
Abstract
Sex differences in the prevalence of psychiatric disorders are well documented, with exposure to stress during gestation differentially impacting females and males. We explored sex-specific DNA methylation in the cord blood of 39 females and 32 males born at term and with appropriate weight at birth regarding their potential connection to psychiatric outcomes. Mothers were interviewed to gather information about environmental factors (gestational exposure) that could interfere with the methylation profiles in the newborns. Bisulphite converted DNA was hybridized to Illumina HumanMethylation450 BeadChips. Excluding XYS probes, there were 2,332 differentially methylated CpG sites (DMSs) between sexes, which were enriched within brain modules of co-methylated CpGs during brain development and also differentially methylated in the brains of boys and girls. Genes associated with the DMSs were enriched for neurodevelopmental disorders, particularly for CpG sites found differentially methylated in brain tissue between patients with schizophrenia and controls. Moreover, the DMS had an overlap of 890 (38%) CpG sites with a cohort submitted to toxic exposition during gestation. This study supports the evidences that sex differences in DNA methylation of autosomes act as a primary driver of sex differences that are found in psychiatric outcomes.
Similar content being viewed by others

Introduction
Epigenetics is a dynamic biological mechanism involved in the modulation of an individual’s genome to environmental stimuli. Much of the epigenome is established during embryogenesis and early gestation and is transmitted in a relatively stable fashion1. Among the epigenetic factors, DNA methylation is thought to contribute to stable long-term regulation of gene expression. A large study in families with twins reported methylation differences dependent on genetic factors and environment2. Due to the stability, DNA methylation might be related to the development of psychiatric diseases later in life3. Several studies pointed out to a correspondence at methylation levels between blood and brain within individuals, suggesting that, although with some restrictions, blood can be used as a source to study psychiatric diseases4,5.
During perinatal sensitive periods, the environment exerts a critical impact on the maturation of brain structure and function6,7,8,9,10. It has been well characterized in humans that exposure to external damaging agents, such as smoking, mercury and arsenic during gestation induced epigenetic alterations in newborns11,12,13. Likewise, early life adversity, including pre- and post-natal stress, increased the risk of psycopathologies in animal models14. In humans, the Dutch Hunger Winter studies demonstrated the relationship between famine exposure at different times of prenatal development and phenotypic outcomes. Famine exposure around the period of conception was associated with increased risk of adult schizophrenia15, and with persistent changes in DNA methylation for INSIGF, GNASAS, LEP, MEG3, IL10 and ABCA116. Furthermore, epidemiological studies indicated that psychological stress such as bereavement, unwanted pregnancies, military invasion and natural disasters increased the risk for the offspring to develop neurodevelopmental diseases later in life, such as schizophrenia, depression and anxiety17,18,19,20.
The relation between psychiatric diseases and adverse experience in early life were included in the Developmental Origins of Health and Disease (DOHaD) hypothesis. Most DOHaD diseases have a sex-biased susceptibility21,22, suggesting that the response to stress is different in males and females. Accordingly, when exposed to stress, males suffer from behavioral problems earlier in development - at one year of age23, whilst females show the effects at later life periods24,25. In addition, there is a clear temporal specificity and sexually dichotomous influence of the effects of prenatal stress26,27,28. Males seem to be more vulnerable to develop schizophrenic, autism and attention-deficit/hyperactivity disorder (ADHD) symptoms in response to perinatal stress, all these were associated with intra uterus growth restriction (IUGR)29,30.
Animal models also contributed to association between methylation alterations and different response to stress exposition in females and males. For instance, intrauterine exposure to Bisphenol A (BPA), an estrogenic endocrine disruptor, seems to induce DNA methylation changes in male cortex and in female hypothalamus as result of expression changes in ESR1 and DNMT131. In mice, treatment with valproic acid during gestation induced sex-specific morphological alterations at a discreet region of the midbrain, including a higher cell counts in males compared to females14. In mice, exposure to stress in the prenatal period caused elevation of DNMT1 expression levels in females’ placenta, but not males. Contrariwise, males showed response to maladaptive behavioral stress together with change in methylation and expression of the glucocorticoid receptor and corticotropin-releasing factor in the fetal brain28.
A correlation between changes in methylation and gene expression were suggested to be involved with the differential brain development32. Thus, differences in the regulation of gene expression between females and males could explain the variances in specific brain structure8,27,28,32,33, cognitive abilities and stress-coping strategies28 Some studies already evaluated sex differences at methylation levels between females and males in the cord blood34, prefrontal cortex32,35 and pancreatic isolates35, nevertheless, none considered exposition to stress. Whilst it is known that the developing brain has enhanced sex-biased sensitivity to environmental stimuli at specific periods of gestation that contributes to vulnerability to psychiatric diseases, the possibility that DNA methylation differences between females and males could be involved with psychiatric diseases, independently of exposition to stress, remains to be addressed.
