
Abstract
Oxidation of pyrite (FeS2) plays a significant role in the redox cycling of iron and sulfur on Earth and is the primary cause of acid mine drainage (AMD). It has been established that this process involves multi-step electron-transfer reactions between surface defects and adsorbed O2 and H2O, releasing sulfoxy species (e.g., S2O32−, SO42−) and ferrous iron (Fe2+) to the solution and also producing intermediate by-products, such as hydrogen peroxide (H2O2) and other reactive oxygen species (ROS), however, our understanding of the kinetics of these transient species is still limited. We investigated the kinetics of H2O2 formation in aqueous suspensions of FeS2 microparticles by monitoring, in real time, the H2O2 and dissolved O2 concentration under oxic and anoxic conditions using amperometric microsensors. Additional spectroscopic and structural analyses were done to track the dependencies between the process of FeS2 dissolution and the degradation of H2O2 through the Fenton reaction. Based on our experimental results, we built a kinetic model which explains the observed trend of H2O2, showing that FeS2 dissolution can act as a natural Fenton reagent, influencing the oxidation of third-party species during the long term evolution of geochemical systems, even in oxygen-limited environments.
Similar content being viewed by others
Introduction
The chemical and electrical properties of pyrite (FeS2) – the most common iron disulfide in the Earth’s crust – make it a promising material for the construction of photovoltaic panels1, as well as for wastewater remediation2,3,4,5,6,7. In the last decades, a number of experimental and field studies have addressed the oxidative dissolution of pyrite, providing an accurate characterization of the process8,9,10,11,12,13,14,15,16,17. Recent investigations have shown that, aside from the iron and sulfoxy species released during pyrite dissolution, ROS are always present as transient by-products, both under oxic and anoxic conditions18,19,20,21,22,23,24,25,26, and providing an important pathway to oxidize third-party species through oxygen evolution reaction (OER)18,27,28,29,30.
The rupture of S-Fe and S-S bonds over pyrite surface by mechanical fracture or during the dissolution process induces the formation of the non-stoichiometric defect sites (i.e., the dangling bonds) that trigger adsorption reactions31,32,33. The iron surface species formed cause a reduction in the band gap of the pyrite from 0.86 electron volts (eV) in the bulk to 0.55 eV on the surface34, catalyzing the dissociation of adsorbed oxygen (O2) and water (H2O) molecules and leading to the formation of H2O2 (equations 1,2,3,4)19,25,26,35:




At the same time, pyrite releases Fe2+into solution than can catalyze the Fenton and Haber-Weiss reactions, leading to the generation of other ROS (equations 5)4,22,25,26:




Although this sequence of reactions describes the basic features of H2O2 formation during pyrite dissolution (equations 1,2,3,4) and its further degradation to secondary ROS in solution (equations 5,6,7,8), various important aspects remain unclear, for instance: (1) can free radicals be formed by mechanism other than the photoactivation (i.e. by mechanical bond fracture or non-stoichiometry dissolution of pyrite)? (2) can the formation of H2O2 occur in strictly anoxic conditions overcoming the energy required to split the water molecule and the further release of O2? and (3) can the process of ROS generation by pyrite be sustained over long periods of time?.
Real-time measurements of this process are made difficult by ROS reactivity and the subsequent redox transformations of iron and sulfur species. Spectroscopic and fluorescence methods are commonly used to measure the concentration of H2O2; however, these methods usually need the presence of dyes or chelating agents that are not well suited for the kinetic analysis of transient phases. In this study, we measured the real-time generation and decomposition of H2O2 and dissolved O2 induced by pyrite surfaces under different boundary conditions (i.e., dark/light, oxic/anoxic) to investigate the kinetic role of ROS during pyrite dissolution. In addition, we analyzed both the chemical evolution of dissolved species and surface pyrite oxidation with spectroscopy (UV-Vis, XPS), Cyclic Voltammetry (CV) and high-resolution transmission electron microscopy (HTREM) and performed specific experiments to evaluate the persistence of H2O2 formation. Based on our results, we developed a kinetic model for the coupling between pyrite dissolution and H2O2 generation/degradation through the Fenton reaction. When combined with the observed trends this model leads to the definition of constraints on the overall process of ROS oxidation mechanism induced by pyrite surfaces.
Results
H2O2 and O2 evolution: general pathway
Figure 1 shows the concentration of H2O2 as a function of time in aqueous suspensions of pyrite starting at circumneutral pH, under oxic and anoxic conditions. H2O2 concentration increased at the beginning of the experiment until a maximum value was reached, and decreased thereafter asymptotically towards a nearly stationary, residual value ([H2O2]>200 nM) still measurable at the end of the experiment (~22 h). This coupled generation-decay response was generally observed in every experiment, although there were variations in the particular shape of the curves and, some experiments showed characteristic shoulders or secondary maxima at intermediate stages of the process. The overall process followed a sigmoidal trend, suggesting a strong interaction between the H2O2 generation and degradation rates characteristic of an autocatalytic process. H2O2 formation and decomposition evolved more slowly under anoxic than under oxic conditions. We did not observe any correlation between pyrite loading and the H2O2 yield between experimental runs under our experimental conditions (i.e., 0.5–0.3 g/L, unbuffered neutral pH) because of several factors (e.g., kinks, steps, lattice anisotropy) can determine the variability of the reactive surface between samples36,37,38,39.

Inset: particle loading (g/L) (a) oxic conditions, (b) anoxic conditions.
The evolution of O2 under oxic conditions was characterized by an asymptotic decrease followed by a slight increase and a steady stable period at the end of the experiment (Supplementary Fig. S1). The opposite trend was observed under anoxic conditions (i.e., an initial increase in O2 followed by an asymptotic decrease). To clarify the role of O2 over the formation of H2O2, we monitored simultaneously the evolution of O2 and H2O2 under oxic and anoxic conditions (Fig. 2). In oxic conditions (Fig. 2a), the amount of H2O2 increased whereas O2 was rapidly consumed at the beginning of the experiment. Under anoxic conditions O2 and H2O2 formed concomitantly (Fig. 2b).

