
Abstract
Although the copper/zinc superoxide dismutase-1 (SOD1) gene has been identified in both familial ALS (FALS) and sporadic ALS (SALS), it has rarely been studied in Chinese patients with ALS, and there are few studies with large samples. This study sought to assess the prevalence of SOD1 mutations in Chinese ALS patients. We screened a cohort of 499 ALS patients (487 SALS and 12 FALS) from the Department of Neurology at the West China Hospital of Sichuan University and analyzed all coding exons of SOD1 by Sanger sequencing. In addition, we reviewed the mutation frequencies of common ALS causative genes in Chinese populations. Eight missense mutations in SOD1 were found in 8 ALS individuals: two novel mutations (p.G73D and p.V120F) and six previously reported mutations. The frequencies of SOD1 mutations were 1.03% (5/487) in SALS and 25% (3/12) in FALS from Southwest China. A literature review indicated that the mutation rates of major ALS causative genes were 53.55% in FALS and 6.29% in SALS. In Chinese SALS and FALS, the highest mutation frequency was in the SOD1 gene. Our results suggest that SOD1 mutation is the most common cause of ALS in Chinese populations and that the mutation spectrum of ALS varies among different ethnic populations.
Similar content being viewed by others
Introduction
Amyotrophic lateral sclerosis (ALS) is a genetically heterogeneous neurodegenerative disorder that is clinically characterized by progressive neurological deterioration, the coexistence of upper and lower motor neuron signs, and death from respiratory failure, typically 3–5 years after the onset of symptoms1. Most cases of ALS are sporadic, but approximately 10% of cases are familial (FALS)1. To date, mutations in 29 genes have been linked to the pathogenesis of ALS2. In Caucasians, these mutations are found in a large fraction (approximately 2/3) of FALS patients; however, they are found in only approximately 11% of sporadic ALS (SALS) patients3.
The copper/zinc superoxide dismutase-1 (SOD1) gene is composed of 5 exons coding for a homodimeric enzyme in which each subunit is composed of 154 evolutionarily conserved amino acids and binds with a catalytic Cu+ ion and a stabilizing Zn2+ ion. The SOD1 gene was first found to be a causative gene for FALS in 19934. Mutations in the SOD1 gene account for 12–23.5% of FALS and ~7.3% of apparent SALS, on the basis of multiple large population studies in Caucasians5. SOD1 was reported to be the most commonly mutated ALS gene until a large hexanucleotide (GGGGCC) repeat expansion (HRE) in the chromosome 9 open reading frame 72 (C9orf72) gene was identified in 2011. This HRE is found in ~46.4% of FALS cases and ~21.1% of SALS cases, and it is the most common cause of ALS among Caucasian populations6. In the past twenty years, other ALS genes have been discovered, including TAR DNA-binding protein (TARDBP), Fused in Sarcoma (FUS), Angiogenin (ANG), Valosin-containing protein (VCP), Sequestosome 1 (SQSTM1), and Profilin 1 (PFN1). However, these genes rarely account for FALS or SALS7,8. The relative contributions of these ALS causative genes to ALS vary among different populations. Patients with ALS who are from Asia, especially from China, are important components of the rare disease database. However, large-scale screening for mutations of the common causative genes is less common in Chinese patients than in Caucasians. Previous studies have shown that in Chinese SALS patients, the frequency of TARDBP is 0–0.93%9,10,11,12,13,14, that of FUS is 1.55–1.85%9,11, that of ANG is 0.31%9, that of VCP is 0%9, that of SQSTM1 is 0.98–1.38%15,16, that of PFN1 is 0–0.19%9,17, that of matrin 3 (MATR3) is 0.20%18, that of TANK-binding kinase 1 (TBK1) is 0.57%19 and that of coiled-coil-helix-coiled-coil-helix domain-containing protein 10 (CHCHD10) is 0–0.41%20,21. For the C9orf72 HRE, five independent studies have revealed that its frequency in SALS is 0–1.53%9,11,22,23,24. Therefore, these nine common causative genes may account for approximately 6% of Chinese SALS cases. However, regarding SOD1, the first identified causative gene, only two studies from northern and central-southern China with a total of 482 SALS patients have investigated the mutation frequency (6/482) of SOD1 in mainland China9,25. Thus, the assumption that there are large differences in the frequencies of these causative genes in SALS between Chinese and Caucasian patients may be premature. In the current study, we sought to evaluate the prevalence of SOD1 mutations in southwest Chinese ALS patients by screening for mutations of the SOD1 gene in 499 ALS patients and reviewing the mutation frequencies of common ALS causative genes in Chinese populations.
