
Abstract
Background:
Clear cell renal cell carcinoma (CCRCC) is the commonest form of kidney cancer. Up to 91% have biallelic inactivation of VHL, resulting in stabilisation of HIF-α subunits. Factor inhibiting HIF-1 is an enzyme that hydroxylates HIF-α subunits and prevents recruitment of the co-activator CBP/P300. An important question is whether FIH-1 controls HIF activity in CCRCC.
Methods:
Human VHL defective CCRCC lines RCC10, RCC4 and 786–O were used to determine the role of FIH-1 in modulating HIF activity, using small interfering RNA knockdown, retroviral gene expression, quantitative RT–PCR, western blot analysis, Annexin V and propidium iodide labelling.
Results:
Although it was previously suggested that FIH-1 is suppressed in CCRCC, we found that FIH-1 mRNA and protein are actually present at similar levels in CCRCC and normal kidney. The FIH-1 inhibition or knockdown in the VHL defective CCRCC lines RCC10 and RCC4 (which express both HIF-1α and HIF-2α) resulted in increased expression of HIF target genes. In the 786-O CCRCC cell line, which expresses only HIF-2α, FIH-1 attenuation showed no significant effect on expression of these genes; introduction of HIF-1α resulted in sensitivity of HIF targets to FIH-1 knockdown. In RCC4 and RCC10, knockdown of FIH-1 increased apoptosis. Suppressing HIF-1α expression in RCC10 prevented FIH-1 knockdown from increasing apoptosis.
Conclusion:
Our results support a unifying model in which HIF-1α has a tumour suppressor action in CCRCC, held in check by FIH-1. Inhibiting FIH-1 in CCRCC could be used to bias the HIF response towards HIF-1α and decrease tumour cell viability.
Similar content being viewed by others


Main
Clear cell renal cell carcinoma (CCRCC) often presents late and remains a significant cause of morbidity and mortality. Recently there have been advances in identifying signalling pathways that are activated in CCRCC, and the introduction of treatments targeting these pathways has shown useful activity in patients with the disease (Linehan et al, 2009). At a genetic level, biallelic inactivation of VHL occurs in the majority of CCRCC, and also underlies the tumours that develop in families with von Hippel–Lindau disease caused by a germline mutation in VHL. Re-expression of VHL efficiently suppresses tumour growth of CCRCC in xenograft assays, establishing that VHL acts as a gatekeeper tumour suppressor gene in the renal epithelium (Iliopoulos et al, 1995). However, understanding of the mechanism(s) by which VHL does this is challenging because it is a multifunctional protein (Frew and Krek, 2008). The best characterised function of VHL is its role as a negative regulator of hypoxia-inducible factor (HIF), through oxygen-dependent degradation of the HIF-α subunit, of which there are two isoforms HIF-1α and HIF-2α (Maxwell et al, 1999). Although it is likely that other actions of VHL contribute to its tumour suppressor action in the kidney, activation of HIF (and more specifically HIF-2α) has been shown to be necessary and sufficient for growth of VHL defective CCRCC cells in xenograft assays (Kondo et al, 2002, 2003). The HIF activation has a range of effects, which could contribute to tumour progression, including enhancing glucose uptake, and increasing expression of glycolytic enzymes and angiogenic mediators (Semenza, 2007). When VHL is present, HIF activation is dramatically downregulated in the presence of oxygen through oxygen-dependent enzymatic hydroxylation of specific prolyl residues by the prolyl hydroxylase domain (PHD) enzymes in the central part of HIF-α subunits, which leads to capture by VHL and ubiquitylation (Epstein et al, 2001). In VHL defective cells, HIF-α subunits are stable in the presence of oxygen (Maxwell et al, 1999). Studies in mice and humans have established that VHL loss-of-function alone is not sufficient for tumourigenesis (Mandriota et al, 2002; Rankin et al, 2006; Frew et al, 2008). The additional events that are required for tumour development are incompletely understood, but there is evolution from exclusive HIF-1α expression in normal renal epithelium and very early lesions to a predominant or exclusive HIF-2α response in tumours, which is likely to be important (Raval et al, 2005).
Co-activator recruitment by HIF-α subunits is regulated by oxygen via FIH-1. This hydroxylates a conserved asparagine residue (Asn 803 in human HIF-1α) within the C-terminal transactivation domain (CTAD) of HIF-α, thereby preventing binding of the co-factor p300 and inhibiting HIF transcriptional activation (Freedman et al, 2002; Hewitson et al, 2002; Lando et al, 2002a, 2002b; Elkins et al, 2003). The role of FIH-1 in regulating the HIF response has been less extensively investigated than that of the PHD enzymes and VHL, but it is established that attenuating FIH-1 increases expression of HIF target genes across a wide range of oxygen tensions (Stolze et al, 2004). Importantly, it has recently been established that FIH-1 hydroxylates ankyrin repeats in other proteins besides HIF-α subunits (Cockman et al, 2006; Linke et al, 2007).
Here we investigate the role of FIH-1 in modulating HIF activity in VHL defective CCRCC. Previous studies of two renal cancer cell lines, A498 and 786-O, suggested that FIH-1 expression was specifically repressed by a mechanism involving phosphatidylinositol 3-kinase (PI3K) and the atypical protein kinase C, PKCζ (Datta et al, 2004; Li et al, 2007). Importantly, we found that FIH-1 is in fact present in CCRCC at similar levels to normal kidney. Further, we show that FIH-1 modulates HIF activity in VHL defective CCRCC lines that contain HIF-1α and HIF-2α, and that inhibiting FIH-1 decreases expansion of these cells in culture and increases apoptosis, in a HIF-1α-dependent manner.