The bottom figure shows the first two hours of the reaction, under (a) oxic (particle loading = 1.75 g/l) and (b) anoxic conditions (particle loading = 1.09 g/L).
During the experiments, pH became acidic under both oxic and anoxic conditions. Overall, pH dropped rapidly towards a nearly constant value approximately 2 to 3 pH units lower then at the initial pH value. The initial drop was more pronounced under anoxic than under oxic conditions (2 hours vs 10 hours, respectively), and in both cases the initial decline in pH values was accelerated with increasing pyrite loading (Supplementary Fig. S2).
The Fenton reaction in anoxic conditions: spectroscopic experiments
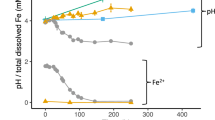
To determine if the Fenton reaction was actually the mechanism to degrade the H2O2 forming under anoxic conditions, we monitored the production of Fe3+and OH· species in pyrite suspensions under anoxic conditions and in the dark (Fig. 3). First, we evaluated the intensity of the absorbance bands in the UV-Vis range of 260–700 nm to identify the formation of dissolved Fe3+-complexes that coexist in solution in the range 300–450 nm40,41,42,43 (Fig. 3a). The first absorbance peak (λmax ~375 nm) evolved after three hours of reaction and was shifted to higher wavelengths (around 390–400 nm), as the reaction proceeded. Second, we monitored the formation of short-lived radicals, mainly OH· by measuring the decrease in the light absorption spectrum (λmax = 590 nm) of Crystal Violet used as a dye probe44 (Fig. 3b). The absorption spectrum of CV showed a rapid decline indicating that OH· formation occurs concomitantly from the start-up of the experiment.

(a) Absorption bands detected from aqueous pyrite suspension in anoxic conditions and in the dark showing Fe3+-complexes signatures (particle loading = 0.28 g/L). The numbers inserted over the absorption bands show the reaction time. Spectra were registered in real time using a liquid waveguide capillary flow cell (LWCC; path length: 250 cm; WPI), connected to the batch reactor by a peristaltic pump; (b) Degradation of CV solution upon pyrite aqueous reaction under anoxic conditions and in the dark (particle loading = 0.12 g/L, [CV]0 = 225.5 μM).
Tracking the reversibility of H2O2 generation: cycling experiments
We performed these experiments to better understand the long-term evolution of H2O2 during pyrite dissolution in an open system. In order to use the same physical pyrite particles and in the same geometry, pyrite microparticles were adhered over silicone strips to form a thin film which was placed on the internal wall of the reactor as in the spectroscopic experiments. We monitored the evolution of H2O2 until the observable amount of H2O2 attained a constant value or a concentration of zero (Fig. 4). At that point, we replaced the solution inside the batch reactor with distilled H2O, and monitored the evolution of H2O2 again (we repeated this procedure twice). After a full cycle of H2O2 generation and decay, the formation of H2O2 was found to resume after the addition of fresh distilled H2O, although the maximum H2O2 yield was consistently lower than in the previous cycle (Fig. 4a). Assuming a zero-order kinetic model, the initial observed rate of the H2O2 formation (k0 obs) showed a slight decrease between the first and the second cycle, which was more prevalent in the third cycle (Fig. 4b). This decrease could be attributed to the formation of oxide patches on pyrite surface that partially block some of the iron reactive centers.

(a) H2O2 evolution from the same pyrite slurry after renewing H2O twice, in oxic-open conditions (pyrite load particle = 0.33 g/L, ΔpH1cycle = 6.8–3.8, ΔpH2cycle = 6.8–6.1, ΔpH3cycle = 6.8–6.7); (b) Initial observed rate of H2O2 formation assuming a zero kinetic order rate.
Surface characterization (CV, XPS and HRTEM)
Results of cyclic voltammetry under anoxic conditions revealed the anodic peaks associated with H2O oxidation by one electron and by two electron transfer (Supplementary Fig. S3). Interestingly, in the cathodic counterpart, the peak assigned to iron reduction is split into two peaks (0.1 V NHE and 0.2VNHE, respectively), indicating that a fraction of ≡Fe3+is reduced in a nearly spontaneous manner. The analyzed XPS spectra of the (001) face of pyrite after aqueous reaction in the presence and absence of dissolved O2 (g) resulted in significant differences, showing major surface oxidation under oxic conditions with the subsequent formation of patches (Supplementary Fig. S4). In fact, XPS analysis of the (100) face of the pyrite after anoxic reaction only showed appreciable changes in the O1s orbital showing a shift to lower binding energies associated with an increase of the hydroxyl contribution (−OH) (Supplementary Fig. S5). In order to facilitate the identification of ≡S2− and ≡Fe3+dangling bonds, one sample was ion-sputtered, which promoted the breakage of the S-S dimers (as in the grinding procedure) (Supplementary Fig. S6). The formation of iron oxidation patches was also observed with HRTEM. Figure 5 shows an image of the pyrite surface after 22 hours of reaction in a micromolar solution of H2O2 in absence of O2(g). The presence of discrete oxidation patches was observed in the uppermost area of the micrograph (Fig. 5a). The FFT (Fast Fourier transform) proved that the lattice fringe spacing of low contrast clusters (~0.25 nm) were consistent with ferrihydrite nanocrystals (Fig. 5b), viewed down [001]. Additional HRTEM images (Supplementary Fig. S7) also showed the interplanar spacing characteristic of two-line ferrihydrite45,46. The FFT of the crystalline part showed the interplanar spacing of pyrite but also of goethite, suggesting that ferrihydrite can be a precursor of goethite formation (Fig. 5c)47.