Methods
Subjects
This study included a total of 499 Chinese patients diagnosed with ALS (487 with SALS and 12 with FALS) according to the EI Escorial revised criteria26 from the Department of Neurology at the West China Hospital of Sichuan University. Detailed patient clinical data, including sex, age of onset, initial symptoms and survival time, were analyzed. Patients with an identifiable family history of ALS among first-, second- or third-degree relatives were classified as having familial ALS (FALS)27. A total of 466 unrelated Chinese healthy control subjects (HCs) matched in sex, age, and residential area were included as the control group (46.57% women; mean age 52.78 ± 12.44 years). A neurologist examined all HCs to rule out neurological disorders. This study was approved by the Ethics Committee of Sichuan University and was conducted in accordance with the relevant guidelines. Written informed consent was obtained from all participants.
Mutation screening
Blood samples were collected from patients and HCs. Genomic DNA was extracted using standard phenol-chloroform protocols. Published primer sequences were used for the amplification of all 5 coding exons and intronic flanking regions of the SOD1 gene (NM_000454.4)28,29. The polymerase chain reaction (PCR) products were directly sequenced using an ABI3100 automated DNA sequencing system (Tsingke, Chengdu, China). To determine whether each novel variant was a causative mutation or a neutral polymorphism, the PCR restriction fragment length polymorphism (PCR-RFLP) method was used. The primer sequences, restriction enzymes, and length of PCR products are summarized in Supplementary Table 1. The RFLP results were confirmed by direct sequencing of the PCR products. The detrimental role of the novel mutation was predicted with the Sorting Intolerant from Tolerant (SIFT)30 and PolyPhen-2 bioinformatics prediction tools31. All the patients who were identified as carrying mutations of the SOD1 gene were screened for mutations of other common ALS causative genes, including C9orf72, TARDBP, FUS, PFN1 and SQSTM1 (Supplementary Tables 2 and 3).
Literature search
A comprehensive literature review was performed using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), Medline (National Library of Medicine), and China National Knowledge Internet (www.cnki.net) with the individual search terms “SOD1”, “superoxide dismutase 1 gene”, “FUS”, “fused in sarcoma”, “TARDBP”, “TAR DNA binding protein”, “ANG”, “angiogenin”, “VCP”, “valosin-containing protein”, “PFN1”, “profilin 1”, “C9orf72”, “CHCHD10”, “TBK1”, and “TANK-binding kinase 1”, each combined with “amyotrophic lateral sclerosis” or “ALS” or “motor neuron disease” or “MND” and “Chinese”. To eliminate repeat data, when 2 or more simultaneously published studies had the same first author’s name and similar participant characteristics and data, we included the study with the most comprehensive description of the data. Single case reports were excluded; only case series and case-control studies were included in the review.
Statistical analysis
Dichotomous variables such as sex and site of onset were analyzed using a standard chi-square test or Fisher’s exact test. Continuous data were compared using Student’s t-test. The results of all continuous data are presented as the mean ± standard deviation (SD), and a two-tailed p < 0.05 was considered to be statistically significant. All analyses were performed using SPSS 19.0 (SPSS, Inc., Chicago, IL).
Results
The demographic and clinical characteristics of patients included in the study are presented in Table 1. In the 499 patients examined, we identified 8 different SOD1 heterozygous point mutations in 8 ALS patients (Table 2), including 3 FALS and 5 SALS patients. For historical reasons, the numbers of SOD1 mutations are coded without the initial methionine32. For consistency with modern approaches without disregarding the traditional naming convention, we designate the technically correct numbering preceded by “p.”, followed by the traditional naming in parentheses. Thus, the mutations were identified as follows: c.115C > G[p.L39V], c.125G > A[p.G42D], c.131A > G[p.H44R], c.199C > T[p.P67S], c.218G > A[p.G73D], c.335G > A[p.C112Y], c.341T > C[p.I114T] and c.358G > T[p.V120F].