Materials and methods
Cell culture, chemicals, and antibodies
Cell lines RCC10, RCC4 and 786-O, and RCC10 stable transfectants with wild-type pVHL have been described previously (Maxwell et al, 1999; Krieg et al, 2000). Cells were cultured in RPMI 1640 (Life Technologies, Gaithersburg, MD, USA) supplemented with penicillin/streptomycin, glutamine, and 10% fetal bovine serum. Cultures were incubated at 37°C in humidified air with 5% CO2.
Dimethyloxalylglycine (DMOG) was purchased from Frontier Scientific (Logan, UT, USA). For hypoxic experiments, cells were exposed to 0.1 and 1% oxygen for the indicated times using either a hypoxic workstation (INVIVO2 100, Ruskinn, Leeds, UK) or a hypoxic incubator Galaxy R (Biotech, Palo Alto, CA, USA).
The antibodies used were as follows: FIH-1 (Novus biologicals, Littleton, CO, USA), HIF-1α (clone 54; Transduction Labs, Lexington, KY, USA), HIF-2α (p190b; Cancer Research UK, London, UK), GLUT1 and β-actin (Abcam, Cambridge, UK), α-tubulin (Sigma-Aldrich, Poole, UK), Pk-tag (AbD, Serotec, UK).
Clinical material
The study was approved by the Hammersmith and Queen Charlotte's and Chelsea Research Ethics Committee (2002/6486). Following informed consent, samples of uninvolved kidney tissue and tumour were snap frozen in the operating theatre and stored at −80°C.
Immunoblotting
Tissues and cells were homogenised in protein extraction buffer as described (Wiesener et al, 1998). Immunoblots were visualised with enhanced chemiluminescence (ECL) or ECL Plus (Amersham, Arlington Heights, IL, USA).
Real-time reverse transcription-PCR
Total cellular RNA from cells and tissues was isolated using RNA Bee (Biogenesis, Poole, UK), according to the manufacturer's instructions. Total RNA (2 μg per 20 μl reaction) was retrotranscribed using an avian myeloblastosis virus retrotranscription kit (Roche, Indianapolis, IN, USA). PCR was carried out using an Opticon 2 machine (MJ Research, Waltham, MA, USA). Analysis of each experimental sample was in duplicate or triplicate. All real-time reverse transcription-PCR (RT–PCR) data are given as a value normalised to the level of β-actin expression in the same retrotranscription. For tumour samples, values were normalised to the level of 18S expression. The β-actin expression was not significantly altered by hypoxia, or DMOG.
β-Actin, 18S, glucose transporter 1 (GLUT1), BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3), factor inhibiting hypoxia-inducible factor (FIH-1), PHD3 and vascular endothelial growth factor (VEGF) mRNA were measured using SYBR Green (ABgene, Epsom, UK) and the following primers:
β-Actin, 5′-CCCAGAGCAAGAGAGG-3′ (forward) and 5′-GTCCAGACGCAGGATG-3′ (reverse); 18S, 5′-CGCCGCTAGAGGTGAAATTC-3′ (forward) and 5′-TTGGCAAATGCTTTCGCTC-3′ (reverse); GLUT1, 5′-TGGCATGGCGGGTTGT-3′ (forward) and 5′-CCAGGGTAGCTGCTCCAGC-3′ (reverse); PHD3, 5′-GATGCTGAAGAAAGGGC-3′ (forward) and 5′-CTGGCAAAGAGAGTATCTG-3′ (reverse); VEGF, 5′-TGCCAAGTGGTCCCAG-3′ (forward) and 5′-GTGAGGTTTGATCCGC-3′ (reverse); BNIP3, 5′-GATATGGGATTGGTCAAGTCGG-3′ (forward) and 5′-CGCTCGTGTTCCTCATGCT-3′ (reverse); FIH-1, 5′-AAAATGTGGTTGGTTACGAAACAG-3′ (forward) and 5′-GACTCTATGTGATGCCACCAGTACA-3′ (reverse).
Small interfering (si)RNA transfections
Sequences for siRNA targeting FIH-1 were generated using Target Finder (Ambion Bioscience, Austin, TX, USA) and purchased from Eurogentec (Southampton, UK). Transfections were performed in p60 culture dishes using LipofectAMINE 2000 (Invitrogen, San Diego, CA, USA) with siRNA oligos at a concentration of 50 nmol l−1. Cells were transfected as a pool, and after 15–20 h, were divided onto six-well dishes. Cells were analysed 2 days after transfection.
Small interfering RNA oligo sequences were as follows: FIH #1: F-5′-AUGAGGAGCCUGUGGUGCUdTdT-3′ R-5′-AGCACCACAGGCUCCUCAUdTdT-3′; FIH #2: F-5′-GAUGCUUGGAGAGGCCUUGdTdT-3′ R-5′-CCAAGGCCUCUCCAAGCAUdTdT-3′; HIF-1 α: F-5′-CUGAUGACCAGCAACUUGAdTdT-3′ R-5′-UCAAGUUGCUGGUCAUCAGdTdT-3′; HIF-2 α: F-5′-CAGCAUCUUUGAUAGCAGUdTdT-3′ R-5′-ACUGCUAUCAAAGAUGCUGdTdT-3′; Firefly Luciferase (LUC): F-5′-CGUACGCGGAAUACUUCGAdTdT-3′ R-5′-AAGCUAAAGGUACACAAUUdTdT-3′.
