
Abstract
Background:
Olaparib, an oral PARP inhibitor, has shown antitumour activity as monotherapy in patients with germline BRCA1/2 (gBRCA)-mutated breast and ovarian cancer. This study evaluated olaparib capsules in combination with liposomal doxorubicin (PLD) in patients with advanced solid tumours (NCT00819221).
Methods:
Patients received 28-day cycles of olaparib, continuously (days 1–28) or intermittently (days 1–7), plus PLD (40 mg m−2, day 1); seven olaparib dose cohorts (50–400 mg bid) were explored to determine the recommended dose. Assessments included safety, pharmacokinetics, pharmacodynamics and preliminary efficacy (objective response rate (ORR)).
Results:
Of 44 patients treated (ovarian, n=28; breast, n=13; other/unknown, n=3), two experienced dose-limiting toxicities (grade 3 stomatitis and fatal pneumonia/pneumonitis (200 mg per 28-day cycle); grade 4 thrombocytopenia (400 mg per 7-day cycle)). The maximum tolerated dose was not reached using continuous olaparib 400 mg bid plus PLD. Grade ⩾3 and serious AEs were reported for 27 (61%) and 12 (27%) patients, respectively. No major pharmacokinetic interference was observed between olaparib and PLD. The ORR was 33% (n=14 out of 42; complete response, n=3). A total of 13 responders had ovarian cancer: 10 were platinum-sensitive, 11 had a gBRCA mutation.
Conclusions:
Continuous/intermittent olaparib (up to 400 mg bid) combined with PLD (40 mg m−2) was generally tolerated and showed evidence of antitumour activity in ovarian cancer.
Similar content being viewed by others

Main
Poly(ADP-ribose) polymerases (PARPs), which repair single-strand DNA breaks through the base-excision repair (BER) pathway, have emerged as important targets for cancer therapies in patients with an homologous recombination repair deficiency (HRD), because PARP inhibition leads to the formation of double-stranded DNA breaks that cannot be accurately repaired in tumours with an HRD, such as a BRCA1/2 mutation; this concept is known as synthetic lethality. In preclinical studies, PARP inhibitors have demonstrated efficacy in tumours with BRCA1/2 mutations (Moynahan et al, 1999, 2001; Bryant et al, 2005; Farmer et al, 2005).
Olaparib is a potent oral PARP inhibitor that has demonstrated efficacy as monotherapy in trials involving ovarian and breast cancer patients with germline BRCA1/2 (gBRCA) mutations and/or sensitivity to platinum-based therapies (Fong et al, 2009; Audeh et al, 2010; Fong et al, 2010; Tutt et al, 2010; Gelmon et al, 2011; Kaye et al, 2012; Ledermann et al, 2012). The maximum tolerated dose (MTD) of olaparib monotherapy (capsule formulation) was identified as 400 mg twice daily (bid) (Fong et al, 2009). In Phase II trials in patients with recurrent ovarian cancer and gBRCA mutations, olaparib 400 mg bid monotherapy led to response rates of 31–33% (Audeh et al, 2010; Kaye et al, 2012). Furthermore, olaparib monotherapy demonstrated activity in high-grade serous or poorly differentiated ovarian cancer patients with and without gBRCA mutations (objective response rate (ORR): 41% and 24%, respectively) (Gelmon et al, 2011). In a randomized Phase II study targeting patients highly enriched for HRDs, olaparib maintenance treatment significantly improved progression-free survival (PFS) in patients with platinum-sensitive, relapsed, high-grade serous ovarian cancer compared with placebo (hazard ratio=0.35; 95% CI, 0.25–0.49; P<0.00001; median PFS 8.4 vs 4.8 months, respectively) (Ledermann et al, 2012).
Monotherapy studies have shown that olaparib is relatively well tolerated; the most common adverse events (AEs) being nausea, fatigue, vomiting and anaemia (Fong et al, 2009; Audeh et al, 2010; Fong et al, 2010; Tutt et al, 2010; Gelmon et al, 2011; Kaye et al, 2012; Ledermann et al, 2012). Combination studies with standard chemotherapeutic agents in patients with advanced solid tumours (ASTs) have resulted in sub-therapeutic recommended doses (RD) because of haematologic toxicities (Giaccone et al, 2010; Khan et al, 2011; Samol et al, 2012).