The age of onset in ALS patients carrying SOD1 mutations ranged from 33.1 years to 59.4 years, and the mean was 45.5 ± 8.5 years. The mean disease duration of five patients with SOD1 mutations was 25.20 ± 21.81 months (range of 9 to 63 months), excluding the three patients with SOD1 mutations who were still alive. The eight patients carrying the mutations of the SOD1 gene in the current study had no mutations in the C9orf72, TARDBP, FUS, PFN1 and SQSTM1 genes. The clinical features of patients carrying mutations of the SOD1 gene are summarized in Table 2.
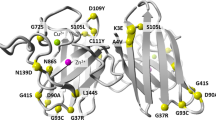
The pathogenic roles of p.L39V (L38V), p.G42D (G41D), p.H44R (H43R) and p.I114T (I113T) have been confirmed in ALS with reference to the Single Nucleotide Polymorphism database (dbSNP, http://www.ncbi.nlm.nih.gov/SNP/). In addition, previous studies have reported that p.P67S (P66S) and p.C112Y (C111Y) are likely to be pathogenic33,34. However, p.G73D and p.V120F had not been reported previously (Fig. 1).

(A) Forward sequence chromatograms of the portions of these PCR products show the heterozygous G > A (p.G73D) in the patient (case 1) but not in the HC. (B) Forward sequence chromatograms of the portions of these PCR products show the heterozygous G > T (p.V120F) in the patient (case 2) but not in the HC. (C) Protein sequence alignment of SOD1 in vertebrate species and the evolutionary conservation of the SOD1 mutations p.G73D and p.V120F.
To investigate the contributions of the causative genes for ALS in Chinese patients, we reviewed the previously reported mutation frequencies of common causative genes, including SOD1, TARDBP, FUS, ANG, VCP, SQSTM1, PFN1, TBK1, CHCHD10 and C9orf72, in Chinese populations. The mutation rates of these ALS causative genes were 53.55% in FALS and 6.29% in SALS. In the SALS patients, we found that the highest mutation frequency was in the SOD1 gene, followed by the FUS, SQSTM1, TBK1, C9orf72, TARDBP, ANG, CHCHD10, PFN1 and VCP genes. In the FALS patients, the highest mutation frequency was also found in the SOD1 gene, followed by the TARDBP, FUS and C9orf72 genes (Supplementary Table 4).
Discussion
Although studies on SOD1 gene mutations in ALS are numerous, to our knowledge, this is the first study on the mutation frequency and clinical features of patients with SOD1 gene mutations in a large Chinese cohort. In this study, the frequency of SOD1 gene mutations was found to be 25% (3/12) in FALS and 1.03% (5/487) in SALS patients from southwest China.
Together with the findings of the current study, we found that the SOD1 gene was the most common causative gene, accounting for 1.45% of SALS and 25.33% of FALS in Chinese patients. However, the proportion was lower than that in Caucasians, especially in SALS (0–7.3%, average 3.06% [35/1142])5. In addition, C9orf72 was the most common gene in Caucasians (SALS: 0–21.1%, average 7.11%[230/3237]; FALS: 21.7–57.9%, average 39.31%[217/562])6, but it contributed to only 0.53% of SALS and 5.98% of FALS in Chinese patients, a frequency even lower than that of TARDBP or FUS (Supplementary Table 4). Therefore, the two most common causative genes, SOD1 and C9orf72, accounted for approximately 2% of SALS and 31% of FALS in Chinese patients and for approximately 10% of SALS and 60% of FALS in Caucasians. This difference in the genetic backgrounds prompted us to search for potential causative genes that account for a large proportion of ALS cases in Chinese patients. Based on these findings, we screened the sequences of the most common causative genes other than C9orf72 and SOD1 – FUS and SQSTM1 – in Chinese ALS patients. In addition, due to the small sample of FALS subjects recruited in all included Chinese studies, the mutation frequency may have some bias. Therefore, a large sample of familial Chinese ALS patients should be assessed for confirmation. Overall, it is clear that the mutation spectrum of ALS varies among different ethnic populations8.