Lentiviral short hairpin RNA (shRNA) transfections
The RCC10 cells were plated at a density of 4 × 104 per well in 24-well plates and cultured in complete media for 24 h. The following day, fresh media, supplemented with 6 μg ml−1 polybrene (Sigma-Aldrich), were applied and the cells were transfected with lentiviral particles targeting HIF-1α (Sigma-Aldrich, MISSION shRNA, NM_001530, clone ID TRCN0000003810) or luciferase (Sigma-Aldrich, vector ID SHC007V, F-5′-CCGGCGCTGAGTACTTCGAAATGTCCTC-3′ R-5′-GAGGACATTTCGAAGTACTCAGCGTTTTT3′) at a multiplicity of infection of five. The following day fresh medium was added and 48 h post infection complete medium containing 2 μg ml−1 puromycin was applied to select for transfected cells.
Infection of HIF and FIH-1 retroviral vectors
Viral supernatants were prepared by transfecting the Phoenix packaging cell line (Orbigen, San Diego, CA, USA) using LipofectAMINE 2000. After the initial transfection, Phoenix cells were grown at 32°C. The supernatant was collected and filtered (0.45 μm), then supplemented with 0.25-volume fresh medium with 7.5 μg ml−1 polybrene (Sigma-Aldrich, Poole, UK), and added to cells that had been plated the day before on p100 dishes at 30–40% confluence. After 20 h, cells were washed, and fresh media were added for 20 h before performing a second round of infection.
An active form of HIF-1α carrying the substitutions P402A and P564A, which is resistant to hydroxylation by PHD enzymes, was cloned into pBMNz-HIF1α-neo (Raval et al, 2005). Following infection as described, 786-O cells were selected with G418.
Retroviral vectors of pFIH-1
The coding sequence for human FIH-1 with and without a C terminal Pk tag (V5 epitope from paramyxovirus) was inserted into pCMVR-Neo using standard manoeuvres. Following infection with retroviruses, cells were selected with G418.
Cell proliferation and apoptosis assays
Cell culture expansion was measured over a period of 3 days by MTT assay (3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide; Sigma-Aldrich). Cells were transfected as described above and after 15–20 h, were plated at a density of 3 × 103 cells per well into a 24-well cell tissue culture plate. After 24, 48 and 72 h, 50 μl of 5 mg ml−1 MTT solution were added to the cell cultures in 0.5 ml of medium. After 4 h, media were removed and precipitated formazan crystals formed in viable cells were solubilised with 200 μl of isopropanol-triton (0.1%). Product formation was quantified by absorbance at 550 nm.
Cell culture expansion was also assessed by manual counting. Transfected cells were plated at a density of 2 × 104 cells per well into a six-well tissue culture plate, or 1 × 104 cells per well into a 24-well tissue culture plate and viable cells were counted using a hemocytometer after trypan blue staining.
Apoptosis of siRNA-transfected cells was measured by the Annexin-V-FITC Detection Kit I (BD Biosciences, Oxford, UK) according to the manufacturer's instructions. After staining, cells were analysed on a Becton Dickinson FACS Caliber flow cytometer with CellQuest software (BD Biosciences).
Apoptosis was also measured using the Cell Death Detection ELISA Plus kit (Roche, Burgess Hill, UK). Cell pellets of transfected cells were placed into 200 μl of lysis buffer provided by the manufacturer for 30 min and centrifuged. Aliquots of the supernatant (20 μl) were used in an ELISA with anti-DNA and anti-histone antibodies to detect the presence of cytoplasmic nucleosomes.
Statistical analysis
Data are presented as the mean (±s.e.m.) of three independent experiments. ANOVA or Student's t-test were used to evaluate differences and the level of statistical significance is indicated by the use of asterisks in the figures: *P<0.05, **P<0.01, ***P<0.001.
Results
Factor inhibiting HIF-1 is expressed in renal cancer
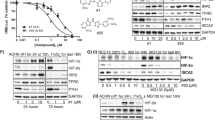
The expression of FIH-1 in CCRCC has not been directly examined to our knowledge. We therefore analysed expression of FIH-1 in CCRCC samples and adjacent uninvolved kidney samples from the same patient. Specimens were obtained at the time of nephrectomy for CCRCC. Although the levels detected were variable, Figure 1 shows that there was no significant difference in FIH-1 mRNA or protein levels between tumour specimens and adjacent kidney (Figures 1A and B). As expected for cancers where the HIF pathway is constitutively activated, GLUT1 mRNA levels were increased in the tumour samples (data not shown).

The FIH-1 is expressed at a similar level in renal cancer and uninvolved kidney. (A) The FIH-1 mRNA levels in renal cancer samples and uninvolved kidney from the same patients. n=10. (B) Representative immunoblots showing FIH-1 protein in renal cancer samples and uninvolved kidney from the same patients.