To unveil the contribution of stress exposition and the sexual antagonism effects in DNA methylation as well as their association with psychiatric vulnerabilities, we conducted a genome-wide methylation analysis of cord-blood in a well-characterized cohort of children born at term and of adequate weight/size at birth. Adjustments for cell heterogeneity and stress exposure co-variables were performed followed by the identification of differentially methylated CpG sites (DMSs) between females and males. These methylation differences were explored in other cohorts, including brain development, neurodevelopmental disorders and a cohort where children were exposed to stress during gestation.
Results
General characteristics of the study population
We used a natural cohort of newborns collected at the University Hospital of São Paulo Medical School, where, typically, only mothers with a not at risk pregnancy do have labor. We randomly collected cord blood samples from 96 individuals. Then, we excluded six pre-term and two post-term newborns, as well as 11 small and seven large for gestational age newborns, since those presentations have already been associated with psychiatric outcomes and stress exposition36. The final cohort included 71 cases, comprised of 39 females and 32 males, all with adequate weigh/size for gestational age and born at term. Moreover, to maximize differences in methylation probably related to sexes differences, we corrected for other exposure variables that could interfere with the analysis (Table 1, Supplemental Table 1) explored by this study (females versus males). Although smoking were different between females and males (p = 0.034), only psychiatric disease in father’s family presented a significant value as given by the sva package analysis (Supplemental Figure 1) and was corrected accordingly. In addition, data were adjusted for batch effects and cell type composition. Only as a proof of concept, the sex of each sample was confirmed by methylation levels of sexual chromosomes. The study design was included as a chart showing the analyses flow (Supplemental Figure 2).
Methylation differences between sexes correlate with gene expression
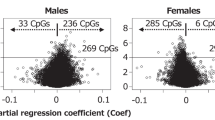
To characterize methylation changes in the cord blood of females and males, methylation levels were derived for 464,964 CpG sites, excluding XYS probes. Supervised comparison between sexes identified 2,332 differentially methylated CpG sites (DMSs) with females presenting twice the number of hypermethylated sites compared to males (adjP < 0.05, 1,546 and 786 CpG sites more methylated in females and males, respectively), with 1,499 CpG sites associated to 1,113 genes (Fig. 1; Supplemental Table S2). Upon clustering of the 2,332 CpG sites (Euclidian distance with average linkage), two clusters formed, completely separating both sexes (Supplemental Figure 3).

Volcano plot of the differential methylation analysis with X and Y axes displaying, respectively, the delta-beta values and the log10 of adjusted p-values for each CpG site. CpGs more methylated in females and males are represented in pink and blue, respectively.
To evaluate if DMSs were associated with differentially expressed genes between females and males, we used an expression dataset of 2,500 samples from 13 tissues37. Excluding genes located in the sexual chromosomes, the 1,113 genes were enrichment among differentially expressed genes between females and males in five tissues, four being brain regions (anterior cingulate cortex, frontal cortex, cerebellum and hippocampus), and the fifth being heart (Supplemental Table 3).
Considering that the 1,113 genes have a sex-biased expression, we used MiSig/WebGestalt to verify if there was an enrichment of binding sites for estrogen and androgen receptors and binding sites located at 2 Kb up or downstream from transcriptional start site (TSS) of each of gene. This analysis resulted in 105 genes regulated by estrogen receptor alpha or beta (five motifs, adjP < 0.05), 24 genes regulated by androgen receptor (three motifs, adjP < 0.05), and ten genes regulated by both transcriptional factors. Then, to explore more distant motifs, we used Opossum38 that searched for transcriptional factors target sites located at 5 Kb or 10 Kb up or downstream from TSS of 838 (out of 1,113) genes, revealing no enrichment for ESR1 or AR binding sites (Supplemental Table 4).
Differentially methylated CpG sites are not randomly distributed within the genome
Females have twice the number of methylated CpGs sites than males, with a non-random distribution in the genome. CpG sites more frequently methylated in females were more often located near transcriptional start sites (TSS1500) and within CpG islands, S_shores and S_shelves. Conversely, CpG sites more frequently methylated in males were enriched within gene bodies and 3′UTR, and were associated with open sea areas (Supplemental Table 5, chi-square test, p < 0.05). Based on the predefined regions, the differentially methylated regions (DMRs) between females and males were distributed as follow: 171 CpG islands (98 and 73 hyper- and hypomethylated in females), 131 promoters (80 and 51 hyper- and hypomethylated in females), 127 genes (64 and 63 hyper- and hypomethylated in females) and 333 genome-wide tiling regions (169 and 164 hyper- and hypomethylated in females) (Supplemental Figure 4, Supplemental Table 6, adjP < 0.05). The top DMRs (adjP < 0.05 and at least 10 CpG sites) are shown in Table 2.