The p.L39V (L38V) mutation has been reported in some FALS patients and is associated with earlier disease onset (mean onset age: 41.5–44.9 years), a mean duration of 2.8 years and classical ALS symptoms4,35,36,37, consistent with the presentation of our patient carrying the p.L39V (L38V) mutation. The p.G42D (G41D) mutation has often been reported in FALS patients and is associated with the initial symptoms of lower limb weakness (mean onset age: 46.0 years), very slowly ascending paresis, and survival for as long as a decade or more37,38,39; this presentation is different from the late onset and shorter survival of our patient carrying the p.G42D (G41D) mutation. This difference might have occurred because the early age of onset predicts longer survival, consistent with the previous observations37. The p.H44R (H43R) mutation was first identified in a 58-year-old Japanese female FALS patient who had initial symptoms of lower limb weakness and who died of respiratory failure 7 months after the onset40, results similar to the observations in our patient carrying the p.H44R (H43R) mutation except for the negative family history. The p.P67S (P66S) mutation has been reported in a Serbian patient who presented with early onset (21 years) and rapid disease progression (died within 17 months)34. Our patient with the p.P67S (P66S) mutation also presented with rapid progression. However, in a South Korean family, patients with p.P67S (P66S) in the SOD1 gene have been reported to show slower progression41. Previous reports have found that FALS patients with p.C112Y (C111Y) and p.I114T (I113T) mutations of the SOD1 gene show extreme variability in the age of onset, clinical manifestations and disease progression33,37,42,43. The p.I114T (I113T) mutation often results in a late age of onset37,44. A male SALS patient with the p.I114T (I113T) mutation was reported to have the initial symptom of bilateral arm weakness at the age of 64, and this patient died of respiratory failure within 7 months45; however, our patient with the p.I114 mutation presented with slow progression with the initial site of the left lower limb.
Aside from these six previously reported mutations, the pathogenic roles of the novel mutations p.G73D and p.V120F were suggested by the following observations. First, the novel variants were absent in our HCs, as assessed using the PCR-RFLP method as well as available repositories including the dbSNP (build 146), the 1000 Genomes database and the Exome Aggregation Consortium (ExAC) database. Second, the amino acids “G” of p.G73 and “V” of p.V120 are highly conserved across species (Fig. 1). We did not find any mutations of other ALS causative genes in patients carrying these heterozygous mutations. Third, two prediction tools, SIFT and PolyPhen-2, predicted detrimental roles of these mutations. Finally, for p.G73D, in the same region, the pathogenic mutation p.G73S (G72S) has been reported in an SALS patient with early onset (28 years) and rapid disease progression (died in 15 months)46. As previously noted, G73 is located in the SOD1 zinc-binding sub-loop, which plays a critical role in stabilizing the conformation of SOD1. Changes in amino acids in this region may lead to alterations in zinc-binding activity. Some studies have proposed that abnormalities in this region of SOD1 may underlie its ALS-related toxicity34,47,48. For p.V120F, the onset was very early, suggesting that this mutation may play an important role in the development of ALS. Nonetheless, further studies are required to clarify the pathogenic effects of these mutations.
Currently, the best-studied mechanism for ALS involves SOD149, and more than 170 missense mutations have been reported in SOD1 (http://alsod.iop.kcl.ac.uk) to date. SOD1 mutations can give rise to almost all described clinical ALS phenotypes, such as progressive muscular atrophy and bulbar palsy, but no clear correlations between the mutated codon and the phenotype have been found. Disease duration may vary among patients harboring the same SOD1 mutations50, although most of our patients who carried mutations of the SOD1 gene presented with clinical manifestations consistent with those of previously reported patients37. Notably, our patient who carried the p.G42D (G41D) mutation showed a relatively rapid disease progression (16 months) and deviated from the usual very slow progression. The disease phenotype and progression may be influenced by epigenetic factors such as sex, modifier genes, environmental factors, and other unknown factors51. Our study found two novel mutations in SOD1 from two SALS patients who presented with relatively slow disease progression. This observation supported the pathogenic roles of these mutations by primary analysis, though these results were not verified. Therefore, our findings expand the mutation spectrum of the SOD1 gene in ALS.