(a) HTREM image showing the formation of secondary products over a lamella of pyrite after 22 hours immersed in a micromolar solution of H2O2 under anoxic conditions; (b) FFT of ferrihydrite patches; (c) FFT of the crystalline part showing spacing characteristic of pyrite and goethite.
Fitting the experimental data: kinetic modeling of H2O2 evolution
Our experimental data suggests that the observed trend of H2O2 is the result of the coupling between the H2O2 generation by iron defect sites on the pyrite surface and the H2O2 decomposition by the Fenton reaction. Based on these results, we built a kinetic model that allowed us to analyze the process in terms of elementary reactions and to determine the specific rate constant of the H2O2 formation in oxic and anoxic conditions by fitting the experimental data. In summary, the model describes the net amount of H2O2 according with the following expression (Supplementary, Modeling approaches):

Where k1, k2 and k3, represent the rate constants of each Fenton reaction step (Supplementary, Table S1). As shown in the equation, the H2O2 formation was calculated assuming a first order dependence on the reactive surface and, both iron defect sites act simultaneously, but in anoxic conditions the H2O2 produced by Fe2+sites is limited by the O2 derived by the Fenton reaction. Minimization between experimental curves and the values calculated from equation 9 were made with a non-linear least squares approach using the Marquardt algorithm, with koxic and kanoxic, the specific rate constants of H2O2 surface generation, as adjustable parameters (Table 1).
Figure 6 presents the fitted values corresponding to the evolution of H2O2 in oxic and anoxic conditions together with the model derived results. Under oxic conditions (Fig. 6a), when pH was higher, the ferrous surface iron was oxidized by dissolved O2 forming H2O2 according the first order rate. Deviations of the linearity started to occur due to a change in the reaction stoichiometry by the simultaneous decrease in pH and increase in dissolved Fe2+and H2O2. From this point, the Fenton reaction became important and the oxidation rate of Fe2+by O2 slowed down leading to a minor O2 consumption. As a result, reactions forming ROS -catalyzed by Fe2+and Fe3+− became effective for H2O2 degradation, which rapidly decreased, while iron species followed an opposite trend. Under anoxic conditions (Fig. 6b), the formation of H2O2 proceeded more slowly. The decomposition of H2O2 was also retarded because the concentration of dissolved [Fe2+] supplied to solution by pyrite dissolution was lower. The Fenton reaction was initiated when pH values dropped below 4.5 and the ratio H2O2/Fe2+increased, catalyzing the H2O2 decomposition similar to the oxic experiments. The analysis of ROS derived from the model in both oxic and anoxic conditions (Fig. 6c and d) shows that, the first reactive species formed was OH· acting as a chain initiator, forming additional free radicals. The majority of the OH· reacted with H2O2, generating HO2·, the conjugate acid of O2·−, which is the limiting reagent to assist the redox cycling of Fe3+/Fe2+forming O2 and helping to buffer the pH drop

(a) experimental and model curves of H2O2 together with Fe2+/ Fe3+, SO42−, pH and O2 model trends, under oxic conditions (pyrite particle loading = 0.71 g/L, A/V0 = 1 m2/L, pH0 = 7, [O2]0 = 232 μM); and, (b) anoxic conditions (pyrite particle loading = 2.16 g/L, A/V0 = 3.16 m2/L, pH0 = 7, [O2]0 = 0 μM). Time evolution of ROS obtained with the previous models under: (c) oxic conditions; and (d) anoxic conditions. *The evolution of O2·− was omitted from the plot because model calculations gave negligible concentrations.
Contrary to the expectation, the maximum amount of H2O2 measured was significantly different between experimental runs, and independent of particle loading. This was likely because the effect of increasing the reactive surface was twofold: (i) an increase in the formation of H2O2, and (ii) an increase in the release of dissolved Fe2+, accelerating the decomposition of H2O2 by the Fenton reaction (Fig. 7). We plotted H2O2 and Fe2+evolution as predicted by the model for a set of different values of pyrite reactive surface area (in Fig. 7a). As a result of this coupling effect, higher values of reactive surface area tend to increase the amount of secondary ROS (i.e., OH·/O−, HO2·/O2·−, O2) rather than stabilize the presence of H2O2 in solution (in Fig. 7b)2,48.

(a) 3D plot showing the H2O2 and Fe2+simulated trends at different values of the surface area of pyrite per volume of H2O (A0/V from 1 to 2.5, m2/L). (b) Summation of ROS generated by the Fenton reaction for the lowest and the highest values of the reactive surface (Initial conditions: koxic = 2.33 × 10−4, kanoxic = 6.46 × 10−9 mol/m2s –average values of the rate constants estimated by the model-, pyrite particle loading = 1 g/L, pH0 = 7, [O2]0 = 0 μM).
Discussion
Aqueous suspensions of pyrite form H2O2, in the presence and absence of dissolved O2, following a generation-decay trend (Fig. 1). The asymmetric shape observed in the experimental curves indicated that the apparent rate of H2O2 generation was significantly faster than the apparent H2O2 degradation. Previous studies22,25 have suggested that in the presence of dissolved O2, pyrite slurries form H2O2 by an electron transfer between the ferrous iron defect sites (Fe2+−S) and the adsorbed O2 through a Habber-Weiss reaction, involving the O2·− radical formation (equations 1 and 2). This hypothesis is consistent with the observation that there is an inverse relationship between H2O2 and O2 at the beginning of the oxic experiment (Fig. 2a). The mechanism of H2O2 formation in anoxic conditions remains more controversial. Some studies have suggested that in absence of O2 the formation of H2O2 is driven by the oxidation of adsorbed H2O catalyzed by the pyrite surface4,19,24,26, whereas other studies have considered that this reaction is unlikely due to energetic considerations15,25. A possible reconciliation comes from considering the presence of ≡Fe3+dangling bonds generated from the cleavage of S-S bonds. Briefly, the rupture of S-S bonds generate S− species which are highly instable and, to compensate its charge disequilibrium, donate one electron to the nearest iron atom by the autoredox reaction26,31,32,49,50,51,52,53,54:

These dangling bonds could decrease the energy requirements for the chemisorption of H2O molecules, some authors even talk about “ferryl” iron dangling bonds ≡Fe4+ 55, leading to H2O splitting into H+and OH·, with the subsequent formation of H2O2, as described by19 equations 3 and 4. Our experiments showed that H2O2 was generated in the absence of both dissolved O2 and light (Fig. 1b). Besides, the estimated ratio of S/Fe (<2) in unreacted pyrite particles (Supplementary Fig. S8) indicated the presence of S vacancies.
Experiments with pyrite slurries under both oxic and anoxic conditions showed a sudden drop of pH during the first hours (Supplementary Fig. S2). This increase of H+concentration together with the progressive accumulation of H2O2 and Fe2+in solution is expected to trigger the Fenton reaction. UV-Vis spectroscopy confirmed the presence of Fe3+-complexes during anoxic pyrite dissolution (Fig. 3a) whose maximum wavelengths were compatible with the following reaction41:

The detection of both Fe3+-complexes and OH· in solution (Fig. 3) indicated that the Fenton reaction occurred even in the absence of dissolved O2, supporting the idea that the suite of Fenton reactions conditioned the decay period of H2O2 curves. Additionally, the formation of OH· concomitantly with the formation of H2O2 from the start-up of the experiment under anoxic conditions (Fig. 3b) together with the splitting of the cathodic peak associated with the nearly spontaneous iron reduction (Supplementary Fig. S3), suggest that ≡Fe3+dangling bonds, actually catalyze the H2O oxidation by one single electron, forming OH· as described by equation 3.
In principle, the overall process of aqueous pyrite oxidation -under oxic conditions- involves only O2 (g) consumption, according to:

However, considering a free radical mechanism, a simultaneous uptake and release of dissolved O2 it to be expected, because it can be both a product and a reactant. In oxic experiments, O2 concentration rapidly dropped and then reached a more constant value (Fig. 2a). Since the amount of Fe2+in solution is presumably low at these early stages, the consumption of O2 during this period could be attributed to Fe2+oxidation at surface sites to form the superoxide anion (O2·−) and to the subsequent production of H2O2 (equations 1 and 2). An interesting result was the formation of O2 (aq) as a by-product in anoxic experiments (Fig. 2b and Supplementary Fig. S1b). Although O2 is not a direct product in the equation 3, since H2O2 is formed, the formation of O2 can occur via several pathways such as the Fenton-like reaction, the “catalase-like reaction”25 or via non-radical disproportionation of H2O2.
XPS and HRTEM results reported herein showed the formation of Fe3+−O patches over pyrite surfaces. The implication of these Fe3+−patches during aqueous pyrite oxidation is not clear. Some studies argued that the electron cycling of Fe2+and Fe3+between unoxidized and oxidized areas favors the electron transfer from the surface of the pyrite to molecular O2, increasing the oxidation rate of pyrite56. However, this mechanism is based on atmospheric oxidation of pyrite and does not explain the formation of H2O2. In contrast, other studies have suggested that the formation of these Fe3+−patches interrupts the redox cycling of Fe2+/Fe3+, thereby inhibiting the formation of H2O2 and decreasing the rate of pyrite dissolution25. Our cycling experiments showed that most of the defect sites of the pyrite microparticles remained active at the end of the experiment, even when H2O2 was no longer observable in solution (Fig. 4). Moreover, it is expected that oxidized patches will desorb at low pH during the pyrite dissolution process. HRTEM images showed an appreciable increment of the pyrite alteration layer (Supplementary Fig. S9), suggesting that these Fe3+−patches failed to completely block the surface renewal during pyrite dissolution.
Based on our experimental results, we propose a model that explains the generation-decay trend of H2O2 in terms of a kinetic competition between (1) the formation of H2O2 by the self-oxidation of iron-sulfur cluster defect sites; and, (2) the degradation of H2O2 by the Fenton reaction triggered by pyrite dissolution. Accordingly, the evolution of H2O2 in solution can be summarized as follows:

At the beginning of the model run (t ≈ 0), the amount of H2O2 in solution was controlled by the surface reaction and the second term tended toward zero. However, when the Fenton reaction started, H2O2 was progressively degraded to secondary ROS, in solution (i.e., OH·, HO2·/O2·−, O2). At the end of the model run the amount of H2O2 remained constant within a value close to zero and equation 13, became:

The model allowed us to estimate the rate constants of H2O2 formation under both oxic and anoxic conditions (Table 1). Peak production of H2O2 was shifted towards longer times in anoxic conditions (Supplementary Fig. S10), pointing to slower oxidation kinetics of pyrite in the absence of O2, a result also supported by our XPS analysis (Supplementary Figs S4 and 5) and by the sulfate and iron released by pyrite dissolution (Supplementary Fig. S11). When compared with experimental data, the model reproduces qualitatively well the observed trends for pH, O2, Fetotal and SO42−, as these parameters are calculated using the overall equations of pyrite dissolution17,57,58 (Fig. 6a–b). An additional feature of the model is that provides a way to reconcile the classical dissolution approach with the free radical assumption, opening a new pathway to analyze the flux of secondary ROS resulting from the degradation of H2O2 through the Fenton reaction (Fig. 6c–d). OH· radicals were the first species produced, rapidly decaying to HO2·/O2·−. These ROS species counterbalances the decrease in pH and promoted the so-called “Fenton like” reaction, which resulted in the formation of O2(g). Fe2+regenerated through this sequence, and also through the dissolution of pyrite, makes the Fenton reaction more efficient59. Therefore, the disappearance of H2O2 in solution was likely due to a fast transformation into ROS, catalyzed by dissolved Fe2+, rather than by the cessation of the generation mechanism itself. This result is consistent with the continuous production of H2O2 and OH·23,26,60 during pyrite dissolution. We hypothesize that as a result of the progressive acidification of the solution together with iron recycling by the Fenton reaction, the abiotic dissolution of pyrite microparticles can be considered as a natural and auto-catalytic Fenton reagent, useful to understanding long-term oxidation processes even in oxygen limited environments.
Methods
Sample preparation and characterization
Natural pyrite samples (Logroño, Spain) were ground to obtain particles with average diameter of 1.4 μm, Laser Diffraction Particle Size Analyzer (LS13320) and BET (Brunauer – Emmett – Teller) surface area of 1.46 m2/g (multi-point N2 adsorption). Prior to use, freshly ground pyrite microparticles were washed by sonication in ethanol (96%) and hydrochloric acid (HCl 0.25 M), to remove organics and oxide surface coatings. Samples were then rinsed with deoxygenated deionized water (MilliQ) and dried in a low vacuum chamber purged with nitrogen (N2) until used. Minor and trace elements in the acid washed samples were evaluated by scanning electron microscopy (SEM) using X-ray mapping (XRM) (Supplementary Fig. S8). X-ray diffraction (XRD) was used to assess the presence of secondary phases and to characterize the degree of structural disorder in the samples using the Rietveld method (Supplementary Fig. S12).
Batch kinetic experiments
The kinetics of H2O2 formation and degradation on pyrite slurries was investigated in batch reactors utilizing amperometric sensors for H2O2 (ISO-HPO-100, World Precision Instruments, Inc.) and dissolved O2 (Unisense DK), and a glass electrode for pH determination (Vernier FPH-BTA). Batch reactors were designed to fit with microsensors, spectroscopic probes, ports for pyrite inlet under N2 atmosphere, and valves. The valves allowed fluid circulation and solution sampling in a closed system configuration.
The production of ferric iron (Fe3+) and hydroxyl radical (OH·) species by pyrite slurries in absence of dissolved O2 were measured using UV-vis spectroscopy by monitoring absorption bands at specific wavelengths. To prevent data masking due to particle absorption, the pyrite microparticles were deposited onto silicone strips as a thin film adhered to the inner reactor walls. Spectroscopic data were collected with a fiber optic UV-Vis spectrometer (Black-comet, Stellarnet or USB4000, Ocean Optics) and acquired with the SpectraWiz® or loggerpro3 codes. In addition, total dissolved iron and sulfate released during pyrite dissolution were measured at different time intervals using inductively coupled plasma mass spectrometry (ICP-MS) and ion chromatography (IC).
All experiments were made under continuous magnetic stirring (500 rpm), at room temperature (T° ∼ 22 °C) and in the dark, unless other conditions are specified. Due to the autocatalytic nature of Fenton chain reactions, kinetic experiments were carried out in unbuffered distilled water. Further details of the batch reactors, electrochemical sensors, experimental procedures and test analysis are given as supplementary material (Supplementary Figs S13–18).
Surface analysis