Differentiated methylated sites between males and females are related to genes associated with normal brain development and neurodevelopment disorders
To gather a global view of the role of these genes, the 1,113 genes were annotated into gene ontology, revealing over-representation of genes related to system development, neurogenesis and neuron differentiation, striated muscle cell differentiation, and single organism behavioral and synaptic transmission. In addition, we found enrichment of genes belonging to the metabolic pathway (60 genes), the MAPK signaling pathway (23 genes), and focal adhesion (19 genes), among others (adjP < 0.01), including the insulin signaling pathway (12 genes). Finally, we observed an enrichment of genes related to neuropsychiatric disorders, stress, cancer, congenital disorders, musculoskeletal including cardiovascular diseases, endocrinological and female and male genitourinary diseases (Supplemental Table 7).
To test if the 1,113 genes were associated with brain development, we searched in the Allen Brain Atlas and found that 930 genes were expressed in at least one of the 26 brain regions (8th post-conception week to 40 years of age). Next, the comparison between the 2,332 DMS and the 7% of CpGs that changes the methylation during brain development39 showed an enrichment, represented by 238 CpGs (p < 0.00001, Supplemental Figure 5). Further, we compared to the 521 CpG sites that, in addition to being modulated during brain development, were also differentially methylated between brains of females and males, resulting in 195 common CpG sites (p < 0.00001, Supplemental Figure 5). Finally, extracting data from the only study in schizophrenia that had used the 450 K Beadchip arrays with publicly available data40 we searched for enrichment between 2,332 DMS between females and males and the 4,641 differentially methylated CpG sites in brain tissue from schizophrenic cases and controls. The analysis reported an overlap of 116 CpG sites between both studies, indicating an enrichment for CpG sites related to schizophrenia among CpG sites that differ between females and males in cord blood (p = 0.0139, Supplemental Figure 5).
Differentiated methylated sites between females and males are enriched for psychiatric disorders in a stress exposition model
To further explore the enrichment of DMSs between sexes after correction for stress exposition among genes associated with psychiatric disorders, we compared our DMSs to the differentially methylated CpG sites in the cord blood between females and males exposed to stress (toxic agents) during gestation from an independent cohort. A study from CHAMACOS (a longitudinal birth cohort study of the effects of exposure to pesticides and environmental chemicals on the health and development of Mexican-American children living in the agricultural region of Salinas Valley, CA) assessed the methylation changes related to sex differences in the cord blood of females and males, taking into account batch effects and cell composition. Excluding XYS CpG sites, they found 3,031 DMS (25.7% of the total) located in the autosomes, with most of them (82%) more methylated in females34. We compared these to our 2,332 DMS and found 890 (38%) common DMSs, all presenting the same pattern regarding hypo- and hypermethylation.
To assess whether there was an enrichment of genes associated with psychiatric diseases in both data sets, CHAMACOS‘ and ours, we used only those DMSs related to genes. Considering that one gene may have more than one CpG site, and different CpG sites may have opposing methylation differences (hypo- or hypermethylated), depending on their location, we excluded genes represented by more than one CpG site. This analysis resulted in 472 common genes between CHAMACOS‘ and ours, 1,309 and 500 genes belonging exclusively to CHAMACOS‘ or our datasets, respectively. Next, an enrichment analysis for diseases were performed with full datasets followed by the enrichment analysis with common and exclusive genes of the two cohorts that showed that, considering all diseases, most of the psychiatric diseases were enriched in all datasets, although there were differences in the ranking as exemplified by the top ten diseases (Supplemental Table 8, diseases and schizophrenia).
These gene sets were tested for enrichment in modules of co-expressed genes along human neocortical development41 (Supplemental Table 9). M8, M13 and M17 were enriched within the full datasets and among common genes. M8 is comprised of genes more expressed in the beginning of gestation; genes from M13 present a decreasing expression until postconception week (PCW) 16, from which point present an increasing pattern; and genes from M17 increase after PCW 16 until birth, from which point it seems to reach a plateau. Genes exclusively from this study (n = 500) were enriched for M9 (p = 0.0009), which contains genes highly expressed in the beginning of gestation that decrease expression levels at around 12th week whereas genes exclusively from CHAMACOS‘ (n = 1,309) were enriched for M15 (p = 0.0246), that presents an increasing level of expression during the whole neocortical development. Genes found only by CHAMACOS study (total or exclusive genes) were enriched for M1, that present a decreasing expression until week 12th, from which point present a constant increasing expression levels (Fig. 2).

Graphs show the distribution of genes related to the studies in enriched modules (p < 0.05). Scatter smooth of module eigengene values was defined by Parikshak et al.41 at different stages of neocortical development. Different periods of lifetime are scaled from light to dark blue colors: early foetal, early-mid foetal, late midfoetal, late foetal, neonatal/early infancy and late infancy. The red line indicates time of birth. Sets of genes enriched for the modules are displayed below of each graph.