In conclusion, mutation of the SOD1 gene appears to be a common cause of ALS in Chinese individuals. Our findings expand the mutation spectrum of the SOD1 gene in ALS. Our findings also support the assumptions that the regions of SOD1 mutations are diverse among different geographical backgrounds and that the mutation spectrum of ALS varies among different ethnic populations.
Additional Information
How to cite this article: Wei, Q. et al. Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: a case-control study and literature review. Sci. Rep. 7, 44606; doi: 10.1038/srep44606 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Mitchell, J. D. & Borasio, G. D. Amyotrophic lateral sclerosis. Lancet (London, England) 369, 2031–2041, doi: 10.1016/s0140-6736(07)60944-1 (2007).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nature neuroscience 18, 631–636, doi: 10.1038/nn.4000 (2015).
Caballero-Hernandez, D. et al. The ‘Omics’ of Amyotrophic Lateral Sclerosis. Trends in molecular medicine 22, 53–67, doi: 10.1016/j.molmed.2015.11.001 (2016).
Rosen, D. R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364, 362, doi: 10.1038/364362c0 (1993).
Andersen, P. M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Current neurology and neuroscience reports 6, 37–46 (2006).
Majounie, E. et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. The Lancet. Neurology 11, 323–330, doi: 10.1016/s1474-4422(12)70043-1 (2012).
Chio, A. et al. Extensive genetics of ALS: a population-based study in Italy. Neurology 79, 1983–1989, doi: 10.1212/WNL.0b013e3182735d36 (2012).
Renton, A. E., Chio, A. & Traynor, B. J. State of play in amyotrophic lateral sclerosis genetics. Nature neuroscience 17, 17–23, doi: 10.1038/nn.3584 (2014).
Zou, Z. Y., Liu, M. S., Li, X. G. & Cui, L. Y. The distinctive genetic architecture of ALS in mainland China. Journal of neurology, neurosurgery, and psychiatry, doi: 10.1136/jnnp-2015-311654 (2015).
Xiong, H. L. et al. Association between novel TARDBP mutations and Chinese patients with amyotrophic lateral sclerosis. BMC medical genetics 11, 8, doi: 10.1186/1471-2350-11-8 (2010).
Soong, B. W. et al. Extensive molecular genetic survey of Taiwanese patients with amyotrophic lateral sclerosis. Neurobiology of aging 35, 2423.e2421–2426, doi: 10.1016/j.neurobiolaging.2014.05.008 (2014).
Tsai, C. P. et al. FUS, TARDBP, and SOD1 mutations in a Taiwanese cohort with familial ALS. Neurobiology of aging 32, 553.e513–521, doi: 10.1016/j.neurobiolaging.2010.04.009 (2011).
Huang, R. et al. TARDBP gene mutations among Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiology of aging 33, 1015.e1011–1016, doi: 10.1016/j.neurobiolaging.2010.07.007 (2012).
Ju, X. et al. Two distinct clinical features and cognitive impairment in amyotrophic lateral sclerosis patients with TARDBP gene mutations in the Chinese population. Neurobiology of aging 38, 216.e211–216, doi: 10.1016/j.neurobiolaging.2015.10.032 (2016).
Chen, Y. et al. SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiology of aging 35, 726.e727–726.e729, doi: 10.1016/j.neurobiolaging.2013.09.008 (2014).
Yang, Y. et al. Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotrophic lateral sclerosis & frontotemporal degeneration 16, 378–384, doi: 10.3109/21678421.2015.1009466 (2015).
Chen, Y. et al. PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiology of aging 34, 1922.e1921–1925, doi: 10.1016/j.neurobiolaging.2013.01.013 (2013).
Xu, L., Li, J., Tang, L., Zhang, N. & Fan, D. MATR3 mutation analysis in a Chinese cohort with sporadic amyotrophic lateral sclerosis. Neurobiology of aging 38, 218.e213–214, doi: 10.1016/j.neurobiolaging.2015.11.023 (2016).