Pegylated liposomal doxorubicin (PLD) is an approved treatment for ovarian cancer patients failing platinum and taxane chemotherapies (Gordon et al, 2001; Rose, 2005), and has shown efficacy in a Phase II trial of ovarian cancer patients with gBRCA mutations (Kaye et al, 2012) and a Phase III trial in recurrent ovarian cancer patients (Gordon et al, 2001). In practice, the placement of PLD in the treatment algorithm varies between countries, with use in the second-line setting for patients with platinum-sensitive epithelial ovarian cancer in combination with carboplatin or trabectedin according to the duration of the platinum-free interval since last chemotherapy cycle. However, other second-line options exist and other regimens, such as carboplatin, gemcitabine and bevacizumab, may be applied (Aghajanian et al, 2012). The combination of PARP inhibition with PLD may provide a synergistic effect in patients with advanced ovarian cancer, especially those with HRDs, because of the decreased ability to repair chemotherapy-induced DNA damage. Preclinical studies with PARP inhibitors have shown potentiation of the cytotoxic effects of chemotherapeutic agents (Drew and Plummer, 2009). In particular, PARP inhibition has been shown to sensitise human hepatocellular carcinoma cell lines to doxorubicin treatment in a dose-dependent manner (Muñoz-Gámez et al, 2011). In another study, performed in HeLa cells, the combination of a PARP inhibitor with doxorubicin treatment led to a 50% increase in doxorubicin-mediated cell death compared with doxorubicin treatment alone (Magan et al, 2012). The toxicity profile of PLD appears to be distinct from that of olaparib, with the most common AEs associated with PLD being palmar–plantar erythrodysesthesia syndrome (PPES), stomatitis and nausea (Kaye et al, 2012). PARP inhibition should be sustained throughout the DNA damage and repair processes but, when combining PARP inhibitors with chemotherapy, prolonged inhibition may be unnecessary provided that a critical inhibitory level is maintained during DNA repair. Consequently, intermittent olaparib treatment schedules may show comparable activity, but better tolerability, vs continuous regimens and represent an interesting option for combination studies.
The aim of this study was to determine the optimal treatment schedule and RD of oral olaparib capsules when administered bid for either 1 week (intermittent) or 4 weeks (continuous), in combination with PLD, in patients with ASTs.
Materials and Methods
Patients
Eligible patients were aged ⩾18 years with histologically/cytologically confirmed metastatic cancer; adequate bone marrow, hepatic and renal functions; ECOG performance status ⩽2; and ⩽3 before chemotherapy regimens for advanced disease. gBRCA mutation status was obtained retrospectively for patients in whom gBRCA testing had been performed before study entry. Exclusion criteria included active treatment or high-dose radiotherapy within the last 28 days, prior cumulative dosing (>300 mg m−2) of doxorubicin equivalent, anthracycline resistance and persistent Common Terminology Criteria (CTC) grade ⩾2 toxicities caused by prior therapy. Please see the Supplementary File for further details. All patients provided written informed consent.
Study design
This Phase I, open-label, multicentre dose-finding study (NCT00819221) was designed to evaluate the safety/tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of olaparib capsules in combination with PLD (40 mg m−2 every 28 days) in patients with ASTs. Olaparib was administered for either 7 days (intermittent schedule) or 28 days (continuous schedule) per 28-day treatment cycle. Up to seven olaparib dose levels were to be explored; the dose schedule for the initial cohort (cohort 0) was 50 mg once daily on day 1, followed by 50 mg bid for 7 days. Further patient cohorts were: 100, 200 and 400 mg bid for 7 days (per treatment cycle), and 100, 200 and 400 mg bid for 28 days (per treatment cycle). The MTD was determined as the dose for the cohort in which ⩾2 out of 6 patients experienced a dose-limiting toxicity (DLT). The two cohorts below the MTD would be expanded to ⩽12 patients each (expansion phase) to confirm the RD. The study was based on a standard 3+3 design, with three patients recruited initially to a cohort (starting with cohort 0); if no DLTs were observed, recruitment to the next dose level commenced. If one patient experienced a DLT, the cohort would be expanded to six patients. The intermittent schedule for each dose level would be tested initially; if tolerable, the continuous schedule for that dose level would be assessed in a separate cohort concomitantly with 7-day dosing at the next dose level. This concerted escalation of dose, and dosing duration, was designed to increase patient accrual and potentially shorten the study duration, while preserving patient safety (Sessa et al, 2007). In the event of toxicity, a maximum of two-dose reductions were allowed provided the olaparib dose was ⩾50 mg bid. After two cycles, patients who had not met a withdrawal criterion (voluntary discontinuation, severe non-compliance, disease progression, AE or safety concern) could continue receiving combination therapy for ⩽6 cycles or switch to olaparib monotherapy (according to efficacy).
The study was performed in accordance with the Declaration of Helsinki, the International Conference on Harmonization/Good Clinical Practice and the AstraZeneca policy on Bioethics (AstraZeneca, 2011).
End points and assessments
The primary objective was to determine the RD of twice-daily olaparib combined with PLD, based on the incidence of DLTs. Secondary objectives were to determine the PK of olaparib alone and in combination with PLD, and to evaluate two different schedules of olaparib administration. Exploratory objectives included an evaluation of preliminary efficacy of olaparib plus PLD; an assessment of antitumour activity in patient subgroups with ovarian and breast cancer; and an assessment of the effects of the combination on DNA repair by evaluating phosphorylation of histone H2AX (γH2AX) in peripheral blood mononuclear cells (PBMCs) as a biomarker of double-strand DNA breaks.