Discussion
Differences underlying sexes have been associated with distinct incidence and severity of a group of psychiatry diseases, although the mechanisms are not well understood, moreover, differential methylation has been involved with this sex bias prevalence of psychiatry disorders. The immune system response during certain periods of development has sex-specific effects on regulation of various neurotransmitters important for the stress response throughout life, with consequences for physical and psychiatric disorders in later life36. Besides, there is a clear sex dimorphism in structures during brain development that were implicated to a sex-bias vulnerability to psychiatric disorders independent of stress exposition42. The potential underlying mechanisms driving these sex differences regarding stress responses and their relevance to disease were deeply discussed. The authors concluded that the continued focus and appreciation for how sex differences in stress responses may predict disease risk and resiliency is critical for developing preventive strategies and treatments43. In this study, we conducted a genome-wide DNA methylation profile of cord blood of a well-characterized cohort composed of babies born at term and with adequate weight, after correcting for stress exposition factors that could interfere with DNA methylation. We showed that differential methylation between females and males found in the cord blood can represent gene expression differences in tissues with sex dimorphism.
The DMS were equally distributed across all 22 autosomes, as previously described34, but their locations were not random. Whilst females had more methylated CpGs associated with repressed gene expression, males had more methylated CpGs in the open sea, which is more often positively correlated with expression44,45.
Pathways enriched for the 1,113 genes related to the DMS between sexes included metabolic and MAPK pathways. Changes in the metabolism of 1-carbon and methyl donor availability in the fetal liver, a major metabolic organ, were suggested to have a sex influence in a subset of genes46. Several pathways related to muscle were over-represented, such as the hypertrophic cardiomyopathy and vascular smooth muscle contraction pathways. The development and establishment of musculature is thought to be a major difference between adult males and females. At molecular level, these differences seem to be present already at birth. Accordingly, cardiovascular and musculoskeletal disorders were enriched in the diseases analyses.
The enrichment analysis revealed a possible role of our gene set to the brain as CpG sites and genes are modulated during brain development on top of being differently methylated between sexes39. Furthermore, these genes have been associated to psychiatry diseases as demonstrated by the enrichment of the 2,332 DMSs between females and males among the 4,641 differentially methylated CpG sites in brain tissue between patients with schizophrenia and controls40. Among the common genes, SHANK genes participate in synaptic formation, including in SHANK2 and SHANK3, in which mutations were described in a wide spectrum of neurodevelopmental and neuropsychiatric diseases47.
When analyzing the near and distant motifs for sex hormones, we suggested that: (1) the differential methylation found in CpG sites could interfere with the binding in motifs close to TSS of transcriptional factors classically related to females and males and; (2) other factors that are not exclusively associated with the regulation performed by sexual hormones are contributing to differentially expressed and methylated genes between females and males. Also, most of the DMS (66%) showed higher DNA methylation in females compared to males. It has been reported that females have higher methylation levels than males, although most studies used the X and Y chromosomes in their analyses, where a substantial portion of the DMSs are located34. Importantly some of these studies were performed in tissues other than cord blood, such as brain or pancreas, and with older subjects, that present changes in methylation because of the aging process35,48,49.
X dosage and the presence of Y chromosome exerts a role for the methylation patterns on the autosomes. One X was associated to hypomethylation on the autosomes but the presence of Y changes this pattern, suggesting the existence of an interaction between X and Y dosages50, meaning that, in addition to the organization effects of gonadal hormones, the sex chromosomes contribute to the differential methylation between females and males and these differences might contribute the sex bias psychiatric vulnerabilities, agreeing with a previous published discussion43.