Tsai, P. C. et al. Mutational analysis of TBK1 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiology of aging 40, 191.e111–196, doi: 10.1016/j.neurobiolaging.2015.12.022 (2016).
Jiao, B. et al. High prevalence of CHCHD10 mutation in patients with frontotemporal dementia from China. Brain: a journal of neurology 139, e21, doi: 10.1093/brain/awv367 (2016).
Zhou, Q. et al. Mutation Screening of the CHCHD10 Gene in Chinese Patients with Amyotrophic Lateral Sclerosis. Molecular neurobiology, doi: 10.1007/s12035-016-9888-0 (2016).
Jiao, B. et al. Identification of C9orf72 repeat expansions in patients with amyotrophic lateral sclerosis and frontotemporal dementia in mainland China. Neurobiology of aging 35, 936.e919–922, doi: 10.1016/j.neurobiolaging.2013.10.001 (2014).
He, J. et al. C9orf72 hexanucleotide repeat expansions in Chinese sporadic amyotrophic lateral sclerosis. Neurobiology of aging 36, 2660.e2661–2668, doi: 10.1016/j.neurobiolaging.2015.06.002 (2015).
Chen, Y. et al. Large C9orf72 repeat expansions are seen in Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiology of aging 38, 217.e215–222, doi: 10.1016/j.neurobiolaging.2015.11.016 (2016).
Hou, L. et al. Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in central-southern China. Sci Rep 6, 32478, doi: 10.1038/srep32478 (2016).
Brooks, B. R., Miller, R. G., Swash, M. & Munsat, T. L. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases 1, 293–299 (2000).
Kenna, K. P. et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. Journal of medical genetics 50, 776–783, doi: 10.1136/jmedgenet-2013-101795 (2013).
Xu, R. et al. Identification of Tau and SOD1 gene mutation in a small Chinese Han pedigree of adult amyotrophic lateral sclerosis. Neurocase 19, 497–504, doi: 10.1080/13554794.2012.701639 (2013).
Niu, Y.-F. et al. Screening of mutations in SOD1 gene and analysis of geno-type-phenotype correlation in Chinese patients with amyotrophic lateral sclerosis. Hereditas (Beijing) 33, 720–724, doi: 10.3724/sp.j.1005.2011.00720 (2011).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081, doi: 10.1038/nprot.2009.86 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249, doi: 10.1038/nmeth0410-248 (2010).
Al-Chalabi, A. et al. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta neuropathologica 124, 339–352, doi: 10.1007/s00401-012-1022-4 (2012).
Nakamura, A. et al. Marked intrafamilial phenotypic variation in a family with SOD1 C111Y mutation. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases 13, 479–486, doi: 10.3109/17482968.2011.656311 (2012).
Keckarevic, D., Stevic, Z., Keckarevic-Markovic, M., Kecmanovic, M. & Romac, S. A novel P66S mutation in exon 3 of the SOD1 gene with early onset and rapid progression. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases 13, 237–240, doi: 10.3109/17482968.2011.627588 (2012).
Aguirre, T., Matthijs, G., Robberecht, W., Tilkin, P. & Cassiman, J. J. Mutational analysis of the Cu/Zn superoxide dismutase gene in 23 familial and 69 sporadic cases of amyotrophic lateral sclerosis in Belgium. European journal of human genetics: EJHG 7, 599–602, doi: 10.1038/sj.ejhg.5200337 (1999).
Robberecht, W. et al. Cu/Zn superoxide dismutase activity in familial and sporadic amyotrophic lateral sclerosis. Journal of neurochemistry 62, 384–387 (1994).
Cudkowicz, M. E. et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Annals of neurology 41, 210–221, doi: 10.1002/ana.410410212 (1997).
Niu, Q. et al. The G41D mutation in the superoxide dismutase 1 gene is associated with slow motor neuron progression and mild cognitive impairment in a Chinese family with amyotrophic lateral sclerosis. Journal of neurology, neurosurgery, and psychiatry, doi: 10.1136/jnnp-2015-310545 (2015).
Andersen, P. M. et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotrophic lateral sclerosis and other motor neuron disorders: official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases 4, 62–73 (2003).