Toxicity was graded using NCI-CTCAE v3.0. DLTs included the following events occurring during the first treatment cycle and considered, by the investigator, to be related to combination treatment: grade 4 neutropenia lasting >5 days, grade 4 thrombocytopenia, grade ⩾3 febrile neutropenia, grade ⩾3 nausea and/or vomiting (despite maximal anti-emetic therapy) or any other CTCAE grade ⩾3 non-haematologic toxicity. Preliminary efficacy was determined by assessing objective responses based on Response Evaluation Criteria In Solid Tumors (RECIST; v1.0) (Therasse et al, 2000) as determined by the study site investigators. Efficacy was analysed by tumour type and gBRCA mutation status. Additional analyses of ovarian cancer patients were performed by subdividing patients into platinum-sensitive (patients who experienced a progression-free interval of ⩾6 months following discontinuation of the last platinum-containing chemotherapy) and platinum-resistant subgroups. Analyses by gBRCA mutation status and platinum sensitivity were not pre-specified in the study design and were performed retrospectively.
Blood samples (4 ml) were collected according to limited (escalation phase) or full (expansion phase) sampling schedules and analysed to determine plasma concentrations of olaparib and PLD (Supplementary Figure 1). Plasma concentrations were used to derive PK parameters following intermittent and continuous dosing. Olaparib concentrations were determined by solid-phase extraction and LC–MS/MS chromatography. Total doxorubicin was measured by a high-performance liquid chromatography (HPLC)/fluorescence method following liposome dispersion with Triton-X and on-line plasma extraction. PK data were analysed by non-compartmental methods.
For the determination of γH2AX, PBMCs were isolated from venous blood samples (8 ml) obtained on days 1, 8, 15 and 28 of cycle 1 (Supplementary Figure 1); fixed, and stained for intracellular γH2AX. Cytofluorimetric detection was performed with an anti-phospho-H2AX (Ser139) antibody (Cell Signaling Technology, Beverly, MA, USA). Further analyses were performed to correlate γH2AX data with preliminary evidence of antitumour activity.
Statistical analyses
No formal statistical analysis of safety/tolerability was planned. A Student’s t-test for paired data (two tailed) was applied to PK data and a Wilcoxon signed-rank test was performed for PD data to determine statistically significant differences.
Results
Patient disposition
All 44 patients enroled from January 2009–December 2010 were treated and evaluable for safety (Table 1). Two patients receiving continuous olaparib 400 mg bid are ongoing. Two patients receiving continuous olaparib (100 and 200 mg, respectively) did not complete a 28-day treatment cycle, owing to tumour-related intestinal obstruction and tumour-related ileus, so were not evaluable for DLT or efficacy evaluations. Pre-existing gBRCA mutation status data only were collected following a protocol amendment; therefore, gBRCA status was unknown for 45% of patients in this study. Patient characteristics were generally balanced across each cohort.
MTD, RD and DLTs
All seven cohorts were assessed because DLTs were not experienced by ⩾2 patients in any cohort. The MTD of olaparib with PLD could not be determined and the olaparib 400 mg cohorts were expanded. Two DLTs were experienced: grade 3 stomatitis plus fatal pneumonia/pneumonitis and dyspnoea in a patient receiving continuous 200 mg and grade 4 thrombocytopenia, leading to treatment discontinuation, in a patient receiving intermittent 400 mg.
Safety and tolerability
Thirty-nine patients (89%) completed the planned 2 cycles of combination therapy and 14 patients (32%) completed ⩾6 cycles. The most common reason for patient withdrawal was malignant disease progression (n=31; 76%). Three patients had dose reductions; all from 400 to 200 mg bid (continuous dosing) because of AEs (oesophagitis, thrombocytopenia, anaemia). The reductions occurred at cycles 2, 7 and 9.
Across all cohorts, the most common treatment-related AEs (any grade) were stomatitis and nausea (Table 2). The overall incidence of CTCAE grade ⩾3 events was 61%, with the most common being decreased neutrophil count. Serious AEs were experienced by 12 patients (27%), although only five patients had treatment-related serious AEs (pneumonitis (n=2); pneumonia/pneumonitis and dyspnoea (n=1); thrombocytopenia (n=1); oesophagitis (n=1)). Except for the patient who experienced oesophagitis, all patients with treatment-related serious AEs discontinued owing to these events; a further three patients withdrew because of non-serious treatment-related AEs (stomatitis (n=1); dysphagia, erythema and PPES (n=1); asthenia and vomiting (n=1)), accounting for seven patients in total.