Factor inhibiting HIF-1 functions in VHL defective CCRCC cell lines
The presence of equivalent amounts of FIH-1 in tumour and adjacent kidney suggested that FIH-1 may actually be reducing HIF activity in this setting; HIF-α subunits would be abnormally stabilised (because VHL is absent), but HIF transcriptional activity would be limited by the action of FIH-1 in the presence of oxygen, which would hydroxylate the CTAD and reduce co-activator recruitment. To examine this we first exposed VHL defective RCC10 cells to reduced oxygen, as a means to reduce FIH-1 enzymatic activity. As Figure 2A shows, we found increased mRNA levels of four well-characterised HIF-α target genes GLUT1, PHD3, VEGF and BNIP3 in RCC10 and RCC4 at 0.1 and 1% oxygen (Figure 2A). Expression of each of these genes was further increased in hypoxia from the high levels observed in the absence of VHL. Importantly, similar results were obtained in a second VHL defective cell line, RCC4 (Figure 2A). As reducing oxygenation could have other effects besides attenuating FIH-1 activity, we used DMOG, a small molecule inhibitor that is an analogue of the co-substrate 2-oxogluarate as an alternative method of inhibiting FIH-1. Figure 2B shows that treatment of CCRCC cell lines with DMOG increases expression of HIF target genes.

Effect of hypoxia and DMOG on gene expression in RCC10 and RCC4 cells. (A) Expression of the indicated HIF target genes was examined by qRT–PCR in cells that were cultured under standard conditions, 1% oxygen or 0.1% oxygen for 16 h. (B) Expression of HIF target genes following exposure to the 2-oxoglutarate analogue DMOG (500 μ M, 16 h). Data are presented as the mean of three independent experiments. (*P<0.05, **P<0.01, ***P<0.001).
These results would be consistent with FIH-1 exerting a negative effect on HIF activation in these VHL defective cell lines. However hypoxia and DMOG are not specific inhibitors of FIH-1; they will inhibit other 2-oxoglutarate-dependent oxygenases including the PHD enzymes (Epstein et al, 2001). In the absence of VHL, prolyl hydroxylation of HIF-1α and HIF-2α by the PHD enzymes has been reported to decrease transactivation by the N-terminal transactivation domain (NTAD) (To and Huang, 2005), providing a potential mechanism by which hypoxia and DMOG would increase expression of HIF targets in a manner independent of FIH-1 and CTAD activity. To directly examine whether FIH-1 inhibits HIF activity we used RNA interference (RNAi). By using either of two different non-overlapping siRNA sequences independently, we achieved a significant reduction of FIH-1 at the mRNA (>70% attenuation) and protein level. The FIH-1 knockdown resulted in a significant increase in HIF target gene mRNA levels as well as GLUT1 protein levels (Figures 3A and B). Taken together, these results provide clear evidence that FIH-1 is acting to reduce expression of HIF target genes in RCC10 and RCC4 cells under normoxic conditions.

The siRNA knockdown of FIH-1 in renal cancer cell lines increases expression of HIF target genes. (A) The RCC4 and RCC10 cells were treated with siRNA for FIH-1 or luciferase (control). The immunoblot shows FIH-1 was decreased at the protein level and that this was associated with an increase in expression of the HIF target gene GLUT1. (B) The effect of FIH-1 knockdown on expression of the indicated HIF target genes in RCC10 and RCC4 cells. Data are presented as the mean of three independent experiments. (*P<0.05, **P<0.01, ***P<0.001).
Increasing FIH-1 expression has little effect on the expression of HIF target genes
The inhibition of HIF transcriptional activity by FIH-1 in RCC10 and RCC4 cells is clearly incomplete as HIF exerts potent effects on gene expression in these cells, as demonstrated by the effects of siRNA for HIF-α subunits (Raval et al, 2005). One explanation for this incomplete inhibition would be that the amount of FIH-1 enzyme in these cells is insufficient to achieve maximal downregulation of HIF. We therefore examined the effect of increasing expression of FIH-1. A retroviral vector containing cDNA encoding for FIH-1 was prepared and used to infect RCC10 cells. The FIH-1 sequence was tagged with the Pk epitope to allow detection of the exogenous FIH-1 (Figure 4A). Analysis of mRNA levels showed a ∼six-fold increase in FIH-1 mRNA when compared with levels in RCC10 cells infected with an empty vector (Figure 4B). In case the Pk tag might reduce enzymatic activity, we also performed this experiment with untagged FIH-1, with similar results (data not shown). This lack of effect of augmenting FIH-1 contrasts with the effect of introduction of VHL, which leads to marked suppression of HIF target genes (Maxwell et al, 1999). This implies that a substantial proportion of HIF activity is resistant to the action of FIH-1; probably this involves transactivation mediated by the NTAD of HIF-α subunits, which is not regulated by FIH-1.