Mochizuki, Y. et al. [Clinical features and neuropathological findings of familial amyotrophic lateral sclerosis with an H43R mutation in Cu/Zn superoxide dismutase]. Rinsho shinkeigaku = Clinical neurology 43, 491–495 (2003).
Baek, W. et al. A novel exon 3 mutation (P66S) in the SOD1 gene in familial ALS. The Canadian journal of neurological sciences. Le journal canadien des sciences neurologiques 39, 245–246 (2012).
Takei, Y. et al. alpha-Synuclein coaggregation in familial amyotrophic lateral sclerosis with SOD1 gene mutation. Human pathology 44, 1171–1176, doi: 10.1016/j.humpath.2012.10.024 (2013).
Andersen, P. M. & Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nature reviews. Neurology 7, 603–615, doi: 10.1038/nrneurol.2011.150 (2011).
Ghani, M. et al. Mutation analysis of patients with neurodegenerative disorders using NeuroX array. Neurobiology of aging 36, 545 e549–514, doi: 10.1016/j.neurobiolaging.2014.07.038 (2015).
Nakamura, S. et al. An autopsy case of sporadic amyotrophic lateral sclerosis associated with the I113T SOD1 mutation. Neuropathology: official journal of the Japanese Society of Neuropathology 34, 58–63, doi: 10.1111/neup.12049 (2014).
Shaw, C. E. et al. Mutations in all five exons of SOD-1 may cause ALS. Annals of neurology 43, 390–394, doi: 10.1002/ana.410430319 (1998).
Estevez, A. G. et al. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 286, 2498–2500 (1999).
Tiwari, A. & Hayward, L. J. Mutant SOD1 instability: implications for toxicity in amyotrophic lateral sclerosis. Neuro-degenerative diseases 2, 115–127, doi: 10.1159/000089616 (2005).
Frutiger, K., Lukas, T. J., Gorrie, G., Ajroud-Driss, S. & Siddique, T. Gender difference in levels of Cu/Zn superoxide dismutase (SOD1) in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis: official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases 9, 184–187, doi: 10.1080/17482960801984358 (2008).
Luigetti, M. et al. SOD1 G93D sporadic amyotrophic lateral sclerosis (SALS) patient with rapid progression and concomitant novel ANG variant. Neurobiology of aging 32, 1924.e1915–1928, doi: 10.1016/j.neurobiolaging.2011.04.004 (2011).
Radunovic, A. & Leigh, P. N. Cu/Zn superoxide dismutase gene mutations in amyotrophic lateral sclerosis: correlation between genotype and clinical features. Journal of neurology, neurosurgery, and psychiatry 61, 565–572 (1996).
Acknowledgements
The authors thank all individuals in the cohort and their families for their participation in this study. The present study was supported by funding from the National Science Fund of China (no. 81371394) and the National Key Research and Development Program of China (no.2016YFC0901504).
Author information
Authors and Affiliations
Contributions
Qianqian Wei: performed the molecular studies, edited the article and collected and analyzed clinical data. Qingqing Zhou: performed the molecular studies and wrote the first draft. Yongping Chen: analyzed clinical data, interpreted data and edited the paper. Ruwei Ou: collected and analyzed clinical data and performed patient follow-ups. Bei Cao: collected and analyzed clinical data. Yaqian Xu: edited the article and collected clinical data. Jing Yang: collected clinical data. Hui-Fang Shang: planned the study and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wei, Q., Zhou, Q., Chen, Y. et al. Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: a case-control study and literature review. Sci Rep 7, 44606 (2017). https://doi.org/10.1038/srep44606
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep44606
This article is cited by
-
Computational screening of damaging nsSNPs in human SOD1 genes associated with amyotrophic lateral sclerosis identifies destabilising effects of G38R and G42D mutations through in silico evaluation
In Silico Pharmacology (2024)
-
NOTCH2NLC GGC repeats are not expanded in Italian amyotrophic lateral sclerosis patients
Scientific Reports (2023)
-
Better survival in female SOD1-mutant patients with ALS: a study of SOD1-related natural history
Translational Neurodegeneration (2019)
-
Unique characteristics of the genetics epidemiology of amyotrophic lateral sclerosis in China
Science China Life Sciences (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