The importance of considering the sex differences relies on the different response from females and males to stress to maintain homeostasis, which may have different consequences for the long-term responses to perturbations from the environment33. Prenatal stress can induce long-term neurodevelopmental diseases, particular those related to the hypothalamic-pituitary-adrenal (HPA) response to stress51. This exposure to maternal stress may alter the stress-vulnerability of the embryo leading to an increased risk of psychopathology36. The cohort from CHAMACOS was subjected to toxic stress (e.g. exposure to pesticides)34 whilst our study excluded factors already associated with psychiatric vulnerabilities (not born at term and not adequate size/weight for gestational age); and corrected for stress factors covariables that potentially interfere with DNA methylation. Psychiatric disorders were associated to genes from both studies. Nevertheless, comparing both data, we found 890 overlapping CpG sites, with our methylation differences (delta-beta) being smaller than theirs were, suggesting that exposition to stress might increase these differences. Comparing both datasets to the co-expressed gene modules during neocortical development revealed that some modules were similarly enriched. In addition, genes from these modules (M8: SOX1, CDKN2C, TRIP10, SALL1, EFNA4, PARD3B, CDH23, PRDM16, NEK9, C1orf61, HSPB1, CCDC8, FLNB and TEAD3; M13: RTKN, SYN2, SYNGR1, NRXN3, PRSS16, CPLX1, ZNF365 and SLC17A7; M17: CYP1A1, GRIN1, GRIN2D, PRPH, CSMD1, BTBD9, UPP2, and USP12) were found among genes enriched for the overrepresented psychiatric diseases. For instance, SYN2, more methylated in females (cg10245988, island), encodes a neuron-specific phosphoprotein that selectively binds to small synaptic vesicles in the presynaptic nerve terminal. Polymorphisms in this gene were associated with abnormal presynaptic function and increased risk for related neuronal disorders, including autism, bipolar disorder and schizophrenia52,53,54. SNPs in UPP2 were associated with a sex-bias factor in children diagnosed with autism spectrum disorder55. Nonetheless, the enrichment analysis of specific datasets reported different groups of genes regarding their expression levels during neurodevelopment, suggesting specific contributions.
We are aware that this study has limitations. First, we controlled for a limited number of stress factors and cannot guarantee that the methylation analyses were not influenced by unexplored stress exposition factors. Second, we could not measure contamination of maternal cells and thus, did not correct for it. Third, comparisons between data from cord blood and brain were not performed with samples collect from the same individuals. Fourth, we used only exposure to pesticides and environmental chemicals as a model toxic stress. Finally, we found only one study with publicly available data that used the H450K to evaluate DNA methylation in the brain of patients with schizophrenia representing psychiatric diseases found enriched in our analyses.
This study corroborated previous findings from the literature that showed that methylation differences between females and males in the cord blood methylation are associated with brain developing tissues. We describe there that genes related to this differential methylation are enriched for genes previously associated with psychiatric diseases, even after correcting the methylation analysis for stress exposition covariables, in additional to the technical effects. These differences are not solely explained by gonadal hormone effects, but also by sex chromosomes effects on the autosomes. Moreover, they present specific pattern of gene co-expression organized in modules of neocortical development, which implies temporal specific effects.
Material and Methods
Sample collection
Samples came from the Western region of Sao Paulo city, where 6,200 women were enrolled between 26th and 34th week gestation between 2012–2014 (Regiao Oeste Cohort, ROC). For this study, a questionnaire was administered to mothers to evaluate levels of stress and toxic exposures during gestation (smoke, use of drugs). These children are being followed up to gather additional information related to neurodevelopmental diseases and behavioral phenotypes as a continuum of this study; this will not be explored in here due to the relatively small time-lapsed. We collected blood from the umbilical cord of 96 women that delivered at the University Hospital of Sao Paulo. All women gave written informed consent to participate in the study. The study itself, as well as the use of samples, were conducted with ethical approval granted by the Hospital’s Internal review board – CEPHU under protocol number 1076/10, with all experiments performed in compliance with the Helsinki Declaration.
Sample preparation and quality control
DNA was isolated from umbilical cord blood samples using QIAamp DNA Blood Midi Kit (Qiagen). NanoDrop (Thermo Fisher Scientific) and Qubit (Thermo Fisher Scientific) was used to assess DNA purity and quantity. DNA quality was checked by electrophoresis in 0.8% agarose gels followed by bisulfite conversion whereby 1 ug DNA was treated using the EZ DNA Mehylation kit (Zymo Research Corp), following manufacture recommendations.
High quality bisulfite-converted DNA from 96 samples were hybridized in the Human Methylation 450 BeadChip microarrays (HM450K, Illumina), following the Illumina Infinium HD methylation protocol. Both experiments were processed by Deoxi Biotecnologia (www.deoxi.com) according to manufacturer’s instructions. Raw data were extracted by the iScan SQ scanner (Illumina) using GenomeStudio software (v.2011.1), with the methylation module v.1.9.0 (Illumina), into IDAT files, which were used for further analyses. The HM450K platform interrogates DNA methylation levels of 485,577 loci distributed across the genome at single-nucleotide resolution. Probes were annotated according regarding their nearest genes using FDb.InfiniumMethylation.hg19 package with an additional annotation for those probes that were located at more than one gene, Hg19 coordinates from UCSC were selected. This resulted in 20,461 unique entrezID genes. The methylation levels for each CpG probe were provided in beta-values (0 indicating unmethylated CpGs, and 1, fully methylated CpGs). All analyses were performed in R environment using Bioconductor packages (http://www.bioconductor.org). All samples had high quality data passing QC using standard parameters.