Overexpressing FIH-1 in RCC10 cells has little effect on expression of HIF target genes. (A) The RCC10 cells were infected with pCMVR-FIHPk, which encodes FIH-1 with a Pk tag in or empty vector followed by selection with G418 (1.5 mg ml−1). The immunoblot shows a signal at ∼40 kDa using a Pk antibody, confirming the expression of tagged FIH-1 protein. (B) Results of qRT–PCR analysis of gene expression showing an ∼six-fold increase in FIH-1 mRNA. The mRNA levels of the indicated HIF targets were not significantly affected by the overexpression of FIH-1 in RCC10. Data are presented as the mean of three independent experiments. (***P<0.001)
Attenuating FIH-1 does not reduce HIF target gene expression in 786-O cells, which only express HIF-2α
To investigate this further we examined the effect of FIH-1 knockdown in another well-characterised VHL defective cell line, 786-O, which expresses HIF-2α, but not HIF-1α (Maxwell et al, 1999). We found that FIH-1 siRNA has no effect on HIF target gene expression in 786-O cells (Figure 5A). This contrasts with our observations in RCC10 and RCC4 cells, but is in line with a previous study (Datta et al, 2004). Possible explanations for this would be either that FIH-1 was inactive in these cells (as was suggested in the previous study), or that the HIF-2α they contain is not susceptible to inactivation by FIH-1. To distinguish these possibilities, we expressed HIF-1α in 786-O cells, and then performed RNAi against FIH-1. Western blot analysis confirmed exclusive expression of the HIF-2α isoform in a pool of parental 786-O cells infected with empty vector (pBMNz) and expression of HIF-1α in cells infected with pBMNz-HIF-1α (Figure 5B). Using siRNA, we attenuated FIH-1 in both pools of 786-O cells (Figure 5B). In the pBMNz transfected pool, in which there is exclusive expression of HIF-2α, attenuation of FIH-1 did not affect HIF target gene expression. In contrast, in pBMNz-HIF-1α, FIH-1 attenuation augmented HIF target gene levels of PHD3, VEGF and the pro-apoptotic gene BNIP3 (Figure 5C). These results show that active FIH-1 is present in 786-O cells, and introducing HIF-1α can reveal this activity. Furthermore, HIF-2α (at least in 786-O cells) is insensitive to inactivation by FIH-1.

Introducing HIF-1α in 786-O cells reveals the ability of FIH-1 to regulate HIF activity. (A) The 786-O cells were treated with siRNA for FIH-1 or control. No increases in HIF target gene mRNA were observed, contrasting with results from RCC10 and RCC4 (Figure 2). Data are presented as the mean of three independent experiments. (B) The 786-O cells were infected with retrovirus-encoding HIF-1α in which the two prolyl residues that are targets for hydroxylation were mutated (left panels), or empty vector (right panels). Following selection with G418, cells were treated with luciferase control siRNA or FIH-1 siRNA. Immunoblots show expression of FIH-1, HIF-1α and HIF-2α. (C) Analysis of expression of the indicated HIF target genes. Inhibition of FIH-1 in cells infected with pBMNz empty vector (EV) does not increase HIF target gene expression. An increase in expression of HIF target genes following FIH-1 inhibition is seen following introduction of HIF-1α. Data are presented as the mean of three independent experiments. (*P<0.05, **P<0.01, ***P<0.001).
Attenuating FIH-1 reduces growth of renal cancer cells expressing HIF-1α and induces apoptosis
Previously it has been shown that HIF-1α has anti-proliferative effects in VHL defective renal cancer cells (Raval et al, 2005). This raises the interesting possibility that in cells that lack VHL and express HIF-1α, FIH-1 may favour tumour growth by decreasing the anti-proliferative consequences of HIF-1α activation and shifting the balance of HIF activation towards HIF-2α. To test this, we examined the effect of FIH-1 siRNA on cell population expansion and apoptosis of RCC10 and RCC4 cell cultures. Knockdown of FIH-1 significantly reduced expansion of RCC10 cell cultures using either of the two siRNAs (Figure 6A). The RCC4 and RCC10 cells showed reduced population expansion as assessed by counting the number of viable cells or by MTT assays (Figure 6B). In contrast, population expansion of 786-O cells that do not express HIF-1α was not reduced. This suggested that the effect of FIH-1 knockdown on population expansion might be mediated via increasing the activity of HIF-1α. To test this possibility, RCC10 were transfected with shRNA targeting HIF-1α before FIH-1 knockdown. This prevented the effect of FIH-1 knockdown on population expansion (Figure 6C). Interestingly, suppression of HIF-1α expression resulted in modest, but statistically significant, increase in cell numbers in comparison with control, consistent with HIF-1α suppressing proliferation and/or enhancing cell death.

Effects of FIH-1 knockdown on population expansion of CCRCC cell lines. (A) The RCC10 cells were cultured in parallel and either untreated, treated with the indicated concentrations of two different siRNAs targeting FIH-1, or control siRNA at 50 nM. After 72 h, cell numbers were counted (*P<0.05). Immunoblot shows expression of FIH-1, HIF-1α, and HIF-2α. (B) Cultures of RCC10, RCC4 and 786-O, which were treated with FIH-1 siRNA or control siRNA were assessed by counting the number of viable cells at the indicated timepoints (top panel). Cultures of RCC10, RCC4 and 786-O were treated with FIH-1 siRNA or control siRNA and assessed by MTT assay (lower panel). Data are presented as the mean of three independent experiments (*P<0.05, **P<0.01). (C) The RCC10 cells were infected with shRNA vectors for luciferase (control) or HIF-1α. Following selection with G418 they were treated either with control or FIH-1 siRNA and the number of cells was counted 72 h later. The effect of FIH-1 knockdown on cell number was abrogated by knockdown of HIF-1α. Note also that HIF-1α knockdown significantly increased cell numbers compared with control (compare bars 1 and 3). Immunoblots show expression of HIF-1α, HIF-2α and FIH-1. (D) Cytoplasmic histone-associated DNA fragments were assessed in the indicated cell lines following treatment with FIH-1 siRNA or control. Increased cell death occurred in RCC10 and RCC4 but not in 786-O cells (which express only HIF-2α) or RCC10/VHL in which re-expression of VHL restores HIF-α to basal levels (+ve). Data are presented as the mean of three independent experiments (*P<0.05, **P<0.01). (E) Effect of FIH-1 knockdown on apoptosis in RCC10 and 786-O cells. Cells were transfected with siLUC or siFIH and analysed for apoptosis using the BD Pharmingen FITC Annexin V apoptosis detection kit. Plots are representative of two independent experiments.