Five patients died during the study. One ovarian cancer patient in the olaparib 200 mg 28-day cohort presented with cancer-related ileus during the first cycle that was not attributed to study treatment. One patient with breast and metastatic lung cancer (200 mg 28-day cohort) discontinued study treatment on day 26 of the first treatment cycle owing to stomatitis; nine days later, the patient experienced severe dyspnoea and pneumonia and later died due to right lung pneumonia (leading to bilateral pneumonitis) and dyspnoea, all of which were considered possibly related to study treatment. One patient with small-cell lung cancer (100 mg 28-day cohort) discontinued treatment on day 22 of the third treatment cycle, having had several AEs including pulmonary fibrosis; 10 days after the discontinuation of study treatment, the patient experienced severe pneumonitis and later died from treatment-related pneumonitis. The remaining two patients (100 mg 28-day and 200 mg 7-day cohorts) died following a general worsening of their condition. Both patients with treatment-related deaths had previous/ongoing medical conditions (one had asthma treated with steroids, infection, suppurating bronchopneumonia; the other had mediastinal radiotherapy, medical history of chronic interstitial lung disease, thromboses, infection) that potentially contributed to pneumonitis.
Clinically significant haematological abnormalities reported as AEs included alterations in neutrophil count (n=13) and haemoglobin (n=4). Grade ⩾3 haematologic alterations were observed in neutrophils (n=9; 20%), platelets (n=3; 7%), haemoglobin (n=2; 5%) and white blood cells (n=2; 5%).
Pharmacokinetics
The maximum plasma concentration (Cmax) and area under the plasma concentration–time curve (AUC0–10 h) of olaparib increased with dose when given alone (day 1) and in the presence of PLD; olaparib exposure tended to be higher in the presence of PLD (Table 3; Supplementary Figure 2). Following a single dose, olaparib was absorbed rapidly with a mean time to maximum observed concentration (Tmax) of 2.1 h. The minimum plasma concentrations (Cmin) of olaparib were maintained during 28 days of treatment (400 mg bid: day 8, 3.6±2.2 μg ml−1; day 28, 3.9±2.6 μg ml−1) indicating that PLD did not interfere with steady-state olaparib plasma concentrations. PLD parameters were generally similar when olaparib was administered for 7 or 28 days; a statistically significant increase in AUC0-inf and a corresponding decrease in total body clearance (CLTB) were observed in patients receiving continuous olaparib 400 mg bid (day 1–28) compared with short-term administration (day 1–7).
Efficacy
The ORR in the overall population was 33% (14 out of 42). Overall, three evaluable patients (7%) achieved a complete response (CR) and 11 (26%) achieved a partial response (PR) (Table 4). Thirteen responders had ovarian cancer; the ORR in this subgroup was 50% (13 out of 26). In the ovarian subgroup, the response rate in platinum-resistant and platinum-sensitive patients was 25% and 71%, respectively (Table 5). Eleven (61%) gBRCA-mutated patients in the ovarian subgroup achieved a response. Of the two additional patients in the ovarian subgroup who experienced a response, one patient with no gBRCA mutation had a CR and one patient with unknown gBRCA status had a PR; both were platinum-sensitive. The remaining response (PR) was in a gBRCA-mutated patient with breast cancer.
Pharmacodynamics
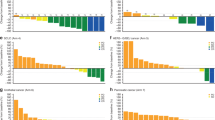
Cytofluorimetric determination of γH2AX phosphorylation level was performed in 41 out of 44 (93%) patients receiving intermittent or continuous olaparib. For both regimens, downregulation of phospho-γH2AX was particularly evident in ovarian patients on days 8 and 15 during the first treatment cycle. Decreases were statistically significant for patients with platinum-resistant ovarian cancer on day 8 (P=0.046), and for patients receiving intermittent olaparib treatment, and was independent of the olaparib dose. However, in platinum-sensitive ovarian patients, the phospho-γH2AX level was stable throughout 28 days of treatment (Figure 1). A rebound of phospho-γH2AX levels occurred between days 15 and 28 in the platinum-resistant ovarian subgroup; this effect was most noticeable in patients with PR or stable disease (Supplementary Figure 3) and in those receiving intermittent dosing (data not shown). A trend towards higher basal phospho-γH2AX levels was observed in the platinum-resistant subgroup compared with the platinum-sensitive subgroup (Supplementary Figure 4). This study only measured phospho-H2AX in surrogate tissue. PBMCs, as tumour samples were not available. The measured levels of phospho-H2AX may therefore not reflect any DNA damage in the tumour target lesions induced by the combination of olaparib and doxorubicin treatment.

Analysis of γ H2AX phosphorylation in ovarian cancer patients. Both treatment regimens are shown: continuous olaparib dosing, days 1–28 in platinum-resistant (A) and platinum-sensitive (C) patients, and intermittent olaparib dosing, days 2 8, in platinum-resistant (B) and platinum-sensitive (D) patients. A Wilcoxon signed-rank test was performed (P=0.046; statistical significance was defined as P<0.05). Median values are indicated.
Discussion
This study evaluated olaparib (intermittent and continuous dosing up to 400 mg bid) in combination with the optimal dose of PLD (40 mg m−2 every 28 days) in patients with ASTs. Although the MTD was not reached, our results suggest that continuous dosing with olaparib capsules, at the recommended monotherapy dose of 400 mg bid, combined with PLD (40 mg m−2) could be considered for Phase II trials of longer duration.