Cyclic Voltammetry (CV) was employed as an additional way to assess the sequence of redox reactions involving free radicals during H2O adsorption at pyrite interface. CV was performed using Pt/Pyrite-Np’s/ Nafion©/ electrodes in N2 purged solutions vs Ag/Cl 3 M KCl. A detailed description of the experimental set-up is given as Supplementary Material. X-ray photoelectron spectroscopy (XPS) was used to analyze the surface oxidation states of (001) faces of pyrite (single-crystal, ~1 cm2 × 2 mm) after aqueous reaction in oxic and anoxic conditions. Platelets parallel to (001) faces were cut (1 cm2 × 2 mm) and cleaned following the same procedure described above and allowed to react with water in oxic and anoxic conditions. Samples were dried and stored in N2-filled tubes until introduction into the XPS vacuum chamber. The oxidation states were analyzed at three different stages of the dissolution process: (t1) unreacted sample; the pyrite crystal was acid-washed to generate oxide-free surfaces by removing the normal contaminants, carbon, nitrogen, and oxygen (C, N, O) due to atmospheric oxidation; (t2): sample after 22 hours immersed in oxic water. (t3): sample after 22 hours immersed in anoxic water. Prior to this stepped analysis, the XPS spectra of a pyrite surface was analyzed after argon ion (Ar+) sputtering to verify the formation of Fe3+and S2− surface species as occurs during mechanical fracture by preferential sulfur removal. XPS Spectra were collected from a take-off angle of 90° relative to the sample surface in a Thermo Scientific K-Alpha ESCA analyzer using monochromatic Al Kα (1486.6 eV) radiation and pass energies of 100 eV and 20 eV for survey spectra and narrow region spectra, respectively. Spectra were aligned by setting the C1s peak to a binding energy of 285 eV61. Deconvolution and fitting of experimental data were done with the XPSpeak4.2 software (http://www.phy.cuhk.edu.hk/~surface/XPSPEAK/). The Shirley method was used for background subtraction and the binding energies of the species identified were assigned using values taken from literature (Supplementary Tables S2 and 3).
High resolution Electron Transmission Microscopy (HTREM) analysis were performed to identify nano-domains of secondary oxidation products at the pyrite interface. HTREM images were acquired on a JEOL JEM- 3011 microscope with accelerating voltage of 200 kV using a Gatan Ultrascan 1000 CCD camera and Digital Microgrograph software. Data processing was performed with the GADDS and image-J codes. Pyrite lamellas were prepared using a focused ion beam (FIB) with a high resolution JEOL JSM-6700 f.
Kinetic model
The model was run using the computer code Copasi 4.8 (COmplex PAthway SImulator)62. We assume that the experimental trend of H2O2, in the presence and absence of O2 (g), is shaped by three main processes: (i) the rate of H2O2 generation by the iron defect sites on the surface of pyrite particles; (ii) the production rates of Fe2+and SO42− by pyrite dissolution; and, (iii) the kinetics of H2O2 degradation by the Fenton reaction. Fits to the experimental curves that describe the evolution of H2O2 were performed by using the Marquardt algorithm employing as adjustable parameters the rate constants of the H2O2 formation. A detailed explanation of the modeling set-up is included in the supplementary material.
Additional Information
How to cite this article: Gil-Lozano, C. et al. Quantifying Fenton reaction pathways driven by self-generated H2O2 on pyrite surfaces. Sci. Rep. 7, 43703; doi: 10.1038/srep43703 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Puthussery, J., Seefeld, S., Berry, N., Gibbs, M. & Law, M. Colloidal Iron Pyrite (FeS2) Nanocrystal Inks for Thin-Film Photovoltaics. Journal of the American Chemical Society 133, 716–719, doi: 10.1021/ja1096368 (2010).
Bae, S., Kim, D. & Lee, W. Degradation of diclofenac by pyrite catalyzed Fenton oxidation. Applied Catalysis B: Environmental 134–135, 93–102, doi: 10.1016/j.apcatb.2012.12.031 (2013).
Gil-Lozano, C., Losa-Adams, E., Davila, F. A. & Gago-Duport, L. Pyrite nanoparticles as a Fenton-like reagent for in situ remediation of organic pollutants. Beilstein journal of nanotechnology 5, 855–864, doi: 10.3762/bjnano.5.97 (2014).
Wang, W., Qu, Y., Yang, B., Liu, X. & Su, W. Lactate oxidation in pyrite suspension: A Fenton-like process in situ generating H2O2. Chemosphere 86, 376–382, doi: 10.1016/j.chemosphere.2011.10.026 (2012).
Che, H. & Lee, W. Selective redox degradation of chlorinated aliphatic compounds by Fenton reaction in pyrite suspension. Chemosphere 82, 1103–1108, doi: 10.1016/j.chemosphere.2010.12.002 (2011).
Choi, K., Bae, S. & Lee, W. Degradation of pyrene in cetylpyridinium chloride-aided soil washing wastewater by pyrite Fenton reaction. Chemical Engineering Journal 249, 34–41, doi: 10.1016/j.cej.2014.03.090 (2014).
Choi, K., Bae, S. & Lee, W. Degradation of off-gas toluene in continuous pyrite Fenton system. Journal of Hazardous Materials 280, 31–37, doi: 10.1016/j.jhazmat.2014.07.054 (2014).
Chandra, A. P. & Gerson, A. R. The mechanisms of pyrite oxidation and leaching: A fundamental perspective. Surface Science Reports 65, 293–315, doi: 10.1016/j.surfrep.2010.08.003 (2010).
Descostes, M., Vitorge, P. & Beaucaire, C. Pyrite dissolution in acidic media. Geochimica et Cosmochimica Acta 68, 4559–4569, doi: 10.1016/j.gca.2004.04.012 (2004).
Druschel, G. & Borda, M. Comment on “Pyrite dissolution in acidic media” by M. Descostes, P. Vitorge, and C. Beaucaire. Geochimica et Cosmochimica Acta 70, 5246–5250, doi: 10.1016/j.gca.2005.07.023 (2006).
Heidel, C. & Tichomirowa, M. The isotopic composition of sulfate from anaerobic and low oxygen pyrite oxidation experiments with ferric iron — New insights into oxidation mechanisms. Chemical Geology 281, 305–316, doi: 10.1016/j.chemgeo.2010.12.017 (2011).