To investigate the mechanism(s) by which cell numbers were decreased, we assayed cytoplasmic histone-associated DNA fragments to assess apoptotic cell death (Figure 6D). This was increased by attenuating FIH-1 in VHL defective RCC10 and RCC4 cells. However, no significant increase was observed in 786-O cells (which express only HIF-2α) or in RCC10 cells in which VHL was stably expressed, resulting in suppression of HIF-1α (and HIF-2α). Independent evidence for increased apoptosis was provided by flow cytometry analysis (Figure 6E). The FIH-1 knockdown in RCC10 cells resulted in an increase in early apoptotic (Annexin-V positive) cells, compared with the control siLUC transfection (46.0 vs 9.6%). Taken together, the results are consistent with FIH-1 decreasing apoptosis through a decrease in the activity of HIF-1α. The 786-O cells showed a much less marked effect, but interestingly there was some increase in early apoptotic cells, on FIH-1 knockdown (16.59% with siRNA for FIH vs 5.7% in controls) (Data not shown). This may reflect a HIF-α, independent action of FIH-1 mediated by one of a number of other identified substrates, which includes components of the Notch or NFκB pathways (Cockman et al, 2006; Coleman et al, 2007; Zheng et al, 2008).
Discussion
The major findings of this study are that FIH-1 is present at similar levels in normal kidney and CCRCC, that FIH-1 inhibition in CCRCC cells can increase expression of HIF targets, and that inhibiting FIH-1 can increase apoptosis in these cells. It is noteworthy that FIH-1 expression is maintained in CCRCC compared with normal kidney, as it was previously suggested that an important step in evolution to CCRCC following the loss of VHL function was suppression of FIH-1. In that model, suppression of FIH-1 was considered necessary to achieve HIF activation. Our study shows that FIH-1 is present and active in CCRCC, but it only partially inactivates HIF. This is consistent with the fact that biallelic inactivation of vhl in mouse and human renal epithelium (and other cell types) is associated with marked activation of HIF target genes, even though FIH-1 has not been inactivated (Mandriota et al, 2002; Rankin et al, 2006).
Evidence for the model in which suppression of FIH-1 was a pivotal aspect of CCRCC included the fact that decreasing FIH-1 did not influence HIF activity in 786-O cells. Our study confirms this observation, but implies a different mechanism in which FIH-1 is present but the HIF-2α in these cells is resistant to its action. In the previous studies it was suggested that FIH-1 suppression was achieved in CCRCC via PKCζ suppressing FIH-1 at the level of transcription. Our finding that FIH-1 expression is similar in normal kidney and CCRCC implies that any negative effect of PKCζ is likely to be present in normal renal epithelium as well as CCRCC. This is supported by the fact that similar suppression of FIH-1 via PKC was also observed in HEK 293 cells, a non-malignant renal cell line (Li et al, 2007).
Although we show that FIH-1 is present in CCRCC, it is striking that its action on HIF ranges from partial suppression (RCC10 and RCC4 cells) to no significant effect (in 786-O cells). Several factors are likely to contribute. First, the amount of FIH-1 available may be insufficient—for example, it may be sequestered through binding to ankyrin repeat domains. However, it is notable that overexpression of FIH-1 had only a minor effect on HIF activity. Second, a substantial proportion of transactivation by HIF-α is independent of the interaction with the CH1 pocket of CBP/P300 (Kasper et al, 2005), this would be predicted to be insensitive to FIH-1 and will include the action of the NTAD. Third, a subset of HIF-α subunits may be resistant to the action of FIH-1. As HIF-1α is known to be a substantially better substrate for FIH-1 than HIF-2α (Bracken et al, 2006) it is likely that this resistant fraction is predominantly HIF-2α. This is further supported by data that mutating the FIH-1 target residue in HIF-2α did not increase its effect on HIF target genes or tumour growth in CCRCC cells, whereas substituting the equivalent residue in HIF-1α had a major effect (Yan et al, 2007). This is consistent with 786-O cells not showing evidence of increased HIF activity following FIH-1 knockdown, as they only contain HIF-2α. In addition, it is clear that a proportion of HIF-1α in RCC10 and RCC4 is not inactivated by FIH-1, as RNAi for HIF-1α in these cells has potent effects on gene expression and epithelial behaviour (Esteban et al, 2006). A possibility that merits further investigation is that a post-translational modification renders a proportion of HIF-1α resistant to the action of FIH-1. Reported modifications of HIF-α that could be candidates include phosphorylation, acetylation and sumoylation. An important candidate modification is phosphorylation of Thr-796 in HIF-1α, which can prevent hydroxylation by FIH-1 (Lancaster et al, 2004).