The AEs reported were consistent with known events associated with olaparib and PLD when given as monotherapy (Gordon et al, 2001; Fong et al, 2009; Audeh et al, 2010; Fong et al, 2010; Tutt et al, 2010; Gelmon et al, 2011; Kaye et al, 2012; Ledermann et al, 2012), and the combination was generally tolerated up to the highest doses administered. Two DLTs were reported in patients from separate cohorts (continuous 200 mg; intermittent 400 mg); the MTD was not reached, as the protocol did not permit exploration of olaparib doses above continuous 400 mg bid. The maximum dose permitted in this trial was olaparib 400 mg bid because this dose was determined as the MTD in a previous trial (Fong et al, 2009). The PLD 40 mg m−2 dose investigated in this trial is a commonly used single-agent dose in clinical practice (Julius et al, 2013), despite being lower than the FDA-approved dose for patients with breast and ovarian cancer (50 mg m−2). In addition, this study used the capsule formulation of olaparib, whereas ongoing Phase III studies in ovarian and breast cancer use the tablet formulation of olaparib. It has been shown that exposure with tablet doses ⩾300 mg bid matched or exceeded that of the 400 mg bid capsule, and olaparib 300 mg bid is the recommended tablet dose for Phase III studies (Mateo et al, 2013).
The tolerability profile observed in this study compares favourably to that seen in studies of PLD monotherapy and in combination with carboplatin. The Phase III CALYPSO study compared PLD (30 mg m−2) plus carboplatin (AUC5) every 4 weeks with paclitaxel plus carboplatin in 976 patients with platinum-sensitive ovarian cancer. PLD plus carboplatin treatment was associated with severe non-haematological toxicity in 28.4% of patients and Grade 3−4 neutropenia in 35.2% of patients. Grade ⩾2 fatigue, nausea and hand-foot syndrome occurred in 36.9%, 35.2% and 12% of patients, respectively (Pujade-Lauraine et al, 2010). Treatment with single-agent PLD (mainly 50 mg m−2 every 4 weeks) is associated with fewer events of neutropenia, anaemia, thrombocytopenia, and gastrointestinal toxicity, but increased cutaneous toxicity compared with other monotherapies (Gibson et al, 2013). Compared with other second-line regimens seen in the clinic, olaparib and PLD combination therapy was associated with fewer grade ⩾3 AEs than carboplatin, gemcitabine and bevacizumab as determined in the OCEANS study (Aghajanian et al, 2012), and carboplatin plus paclitaxel regimens (Pignata et al, 2014).
In the current trial, three patients experienced serious AEs of pneumonitis, resulting in death in two patients. The three events occurred in different patient cohorts (100 mg 28-day cohort, 200 mg 28-day cohort and 400 mg 7-day cohort) and were all considered to be related to study treatment by the investigator. Of the two patients who died because of lung toxicities, both had a history of medical conditions that may have contributed to the observed pneumonitis. The third patient had no known risk factors associated with lung toxicity, but developed grade 3 pneumonitis after receiving five cycles of therapy and, following withdrawal of treatment, made a full recovery. Although previous lung conditions may have contributed to both fatal cases of pneumonitis, we cannot exclude the role of olaparib in the observed events. A previous case of presumed treatment-related pneumonitis leading to treatment discontinuation was seen in a Phase I trial of combination olaparib, gemcitabine and cisplatin (Rajan et al, 2012); however, cases of pneumonia have also been seen in previous trials of PLD (Numico et al, 2002; Berenson et al, 2012).
The combination of olaparib with platinum-based chemotherapies has previously been associated with increased myelosuppression; in a Phase I study, 5 out of 23 (22%) patients with ASTs receiving olaparib plus cisplatin and gemcitabine experienced haematological DLTs (Giaccone et al, 2010). Although, in the present study, 30% of patients experienced alterations in neutrophil count, the events appeared not to be dose related, there were no neutropenia-associated DLTs and the overall tolerability profile of olaparib plus PLD appeared more favourable than that observed in most previous olaparib combination studies (Giaccone et al, 2010; Khan et al, 2011; Balmaña et al, 2012; Samol et al, 2012). A Phase I study of olaparib plus weekly paclitaxel showed higher-than-expected rates of neutropenia despite prophylactic administration of granulocyte colony-stimulating factor (Dent et al, 2013).
PK interference between olaparib and PLD was minor and unlikely to have clinical relevance. Cmax and AUC0–10 h of olaparib in the presence of PLD increased with increasing doses, suggesting lack of acute interference on the absorption and distribution of olaparib (Supplementary Figure 2). A trend towards increased olaparib AUC0–10 h and Cmax on day 2 was observed and was statistically significant with the 400 mg dose; this is probably the result of drug accumulation between the doses. The PK parameters of PLD were similar, regardless of whether olaparib was administered for 7 or 28 days (Table 3). Differences in AUC0-inf and CLTB for PLD after olaparib 400 mg administration for 28 days compared with 7 days were probably related to inter-patient variability and not considered clinically significant.