Lowson, R. T. Aqueous oxidation of pyrite by molecular oxygen. Chemical Reviews 82, 461–497, doi: 10.1021/cr00051a001 (1982).
Moses, C. O., Kirk Nordstrom, D., Herman, J. S. & Mills, A. L. Aqueous pyrite oxidation by dissolved oxygen and by ferric iron. Geochimica et Cosmochimica Acta 51, 1561–1571, doi: 10.1016/0016-7037(87)90337-1 (1987).
Rimstidt, J. D. & Vaughan, D. J. Pyrite oxidation: a state-of-the-art assessment of the reaction mechanism. Geochimica et Cosmochimica Acta 67, 873–880, doi: 10.1016/s0016-7037(02)01165-1 (2003).
Rosso, K. M., Becker, U. & Hochella Jr, M. F. The interaction of pyrite {100} surfaces with O2 and H2O: Fundamental oxidation mechanisms. American Mineralogist 84, 1549–1561, doi: 10.2138/am-1999-1008 (1999).
Singer, P. C. & Stumm, W. Acidic mine drainage: The rate-determining step. Science 167, 1121–1123, doi: 10.1126/science.167.3921.1121 (1970).
Williamson, M. A. & Rimstidt, J. D. The kinetics and electrochemical rate-determining step of aqueous pyrite oxidation. Geochimica et Cosmochimica Acta 58, 5443–5454, doi: 10.1016/0016-7037(94)90241-0 (1994).
Borda, M. J., Elsetinow, A. R., Schoonen, M. A. & Strongin, D. R. Pyrite-Induced Hydrogen Peroxide Formation as a Driving Force in the Evolution of Photosynthetic Organisms on an Early Earth. Astrobiology 1, 283–288, doi: 10.1089/15311070152757474 (2001).
Borda, M. J., Elsetinow, A. R., Strongin, D. R. & Schoonen, M. A. A mechanism for the production of hydroxyl radical at surface defect sites on pyrite. Geochimica et Cosmochimica Acta 67, 935–939, doi: 10.1016/s0016-7037(02)01222-x (2003).
Cohn, C. A., Borda, M. J. & Schoonen, M. A. RNA decomposition by pyrite-induced radicals and possible role of lipids during the emergence of life. Earth and Planetary Science Letters 225, 271–278, doi: 10.1016/j.epsl.2004.07.007 (2004).
Cohn, C. A., Pak, A., Strongin, D. R. & Schoonen, M. A. Quantifying hydrogen peroxide in iron-containing solutions using leuco crystal violet. Geochemical Transactions 6, 47, doi: 10.1186/1467-4866-6-47 (2005).
Cohn, C. A. et al. Pyrite-induced hydroxyl radical formation and its effect on nucleic acids. Geochemical Transactions 7, 11, doi: 10.1186/1467-4866-7-3 (2006).
Cohn, C., Fisher, S., Brownawell, B. & Schoonen, M. Adenine oxidation by pyrite-generated hydroxyl radicals. Geochemical Transactions 11, 8, doi: 10.1186/1467-4866-11-2 (2010).
Javadi, N. A. & Hanumantha, R. K. Formation of hydrogen peroxide by sulphide minerals. Hydrometallurgy 141, 82–88, doi: 10.1016/j.hydromet.2013.10.011 (2014).
Schoonen, M. A. A., Harrington, A. D., Laffers, R. & Strongin, D. R. Role of hydrogen peroxide and hydroxyl radical in pyrite oxidation by molecular oxygen. Geochimica et Cosmochimica Acta 74, 4971–4987, doi: 10.1016/j.gca.2010.05.028 (2010).
Zhang, P., Yuan, S. & Liao, P. Mechanisms of hydroxyl radical production from abiotic oxidation of pyrite under acidic conditions. Geochimica et Cosmochimica Acta 172, 444–457, doi: 10.1016/j.gca.2015.10.015 (2016).
Burns, R. G. & Fisher, D. S. Iron-sulfur mineralogy of Mars: Magmatic evolution and chemical weathering products. Journal of Geophysical Research: Solid Earth 95, 14415–14421, doi: 10.1029/JB095iB09p14415 (1990).
Davila, A. F. et al. Subsurface formation of oxidants on Mars and implications for the preservation of organic biosignatures. Earth and Planetary Science Letters 272, 456–463, doi: 10.1016/j.epsl.2008.05.015 (2008).
Eggleston, C. M., Stern, J. R., Strellis, T. M. & Parkinson, B. A. A natural photoelectrochemical cell for water splitting: Implications for early Earth and Mars. American Mineralogist 97, 1804–1807, doi: 10.2138/am.2012.4211 (2012).
Zolotov, M. Y. & Shock, E. L. Formation of jarosite-bearing deposits through aqueous oxidation of pyrite at Meridiani Planum, Mars. Geophys. Res. Lett. 32, L21203, doi: 10.1029/2005gl024253 (2005).
Nesbitt, H. W., Bancroft, G. M., Pratt, A. R. & Scaini, M. J. Sulfur and iron surface states on fractured pyrite surfaces. American Mineralogist 83, 1067–1076, doi: 10.2138/am-1998-9-1015 (1998).
Schaufuß, A. G. et al. Reactivity of surface chemical states on fractured pyrite. Surface Science 411, 321–328, doi: 10.1016/s0039-6028(98)00355-0 (1998).
Murphy, R. & Strongin, D. R. Surface reactivity of pyrite and related sulfides. Surface Science Reports 64, 1–45, doi: 10.1016/j.surfrep.2008.09.002 (2009).
Krishnamoorthy, A., F. W. Herbert, S. Yip, K. J. Van Vliet, B. Yildiz, Herbert, F. W., Sidney, Y., Krystyn, J. V. V. & Bilge, Y. Electronic states of intrinsic surface and bulk vacancies in FeS2. Journal of Physics: Condensed Matter 25, 045004, doi: 10.1088/0953-8984/25/4/045004 (2012).
Wang, J., Chen, D., Yan, D., Wei, H. & Xiang, L. Evolution from an anoxic to oxic deep ocean during the Ediacaran–Cambrian transition and implications for bioradiation. Chemical Geology 306–307, 129–138, doi: 10.1016/j.chemgeo.2012.03.005 (2012).
Guevremont, J. M., Elseinow, A. R., Strongin, D. R., Bebie, J. & Schoonen, M. A. A. Structure sensitivity of pyrite oxidation; comparison of the (100) and (111) planes. American Mineralogist 83, 1353–1356, doi: 10.2138/am-1998-11-1225 (1998).