Genome-wide methylation analysis
The RnBeads package56 was applied to the dataset. Unreliable measurements were identified using Greedycut (p > 0.05) that removed 1,478 probes. Probes located in SNPs (n = 4,776) as well those located in specific contexts (n = 5,926 – non-CpG sites as well as additional SNPs) were also excluded. Samples were retained only if >99% of sites assayed had detection p > 0.05. The background was corrected using noob method, which is based on a normal-exponential convolution using out-of-band probes57. Signal intensities values from probes type I and II were normalized using SWAN method that adjusts the intensities based on a quantile approach58. Probes located in the sexual chromosomes were filtered out remaining 464,964 probes for analyses.
To quantify methylation differences resultant from cellular composition of the blood, a purified dataset containing six blood cell types was used as ref. 59 to estimate the blood cell type contribution for each cord blood sample. Based on the estimated percentages of each cell type, a projection of the methylation levels for each cord blood sample was used in the limma based analysis of differential DNA methylation, as implemented by RnBeads56. Variables from the questionnaire were tested for association with the main group comparison (female versus male) indicating that only psychiatric disease in the father family was acting as co-variables. Moreover, adjustment for technical effects (e.g. batch effects) and cell type composition were performed before the differential methylation analyses.
To identify differentially methylated CpG sites (DMSs), the M-values (loggit of B-values) were used employing an empirical Bayesian framework linear model from limma60. CpG sites with FDR adjusted p-value (adjP) <0.05 were selected as differentially methylated. RnBeads also identified differentially methylated regions (DMRs) in pre-defined CpG islands, promoters, genes and genome-wide tiling regions, that exhibit DNA methylation changes between females and males (FDR adjP < 0.05).
In silico functionally exploration of data
Functional and disease enrichment analysis were performed in WebGestalt61 using the whole genome as background. Significant values were considered to be those features comprised by at least 10 genes and adjusted p-value (adjP) <0.01 adjusted by the multiple test as given by Benjamini–Hochberg procedure.
CpGs were compared to differentially methylated CpG sites found by three studies with publicly available data34,40,62 using all probes from 450 K BeadChip as background. In addition, genes associated to DMSs were compared to 17 modules comprised of co-expressed genes during human cortical development41. For both cases, we applied the Modular Single-set Enrichment Test (MSET)62, which assess enrichment for a gene or CpG site (here named as feature) list of interest within a set of features using a randomization testing. First, the probability of our list of features being significantly represented in each module were compared to 10,000 simulated sets of features generated randomly from the 450 K Beadchip array (for DMS) or RefSeq (for genes) as background. Then, a p-value was calculated based on the number of simulated randomized sets that contains equal or higher number of features belonging to our list of features in the list generated by each of the three studies. We considered as significant, those comparisons with a p-value < 0.05. For the comparison with modules of genes co-expressed during neocortex development, the module eigengene values from the original study41 were plotted in a scatter smooth plot for the significant modules.
Data access
The HumanMethylation450 BeadChip data set from this study is available in NCBI Gene Expression Omnibus (GEO) under accession number GSE85042.
Additional Information
How to cite this article: Maschietto, M. et al. Sex differences in DNA methylation of the cord blood are related to sex-bias psychiatric diseases. Sci. Rep. 7, 44547; doi: 10.1038/srep44547 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–32 (2007).
van Dongen, J. et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 7, 11115 (2016).
Babenko, O., Kovalchuk, I. & Metz, G. A. S. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci. Biobehav. Rev. 48, 70–91 (2015).
Walton, E. et al. Correspondence of DNA Methylation Between Blood and Brain Tissue and Its Application to Schizophrenia Research. Schizophr. Bull. 42, 406–414 (2016).
Kundakovic, M. et al. DNA methylation of BDNF as a biomarker of early-life adversity. Proc. Natl. Acad. Sci. USA 112, 6807–13 (2015).
Weinstock, M. The long-term behavioural consequences of prenatal stress. Neurosci. Biobehav. Rev. 32, 1073–1086 (2008).
Korosi, A. & Baram, T. Z. The pathways from mother’s love to baby’s future. Front. Behav. Neurosci. 3, 27 (2009).
Fox, S. E., Levitt, P. & Nelson, C. A. III . How the timing and quality of early experiences influence the development of brain architecture. Child Dev. 81, 28–40 (2010).
Loman, M. M. & Gunnar, M. R. Early Experience, Stress, and Neurobehavioral Development Center. Early experience and the development of stress reactivity and regulation in children. Neurosci. Biobehav. Rev. 34, 867–76 (2010).
Lucassen, P. J. et al. Perinatal programming of adult hippocampal structure and function; emerging roles of stress, nutrition and epigenetics. Trends Neurosci. 36, 621–631 (2013).
Ivorra, C. et al. DNA methylation patterns in newborns exposed to tobacco in utero. J. Transl. Med. 13, 25 (2015).