Preliminary evidence of antitumour activity was observed in ovarian cancer patients; although the ovarian subgroup in this study was relatively small, the 50% ORR is higher than that reported previously for Phase II trials that assessed olaparib monotherapy (400 mg bid) in recurrent ovarian cancer patients with gBRCA mutations (31–41%) (Audeh et al, 2010; Gelmon et al, 2011; Kaye et al, 2012). The ORR was also higher than reported in randomized trials of single-agent PLD (18–20%) (Gordon et al, 2001; Kaye et al, 2012). As a result, the combination of olaparib (400 mg bid) and PLD (40 mg m−2) may offer an advantage over either agent alone, particularly since both drugs were combined at their full recommended monotherapy dosages. The ORR in patients with ovarian cancer is within the range achieved by other potential second-line regimens seen in the clinic (Monk et al, 2010; Pujade-Lauraine et al, 2010; Aghajanian et al, 2012). Responses were achieved by 25% of platinum-resistant and 71% of platinum-sensitive ovarian patients. Consistent with previous olaparib trials (Fong et al, 2010; Gelmon et al, 2011), the ORR was higher in platinum-sensitive patients with a gBRCA mutation (67%); however, responses were also seen in platinum-sensitive ovarian patients with wild type or unknown gBRCA mutation status (100%). The ORR in platinum-resistant patients with a gBRCA mutation (50%) was in line with that observed in a recent Phase II trial (Gelmon et al, 2011). Consistent with findings by Gelmon et al (2011), few objective responses were observed in the subgroup of evaluable patients with breast cancer (8%), although only 3 out of 13 were known to have a gBRCA mutation. Although a formal comparison of intermittent and continuous olaparib administration schedules was not performed, antitumour activity was observed with both schedules (7 out of 21 and 7 out of 23 patients, respectively), and both appeared similar in terms of tolerability.
Phosphorylation of γH2AX is associated with cytotoxic agents and has been used widely as a marker of DNA damage (Sedelnikova and Bonner, 2006; Bonner et al, 2008; Fong et al, 2009; Redon et al, 2010). We studied γH2AX in isolated, fixed PBMCs to determine the effects on DNA repair. In contrast to results reported by Fong et al (2009), downregulation of phospho-γH2AX was observed with both continuous and intermittent olaparib regimens during the first treatment cycle. This effect was independent of olaparib dose and most noticeable in platinum-resistant ovarian patients, who presented with higher baseline levels of this marker. Although the decrease in phospho-γH2AX levels was unexpected, peak levels have previously been shown to occur within 6–7 h of treatment with PARP inhibitors (Fong et al, 2009; Kummar et al, 2011, 2012), whereas our observations were not conducted until days 8, 15 and 28. As the phosphorylation of γH2AX is a dynamic phenomenon, we studied the late phase of this event (Supplementary Figure 4). Our aim was to assess changes in γH2AX phosphorylation during chronic treatment with olaparib plus PLD combination; therefore, we selected time points from day 8 onwards so that olaparib had reached a steady-state plasma concentration. In accordance with the results reported by Fong et al (2009), which were unavailable when our study was initiated, we cannot exclude the possibility that, in our study, peak levels of γH2AX phosphorylation may have occurred before day 8. Phosphorylation of γH2AX may be a useful marker for future studies provided that samples are collected at early time points (⩽6 h post treatment).
In conclusion, our data suggest that continuous olaparib 400 mg bid (capsule formulation) in combination with PLD 40 mg m−2 would be suitable for assessment in Phase II studies in patients with ovarian cancer. However, it should be noted that, following recent results from a Phase I study, the recommended monotherapy dose for the olaparib tablet formulation is 300 mg bid (continuous dosing). The encouraging efficacy results seen in ovarian cancer patients were not limited by gBRCA mutation status or sensitivity to platinum therapy, and the tolerability profiles appeared distinct, suggesting that the combination of olaparib with PLD should be explored further.
Change history
12 August 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aghajanian C, Blank SV, Goff BA, Judson PL, Teneriello MG, Husain A, Sovak MA, Yi J, Nycum LR (2012) OCEANS: a randomized, double-blind, placebo-controlled Phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol 30: 2039–2045.
AstraZeneca (2011) Global Policy: Bioethics Available at http://www.astrazeneca.com/Responsibility/Code-policies-standards/Our-global-policies.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A (2010) Oral poly (ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376: 245–251.
Balmaña J, Tung NM, Isakoff SJ, Graña B, Ryan PD, Rafi R, Tracy M, Winer E, Baselga J, Garber JE (2012) Phase I, open-label study of olaparib plus cisplatin in patients with advanced solid tumors. J Clin Oncol 30 (15S): ): abst 1009.
