tateno M, Nishida Y, Adachi-Yamada T . Regulation of JNK by Src during Drosophila development. Science 2000; 287: 324–327.
Ma W, Quirion R . Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain 2002; 99: 175–184.
Tang G, Xu Z, Goldman JE . Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J Biol Chem 2006; 281: 38634–38643.
Gadea A, Schinelli S, Gallo V . Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci 2008; 28: 2394–2408.
Kim KC, Hyun Joo S, Shin CY . CPEB1 modulates lipopolysaccharide-mediated iNOS induction in rat primary astrocytes. Biochem Biophys Res Commun 2011; 409: 687–692.
Yang DP, Kim J, Syed N, Tung YJ, Bhaskaran A, Mindos T et al. p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J Neurosci 2012; 32: 7158–7168.
Barrette B, Calvo E, Vallières N, Lacroix S . Transcriptional profiling of the injured sciatic nerve of mice carrying the Wld(S) mutant gene: identification of genes involved in neuroprotection, neuroinflammation, and nerve regeneration. Brain Behav Immun 2010; 24: 1254–1267.
Leiserson WM, Harkins EW, Keshishian H . Fray, a Drosophila serine/threonine kinase homologous to mammalian PASK, is required for axonal ensheathment. Neuron 2000; 28: 793–806.
Ito K, Urban J, Technau GM . Distribution, classification, and development of Drosophila glial cells in the late embryonic and early larval ventral nerve cord. Roux’s Arch Dev Biol 1995; 204: 284–307.
Lee T, Luo L . Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 2001; 24: 251–254.
Dobritsa AA, Van der Goes van Naters W, Warr CG, Steinbrecht RA, Carlson JR . Integrating the molecular and cellular basis of odor coding in the Drosophila antenna. Neuron 2003; 37: 827–841.
Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007; 448: 151–156.
Vosshall LB, Wong AM, Axel R . An olfactory sensory map in the fly brain. Cell 2000; 102: 147–159.
Awasaki T, Tatsumi R, Takahashi K, Arai K, Nakanishi Y, Ueda R et al. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron 2006; 50: 855–867.
Acknowledgements
We thank Barry Dickson, Beth Stronach, Mary Logan, John Carlson, Neal Silverman, and the VDRC for fly strains and antibodies. We also thank A Nicole Fox for critical reading of the manuscript and the entire Freeman laboratory for insightful discussions. This work was supported by NIH RO1 NS053538 (to MRF) and MRF is an Early Career Scientist with the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Blagosklonny
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
About this article
Cite this article
MacDonald, J., Doherty, J., Hackett, R. et al. The c-Jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell Death Differ 20, 1140–1148 (2013). https://doi.org/10.1038/cdd.2013.30
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.30
Keywords
This article is cited by
-
Stem cell-derived brainstem mouse astrocytes obtain a neurotoxic phenotype in vitro upon neuroinflammation
Journal of Inflammation (2023)
-
Glial Draper signaling triggers cross-neuron plasticity in bystander neurons after neuronal cell death in Drosophila
Nature Communications (2023)
-
Transcriptional control of retinal ganglion cell death after axonal injury
Cell Death & Disease (2022)
-
Glial AP1 is activated with aging and accelerated by traumatic brain injury
Nature Aging (2021)
-
Axon degeneration induces glial responses through Draper-TRAF4-JNK signalling
Nature Communications (2017)