Regardless of the mechanism(s) by which a substantial proportion of HIF activity in CCRCC is resistant to FIH-1, we show that FIH-1 is protecting RCC10 cells from apoptosis and this is mediated by its effect on HIF-1α. That increased activity of HIF is associated with increased apoptosis may appear counter-intuitive, in view of the evidence that suppressing of HIF is a major aspect of VHL's action as a tumour suppressor in the kidney. On the other hand, it is well recognised that although many of HIF-1α's actions promote tumour growth it can also promote cell cycle arrest and apoptosis. Activation of HIF-1α subunit increases expression of genes that are pro-apoptotic (Sowter et al, 2001; Greijer and van der Wall, 2004) and overexpression of HIF-1α has been shown to reduce growth of CCRCC cells as xenografts (Volm and Koomagi, 2000; Raval et al, 2005). Furthermore, foci of biallelic inactivation of VHL in human kidney, which show HIF-1α activation, are not associated with a net increase in proliferation (Mandriota et al, 2002). This suggests that evolution to CCRCC involves a number of additional steps, and there is evidence that a progressive increase in HIF-2α relative to HIF-1α is important. Consistent with this, in xenograft assays of CCRCC cells, active HIF-2α is both necessary and sufficient for tumour growth (Kondo et al, 2002, 2003), whereas active HIF-1α is insufficient (Maranchie et al, 2002). An attractive possibility is that by exerting more effect on HIF-1α than HIF-2α, FIH-1 contributes to VHL defective cells evading apoptosis. This is also consistent with the fact that mutations in FIH-1 have not been reported in CCRCC (Morris et al, 2004).
The intersection of the effects of VHL, HIF and FIH-1 on patterns of gene expression and on cell proliferation have recently been examined in murine embryonic fibroblasts with genetic deletions of each gene, and multiple combinations thereof (Zhang et al, 2010). In murine embryonic fibroblasts (MEFs) loss of FIH-1 was shown to have significant and complex effects on expression of HIF target genes in the absence of VHL under normoxic conditions. Loss of VHL in MEFs was associated with reduced plating efficiency and there was an additive negative effect of loss of FIH-1, consistent with the effects that we observe of FIH-1 knockdown on population expansion in CCRCC cells. Strikingly, removal of HIF-1α restored plating efficiency to that of controls.
The crystal structure of FIH-1 has been solved and there are prototype inhibitors implying it will be druggable (Banerji et al, 2005; McDonough et al, 2005; Moon et al, 2010). An important consideration is that other FIH-1 substrates have been identified besides HIF-1α. In particular, it has been shown that asparagine residues in ankyrin repeat domains are hydroxylated by FIH-1 including the intracellular domain (ICD) of the Notch receptor, the IκB family of inhibitory proteins and tankyrase (Cockman et al, 2006, 2009; Coleman et al, 2007; Ferguson et al, 2007; Zheng et al, 2008). Therefore FIH-1 inhibition is likely to have wide-ranging effects that would have to be investigated before FIH-1 can be considered a suitable target for inhibition.
Useful insight into this is provided by the recently reported knockout mouse, which is viable but exhibits hypermetabolism (Zhang et al, 2010). Our study raises the possibility that FIH-1 would be a useful therapeutic target in clear cell renal carcinoma.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Banerji B, Conejo-Garcia A, McNeill LA, McDonough MA, Buck MR, Hewitson KS, Oldham NJ, Schofield CJ (2005) The inhibition of factor inhibiting hypoxia-inducible factor (FIH) by beta-oxocarboxylic acids. Chem Commun (Camb) 43: 5438–5440
Bracken CP, Fedele AO, Linke S, Balrak W, Lisy K, Whitelaw ML, Peet DJ (2006) Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environment. J Biol Chem 281: 22575–22585
Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe PJ (2006) Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci USA 103: 14767–14772
Cockman ME, Webb JD, Kramer HB, Kessler BM, Ratcliffe PJ (2009) Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol Cell Proteomics 8: 535–546
Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M, Cook KM, Cockman ME, Lancaster DE, Kessler BM, Oldham NJ, Ratcliffe PJ, Schofield CJ (2007) Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem 282: 24027–24038
Datta K, Li J, Bhattacharya R, Gasparian L, Wang E, Mukhopadhyay D (2004) Protein kinase C zeta transactivates hypoxia-inducible factor alpha by promoting its association with p300 in renal cancer. Cancer Res 64: 456–462
Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ (2003) Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem 278: 1802–1806
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107: 43–54
Esteban MA, Tran MG, Harten SK, Hill P, Castellanos MC, Chandra A, Raval R, O’Brien TS, Maxwell PH (2006) Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res 66: 3567–3575
Ferguson III JE, Wu Y, Smith K, Charles P, Powers K, Wang H, Patterson C (2007) ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol Cell Biol 27: 6407–6419
Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ (2002) Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA 99: 5367–5372
Frew IJ, Krek W (2008) pVHL: a multipurpose adaptor protein. Sci Signal 1: pe30
Frew IJ, Thoma CR, Georgiev S, Minola A, Hitz M, Montani M, Moch H, Krek W (2008) pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J 27: 1747–1757
Greijer AE, van der Wall E (2004) The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol 57: 1009–1014
Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ (2002) Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem 277: 26351–26355