Berenson JR, Yellin O, Kazamel T, Hilger JD, Chen CS, Cartmell A, Woliver T, Flam M, Bravin E, Nassir Y, Vescio R, Swift RA (2012) A phase 2 study of pegylated liposomal doxorubicin, bortezomib, dexamethasone and lenalidomide for patients with relapsed/refractory multiple myeloma. Leukemia 26: 1675–1680.
Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y (2008) GammaH2AX and cancer. Nat Rev Cancer 8: 957–967.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434: 913–917.
Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL, Carmichael J (2013) Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res 15: R88.
Drew Y, Plummer R (2009) PARP inhibitors in cancer therapy: two modes of attack on the cancer cell widening the clinical applications. Drug Resist Updat 12: 153–156.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JHM, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361: 123–134.
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A'Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JHM, de Bono JS, Kaye SB (2010) Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol 28: 2512–2519.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, MacPherson E, Carmichael J, Oza A (2011) Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 12: 852–861.
Giaccone G, Rajan A, Kelly RJ, Gutierrez M, Kummar S, Yancey M, Ji JJ, Zhang Y, Parchment RE, Doroshow JH (2010) A Phase I combination study of olaparib (AZD2281; KU-0059436) and cisplatin (C) plus gemcitabine (G) in adults with solid tumors. J Clin Oncol 28 (15S): ): abst 3027.
Gibson JM, Alzghari S, Ahn C, Trantham H, La-Beck NM (2013) The role of pegylated liposomal doxorubicin in ovarian cancer: a meta-analysis of randomized clinical trials. Oncologist 18: 1022–1031.
Gordon AN, Fleagle JT, Guthrie D, Parkin DE, Gore ME, Lacave AJ (2001) Recurrent epithelial ovarian carcinoma: a randomized phase III study of pegylated liposomal doxorubicin versus topotecan. J Clin Oncol 19: 3312–3322.
Julius JM, Tanyi JL, Nogueras-Gonzalez GM, Watkins JL, Coleman RL, Wolf JK, Smith JA (2013) Evaluation of pegylated liposomal doxorubicin dose on the adverse drug event profile and outcomes in treatment of recurrent endometrial cancer. Int J Gynecol Cancer 23: 348–354.
Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, Amnon A, Bell-McGuinn KM, Chen LM, Friedlander M, Safra T, Vergote I, Wickens M, Lowe ES, Carmichael J, Kaufman B (2012) Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol 30: 372–379.
Khan OA, Gore M, Lorigan P, Stone J, Greystoke A, Burke W, Carmichael J, Watson AJ, McGown G, Thorncroft M, Margison GP, Califano R, Larkin J, Wellman S, Middleton MR (2011) A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer 104: 750–755.
Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, Jia L, Weil M, Speranza G, Murgo AJ, Kinders R, Wang L, Parchment RE, Carter J, Stotler H, Rubinstein L, Hollingshead M, Melillo G, Pommier Y, Bonner W, Tomaszawski JE, Doroshow JH (2011) Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res 71: 5626–5634.
Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, Gandara DR, Allen D, Kiesel B, Beumer JH, Newman EM, Rubinstein L, Chen A, Zhang Y, Wang L, Kinders RJ, Parchment RE, Tomaszawski JE, Doroshow JH (2012) A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res 18: 1726–1734.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira Frommer R, Safra T, Matei D, MacPherson E, Watkins C, Carmichael J, Matulonis U (2012) Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 366: 1382–1392.
Magan N, Isaacs RJ, Stowell KM (2012) Treatment with the PARP-inhibitor PJ34 causes enhanced doxorubicin-mediated cell death in HeLa cells. Anticancer Drugs 23: 627–637.
Mateo J, Friedlander M, Sessa C, Leunen K, Nicum S, Gourley C, Fielding A, Bowen K, Kaye S, Molife LR (2013) Administration of continuous/intermittent olaparib in ovarian cancer patients with a germline BRCA1/2 mutation to determine an optimal dosing schedule for the tablet formulation. Eur J Cancer 49 (2 suppl): ): abst 801.
Monk BJ, Herzog TJ, Kaye SB, Krasner CN, Vermorken JB, Muggia FM, Pujade-Lauraine E, Lisyanskaya AS, Makhson AN, Rolski J, Gorbounova VA, Ghatage P, Bidzinski M, Shen K, Ngan HY, Vergote I, Nam JH, Park YC, Lebedinsky CA, Poveda AM (2010) Trabectedin plus pegylated liposomal doxorubicin in recurrent ovarian cancer. J Clin Oncol 28: 3107–3114.
Moynahan ME, Chiu JW, Koller BH, Jasin M (1999) BRCA1 controls homology-directed DNA repair. Mol Cell 4: 511–518.
Moynahan ME, Pierce AJ, Jasin M (2001) BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell 7: 263–272.
Muñoz-Gámez JA, Quiles-Pérez R, Ruiz-Extremera A, Martín-Álvarez AB, Sanjuan-Nuñez L, Carazo A, León J, Oliver FJ, Salmerón J (2011) Inhibition of poly (ADP-ribose) polymerase-1 enhances doxorubicin activity against liver cancer cells. Cancer Lett 301: 47–56.