Bouwland-Both, M. I. et al. Prenatal parental tobacco smoking, gene specific DNA methylation, and newborns size: the Generation R study. Clin. Epigenetics 7, 83 (2015).
Cardenas, A. et al. Differential DNA methylation in umbilical cord blood of infants exposed to mercury and arsenic in utero. Epigenetics 10, 508–15 (2015).
Mowery, T. M. et al. Embryological exposure to valproic acid disrupts morphology of the deep cerebellar nuclei in a sexually dimorphic way. Int. J. Dev. Neurosci. 40, 15–23 (2015).
Susser, E. et al. Schizophrenia after prenatal famine. Further evidence. Arch. Gen. Psychiatry 53, 25–31 (1996).
Tobi, E. W. et al. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 18, 4046–53 (2009).
Mueller, B. R. & Bale, T. L. Early prenatal stress impact on coping strategies and learning performance is sex dependent. Physiol. Behav. 91, 55–65 (2007).
Varese, F. et al. Childhood adversities increase the risk of psychosis: a meta-analysis of patient-control, prospective- and cross-sectional cohort studies. Schizophr. Bull. 38, 661–71 (2012).
Koenig, J. I., Kirkpatrick, B. & Lee, P. Glucocorticoid hormones and early brain development in schizophrenia. Neuropsychopharmacology 27, 309–18 (2002).
Scott, K. M. et al. Association of childhood adversities and early-onset mental disorders with adult-onset chronic physical conditions. Arch. Gen. Psychiatry 68, 838–44 (2011).
Intapad, S., Ojeda, N. B., Dasinger, J. H. & Alexander, B. T. Sex differences in the developmental origins of cardiovascular disease. Physiology (Bethesda). 29, 122–32 (2014).
Gilbert, J. S. & Nijland, M. J. Sex differences in the developmental origins of hypertension and cardiorenal disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1941–52 (2008).
Gerardin, P. et al. Depression During Pregnancy. J. Clin. Psychiatry 72, 378–387 (2011).
Buss, C., Davis, E. P., Hobel, C. J. & Sandman, C. A. Maternal pregnancy-specific anxiety is associated with child executive function at 6-9 years age. Stress 14, 665–76 (2011).
Buss, C., Entringer, S., Swanson, J. M. & Wadhwa, P. D. The Role of Stress in Brain Development: The Gestational Environment’s Long-Term Effects on the Brain. Cerebrum 2012, 4 (2012).
Bock, J., Murmu, M. S., Biala, Y., Weinstock, M. & Braun, K. Prenatal stress and neonatal handling induce sex-specific changes in dendritic complexity and dendritic spine density in hippocampal subregions of prepubertal rats. Neuroscience 193, 34–43 (2011).
Mychasiuk, R., Gibb, R. & Kolb, B. Prenatal stress produces sexually dimorphic and regionally specific changes in gene expression in hippocampus and frontal cortex of developing rat offspring. Dev. Neurosci. 33, 531–8 (2011).
Mueller, B. R. & Bale, T. L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 28, 9055–65 (2008).
van Os, J. & Selten, J. P. Prenatal exposure to maternal stress and subsequent schizophrenia. The May 1940 invasion of The Netherlands. Br. J. Psychiatry 172, 324–6 (1998).
Leff, S. S. et al. Social cognitions, distress, and leadership self-efficacy: Associations with aggression for high-risk minority youth. Dev. Psychopathol. 26, 759–772 (2014).
Kundakovic, M. et al. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc. Natl. Acad. Sci. 110, 9956–9961 (2013).
Xu, H. et al. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum. Mol. Genet. 23, 1260–70 (2014).
Bale, T. L. Sex differences in prenatal epigenetic programming of stress pathways. Stress 14, 348–56 (2011).
Yousefi, P. et al. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genomics 16, 911 (2015).
Hall, E. et al. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 15, 522 (2014).
Monk, C., Spicer, J. & Champagne, F. A. Linking prenatal maternal adversity to developmental outcomes in infants: the role of epigenetic pathways. Dev. Psychopathol. 24, 1361–76 (2012).
Mayne, B. T. et al. Large Scale Gene Expression Meta-Analysis Reveals Tissue-Specific, Sex-Biased Gene Expression in Humans. Front. Genet. 7, 183 (2016).
Ho Sui, S. J. et al. oPOSSUM: identification of over-represented transcription factor binding sites in co-expressed genes. Nucleic Acids Res. 33, 3154–64 (2005).
Spiers, H. et al. Methylomic trajectories across human fetal brain development. Genome Res. 25, 338–52 (2015).
Wockner, L. F. et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl. Psychiatry 4, e339 (2014).
Parikshak, N. N. et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–21 (2013).
Shi, L. et al. Sex Biased Gene Expression Profiling of Human Brains at Major Developmental Stages. Sci. Rep. 6, 21181 (2016).