Iliopoulos O, Kibel A, Gray S, Kaelin Jr WG (1995) Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1: 822–826
Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK (2005) Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J 24: 3846–3858
Kondo K, Kim WY, Lechpammer M, Kaelin Jr WG (2003) Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 1: E83
Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin Jr WG (2002) Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 1: 237–246
Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH (2000) Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19: 5435–5443
Lancaster DE, McNeill LA, McDonough MA, Aplin RT, Hewitson KS, Pugh CW, Ratcliffe PJ, Schofield CJ (2004) Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem J 383: 429–437
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK (2002a) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16: 1466–1471
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML (2002b) Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295: 858–861
Li J, Wang E, Dutta S, Lau JS, Jiang SW, Datta K, Mukhopadhyay D (2007) Protein kinase C-mediated modulation of FIH-1 expression by the homeodomain protein CDP/Cut/Cux. Mol Cell Biol 27: 7345–7353
Linehan WM, Rubin JS, Bottaro DP (2009) VHL loss of function and its impact on oncogenic signaling networks in clear cell renal cell carcinoma. Int J Biochem Cell Biol 41: 753–756
Linke S, Hampton-Smith RJ, Peet DJ (2007) Characterization of ankyrin repeat-containing proteins as substrates of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible transcription factor. Methods Enzymol 435: 61–85
Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, Wykoff CC, Maher ER, Harris AL, Ratcliffe PJ, Maxwell PH (2002) HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 1: 459–468
Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD (2002) The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 1: 247–255
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399: 271–275
McDonough MA, McNeill LA, Tilliet M, Papamicael CA, Chen QY, Banerji B, Hewitson KS, Schofield CJ (2005) Selective inhibition of factor inhibiting hypoxia-inducible factor. J Am Chem Soc 127: 7680–7681
Moon H, Han S, Park H, Choe J (2010) Crystal structures of human FIH-1 in complex with quinol family inhibitors. Mol Cells 29: 471–474
Morris MR, Maina E, Morgan NV, Gentle D, Astuti D, Moch H, Kishida T, Yao M, Schraml P, Richards FM, Latif F, Maher ER (2004) Molecular genetic analysis of FIH-1, FH, and SDHB candidate tumour suppressor genes in renal cell carcinoma. J Clin Pathol 57: 706–711
Rankin EB, Tomaszewski JE, Haase VH (2006) Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 66: 2576–2583
Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ (2005) Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 25: 5675–5686
Semenza GL (2007) Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29: 625–634
Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL (2001) HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 61: 6669–6673
Stolze IP, Tian YM, Appelhoff RJ, Turley H, Wykoff CC, Gleadle JM, Ratcliffe PJ (2004) Genetic analysis of the role of the asparaginyl hydroxylase factor inhibiting hypoxia-inducible factor (HIF) in regulating HIF transcriptional target genes. J Biol Chem 279: 42719–42725
To KK, Huang LE (2005) Suppression of hypoxia-inducible factor 1alpha (HIF-1alpha) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J Biol Chem 280: 38102–38107
Volm M, Koomagi R (2000) Hypoxia-inducible factor (HIF-1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer Res 20: 1527–1533
Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH (1998) Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood 92: 2260–2268
Yan Q, Bartz S, Mao M, Li L, Kaelin Jr WG (2007) The hypoxia-inducible factor 2alpha N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol Cell Biol 27: 2092–2102
Zhang N, Fu Z, Linke S, Chicher J, Gorman JJ, Visk D, Haddad GG, Poellinger L, Peet DJ, Powell F, Johnson RS (2010) The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab 11: 364–378
Zheng X, Linke S, Dias JM, Gradin K, Wallis TP, Hamilton BR, Gustafsson M, Ruas JL, Wilkins S, Bilton RL, Brismar K, Whitelaw ML, Pereira T, Gorman JJ, Ericson J, Peet DJ, Lendahl U, Poellinger L (2008) Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci USA 105: 3368–3373
Acknowledgements
We would like to extend our gratitude to Cenix Bioscience GmbH (Dresden, Germany); in particular CEO Dr Christophe J Echeverri and research assistant Corina Frenzel, for their expert advice regarding our RNAi experiments. We acknowledge Maria Simoes for technical support. We thank Dr Ewa Paleolog, Dr Kyra M Archibald and Dr Simon Hallam for critical reading of our manuscript. The British Heart Foundation and the European Union large-scale collaborative project METOXIA funded this research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Khan, M., Bhattacharyya, T., Andrikopoulos, P. et al. Factor inhibiting HIF (FIH-1) promotes renal cancer cell survival by protecting cells from HIF-1α-mediated apoptosis. Br J Cancer 104, 1151–1159 (2011). https://doi.org/10.1038/bjc.2011.73
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.73
Keywords
This article is cited by
-
Regulation of the hypoxic tumor environment in hepatocellular carcinoma using RNA interference
Cancer Cell International (2017)
-
HIF-1α induction during reperfusion avoids maladaptive repair after renal ischemia/reperfusion involving miR127-3p
Scientific Reports (2017)
-
Genetic modification of hypoxia signaling in animal models and its effect on cancer
Clinical and Translational Oncology (2015)
-
miR-31 is consistently inactivated in EBV-associated nasopharyngeal carcinoma and contributes to its tumorigenesis
Molecular Cancer (2014)