Numico G, Castiglione F, Granetto C, Garrone O, Mariani G, Costanzo GD, Ciura PL, Gasco M, Ostellino O, Porcile G, Merlano M (2002) Single-agent pegylated liposomal doxorubicin (Caelix) in chemotherapy pretreated non-small cell lung cancer patients: a pilot trial. Lung Cancer 35: 59–64.
Pignata S, Scambia G, Katsaros D, Gallo C, Pujade-Lauraine E, De Placido S, Bologna A, Weber B, Raspagliesi F, Panici PB, Cormio G, Sorio R, Cavazzini MG, Ferrandina G, Breda E, Murgia V, Sacco C, Cinieri S, Salutari V, Ricci C, Pisano C, Greggi S, Lauria R, Lorusso D, Marchetti C, Selvaggi L, Signoriello S, Piccirillo MC, Di Maio M, Perrone F (2014) Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol 15: 396–405.
Pujade-Lauraine E, Wagner U, Aavall-Lundqvist E, Gebski V, Heywood M, Vasey PA, Volgger B, Vergote I, Pignata S, Ferrero A, Sehouli J, Lortholary A, Kristensen G, Jackisch C, Joly F, Brown C, Le Fur N, du Bois A (2010) Pegylated liposomal doxorubicin and carboplatin compared with paclitaxel and carboplatin for patients with platinum-sensitive ovarian cancer in late relapse. J Clin Oncol 28: 3323–3329.
Rajan A, Carter CA, Kelly RJ, Gutierrez M, Kummar S, Szabo E, Yancey MA, Ji J, Mannargudi B, Woo S, Spencer S, Figg WD, Giaccone G (2012) A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res 18: 2344–2351.
Redon CE, Nakamura AJ, Zhang YW, Ji JJ, Bonner WM, Kinders RJ, Parchment RE, Doroshow JH, Pommier Y (2010) Histone gammaH2AX and poly(ADP-ribose) as clinical pharmacodynamic biomarkers. Clin Cancer Res 16: 4532–4542.
Rose PG (2005) Pegylated liposomal doxorubicin: optimizing the dosing schedule in ovarian cancer. Oncologist 10: 205–214.
Samol J, Ranson M, Scott E, MacPherson E, Carmichael J, Thomas A, Cassidy J (2012) Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs 30: 1493–1500.
Sedelnikova OA, Bonner WM (2006) GammaH2AX in cancer cells: a potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle 5: 2909–2913.
Sessa C, Cresta S, Cerny T, Baselga J, Rota CE, Malossi A, Hess D, Trigo J, Zucchetti M, D'Incalci M, Zaniboni A, Capri G, Gatti B, Carminati P, Zanna C, Marsoni S, Gianni L (2007) Concerted escalation of dose and dosing duration in a phase I study of the oral camptothecin gimatecan (ST1481) in patients with advanced solid tumors. Ann Oncol 18: 561–568.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376: 235–244.
Acknowledgements
This study was sponsored by AstraZeneca AG. We thank Ben Clarke, from Mudskipper Business Ltd, for medical writing support funded by AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Advisory/consulting roles have been held by RVM for Amgen, GlaxoSmithKline, Novartis, Merck Sharp & Dohme and Bristol-Myers Squibb; by CS and RC for AstraZeneca; and by LG for Roche, Genentech, GlaxoSmithKline, Novartis, Pfizer, Boehringer Ingelheim, Celgene and Tahio Pharmaceutical. No potential conflict of interest were disclosed by the other authors.
Additional information
Previous presentations: ESGO Congress, Milan, 11–14 September 2011 (oral presentation); EMCC (ECCO-ESMO-ESTRO) Congress, Stockholm, 23–27 September 2011 (poster presentation).
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Del Conte, G., Sessa, C., von Moos, R. et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer 111, 651–659 (2014). https://doi.org/10.1038/bjc.2014.345
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.345
Keywords
This article is cited by
-
Folate receptor targeted nanoparticles containing niraparib and doxorubicin as a potential candidate for the treatment of high grade serous ovarian cancer
Scientific Reports (2023)
-
Class I HDAC inhibition reduces DNA damage repair capacity of MYC-amplified medulloblastoma cells
Journal of Neuro-Oncology (2023)
-
Associations with response to Poly(ADP-ribose) Polymerase (PARP) inhibitors in patients with metastatic breast cancer
npj Breast Cancer (2022)
-
A phase I dose-finding, pharmacokinetics and genotyping study of olaparib and lurbinectedin in patients with advanced solid tumors
Scientific Reports (2021)
-
Downregulation of APE1 potentiates breast cancer cells to olaparib by inhibiting PARP-1 expression
Breast Cancer Research and Treatment (2019)

{kind=link}
{kind=link}
{kind=link}
{kind=link}