Bale, T. L. & Epperson, C. N. Sex differences and stress across the lifespan. Nat. Neurosci. 18, 1413–20 (2015).
Ball, M. P. et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 27, 361–8 (2009).
Maunakea, A. K., Chepelev, I., Cui, K. & Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–69 (2013).
Drake, A. J. et al. In utero exposure to cigarette chemicals induces sex-specific disruption of one-carbon metabolism and DNA methylation in the human fetal liver. BMC Med. 13, 18 (2015).
Sala, C., Vicidomini, C., Bigi, I., Mossa, A. & Verpelli, C. Shank synaptic scaffold proteins: keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 135, 849–58 (2015).
Liu, J., Morgan, M., Hutchison, K. & Calhoun, V. D. A Study of the Influence of Sex on Genome Wide Methylation. PLoS One 5, e10028 (2010).
Bacalini, M. G. et al. A meta-analysis on age-associated changes in blood DNA methylation: results from an original analysis pipeline for Infinium 450k data. Aging (Albany. NY). 7, 97–109 (2015).
Sharma, A. et al. DNA methylation signature in peripheral blood reveals distinct characteristics of human X chromosome numerical aberrations. Clin. Epigenetics 7, 76 (2015).
Weinstock, M. in 3–25, doi: 10.1007/978-1-4939-1372-5_1 (2015).
Le-Niculescu, H. et al. Towards understanding the schizophrenia code: an expanded convergent functional genomics approach. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 144B, 129–58 (2007).
Saviouk, V., Moreau, M. P., Tereshchenko, I. V. & Brzustowicz, L. M. Association of synapsin 2 with schizophrenia in families of Northern European ancestry. Schizophr. Res. 96, 100–11 (2007).
Steeb, H. et al. Serum proteomic analysis identifies sex-specific differences in lipid metabolism and inflammation profiles in adults diagnosed with Asperger syndrome, doi: 10.1186/2040-2392-5-4.
Chen, C., Van Horn, J. D. & GENDAAR Research Consortium. Developmental neurogenetics and multimodal neuroimaging of sex differences in autism. Brain Imaging Behav., doi: 10.1007/s11682-015-9504-3 (2016).
Assenov, Y. et al. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 11, 1138–1140 (2014).
Triche, T. J., Weisenberger, D. J., Van Den Berg, D., Laird, P. W. & Siegmund, K. D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 41, e90 (2013).
Maksimovic, J., Gordon, L. & Oshlack, A. SWAN: Subset-quantile Within Array Normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012).
Houseman, E. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Zhang, B., Kirov, S. & Snoddy, J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741–8 (2005).
Zhao, C., Eisinger, B. E., Driessen, T. M. & Gammie, S. C. Addiction and reward-related genes show altered expression in the postpartum nucleus accumbens. Front. Behav. Neurosci. 8, 388 (2014).
Acknowledgements
We thank our colleagues Alda Valéria Neves Soares Gomes and Chang Yi Wei from Obstetric Center of University Hospital of São Paulo, Brasil, for allowing the umbilical cord blood collection. We also thank Kyle Wiley from Yale University of USA for contributing to the writing of the manuscript. We thank funding agencies FAPESP (2015/06281-7, 2012/19472-7 (ROC), 2008/57896-8), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (573974/2008-0), Instituto de Psiquiatria do Desenvolvimento da Infância e da Adolescência (INPD), Saving Brains. Finally, we extend our gratitude to the mothers and their families who volunteered their time and efforts during this research.
Author information
Authors and Affiliations
Contributions
Wrote the manuscript: M.M., H.B. Collected information, material and performed experiments: L.B., A.B., G.G., R.F., A.E. and S.G., A.S. Analyzed data: M.M., A.T., V.E., H.B. Conceived and designed the study: H.B., A.B., G.F., C.M., G.P., E.M. Prepared figures, tables and supplemental material: M.M., E. B., V.E. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Maschietto, M., Bastos, L., Tahira, A. et al. Sex differences in DNA methylation of the cord blood are related to sex-bias psychiatric diseases. Sci Rep 7, 44547 (2017). https://doi.org/10.1038/srep44547
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep44547
This article is cited by
-
Sex effects on DNA methylation affect discovery in epigenome-wide association study of schizophrenia
Molecular Psychiatry (2024)
-
Sex differences in DNA methylation across gestation: a large scale, cross-cohort, multi-tissue analysis
Cellular and Molecular Life Sciences (2024)
-
Sexually dimorphic methylation patterns characterize the placenta and blood from extremely preterm newborns
BMC Biology (2023)
-
The role of genetics and epigenetics in sex differences in human survival
Genus (2023)
-
A higher dysregulation burden of brain DNA methylation in female patients implicated in the sex bias of Schizophrenia
Molecular Psychiatry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