Guevremont, J. M., Strongin, D. R. & Schoonen, M. A. A. Thermal chemistry of H2S and H2O on the (100) plane of pyrite: unique reactivity of defect sites Vol. 83 (Mineralogical Society of America, 1998).
de Leeuw, N. H., Parker, S. C., Sithole, H. M. & Ngoepe, P. E. Modeling the Surface Structure and Reactivity of Pyrite: Introducing a Potential Model for FeS2. The Journal of Physical Chemistry B 104, 7969–7976, doi: 10.1021/jp0009498 (2000).
Fischer, C., Kurganskaya, I., Schäfer, T. & Lüttge, A. Variability of crystal surface reactivity: What do we know? Applied Geochemistry 43, 132–157, doi: 10.1016/j.apgeochem.2014.02.002 (2014).
Feng, W. & Nansheng, D. Photochemistry of hydrolytic iron (III) species and photoinduced degradation of organic compounds. A minireview. Chemosphere 41, 1137–1147, doi: 10.1016/s0045-6535(00)00024-2 (2000).
Lente, G. Reactions of the iron(III) hydroxo dimer with inorganic ligands, University of Debrecen, (2001).
Ma, J., Weng, D., Wu, X., Si, Z. & Wu, Z. Highly dispersed iron species created on alkali-treated zeolite for ammonia SCR. Progress in Natural Science: Materials International 23, 493–500, doi: 10.1016/j.pnsc.2013.08.005 (2013).
Stefánsson, A. Iron(III) Hydrolysis and Solubility at 25 °C. Environmental Science & Technology 41, 6117–6123, doi: 10.1021/es070174h (2007).
Fan, H.-J. et al. Degradation pathways of crystal violet by Fenton and Fenton-like systems: Condition optimization and intermediate separation and identification. Journal of Hazardous Materials 171, 1032–1044, doi: 10.1016/j.jhazmat.2009.06.117 (2009).
Banfield, J. F., Welch, S. A., Zhang, H. & Ebert, T. T. & Penn, R. L. Aggregation-Based Crystal Growth and Microstructure Development in Natural Iron Oxyhydroxide Biomineralization Products. Science 289, 751–754, doi: 10.1126/science.289.5480.751 (2000).
Drits, V. A., Sakharov, B. A., Salyn, A. L. & Manceau, A. Structural model for ferrihydrite. Clay Minerals 28, 185–207, doi: 10.1180/claymin.1993.028.2.02 (1993).
Das, S., Hendry, M. J. & Essilfie-Dughan, J. Transformation of Two-Line Ferrihydrite to Goethite and Hematite as a Function of pH and Temperature. Environmental Science & Technology 45, 268–275, doi: 10.1021/es101903y (2011).
Che, H., Bae, S. & Lee, W. Degradation of trichloroethylene by Fenton reaction in pyrite suspension. Journal of Hazardous Materials 185, 1355–1361, doi: 10.1016/j.jhazmat.2010.10.055 (2011).
Nesbitt, H. W. et al. Synchrotron XPS evidence for Fe2+−S and Fe3+−S surface species on pyrite fracture-surfaces, and their 3D electronic states. American Mineralogist 85, 850–857, doi: 10.2138/am-2000-5-628 (2000).
Leiro, J. A., Mattila, S. S. & Laajalehto, K. XPS study of the sulphur 2p spectra of pyrite. Surface Science 547, 157–161, doi: 10.1016/j.susc.2003.09.033 (2003).
Andersson, K. et al. Experimental and theoretical characterization of the structure of defects at the pyrite FeS2 (100) surface. Physical Review B 70, 195404, doi: 10.1103/PhysRevB.70.195404 (2004).
Zhang, Y. N., Hu, J., Law, M. & Wu, R. Q. Effect of surface stoichiometry on the band gap of the pyrite FeS2 (100) surface. Physical Review B 85, 085314, doi: 10.1103/PhysRevB.85.085314 (2012).
Herbert, F. W., Krishnamoorthy, A., Van Vliet, K. J. & Yildiz, B. Quantification of electronic band gap and surface states on FeS2(100). Surface Science 618, 53–61, doi: 10.1016/j.susc.2013.08.014 (2013).
Sanchez-Arenillas, M. & Mateo-Marti, E. Pyrite surface environment drives molecular adsorption: cystine on pyrite(100) investigated by X-ray photoemission spectroscopy and low energy electron diffraction. Physical Chemistry Chemical Physics 18, 27219–27225, doi: 10.1039/C6CP03760G (2016).
Stirling, A., Bernasconi, M. & Parrinello, M. Defective pyrite (100) surface: An ab initio study. Physical Review B 75, 8, doi: 10.1103/PhysRevB.75.165406 (2007).
Eggleston, C. M., Ehrhardt, J.-J. & Stumm, W. Surface structural controls on pyrite oxidation kinetics; an XPS-UPS, STM, and modeling study. American Mineralogist 81, 1036–1056, doi: 10.2138/am-1996-9-1002 (1996).
Liu, R., Wolfe, A., Dzombak, D., Stewart, B. & Capo, R. Comparison of dissolution under oxic acid drainage conditions for eight sedimentary and hydrothermal pyrite samples. Environ Geol 56, 171–182, doi: 10.1007/s00254-007-1149-0 (2008).
McKibben, M. A. & Barnes, H. L. Oxidation of pyrite in low temperature acidic solutions: Rate laws and surface textures. Geochimica et Cosmochimica Acta 50, 1509–1520, doi: 10.1016/0016-7037(86)90325-X (1986).
Duesterberg, C. K., Mylon, S. E. & Waite, T. D. pH Effects on Iron-Catalyzed Oxidation using Fenton’s Reagent. Environmental Science & Technology 42, 8522–8527, doi: 10.1021/es801720d (2008).
Fisher, S., Schoonen, M. & Brownawell, B. Phenylalanine as a hydroxyl radical-specific probe in pyrite slurries. Geochemical Transactions 13, 1–18, doi: 10.1186/1467-4866-13-3 (2012).
Briggs, D. & Seah, M. P. Practical Surface Analysis: Auger and X-ray photoelectron spectroscopy. 2 nd edn, Vol. 1 657 (John Wiley & Sons, 1990).
Hoops, S. et al. COPASI—a COmplex PAthway SImulator. Bioinformatics 22, 3067–3074, doi: 10.1093/bioinformatics/btl485 (2006).
Acknowledgements
This research was funded by the Spanish Ministry of Science and Innovation in the framework of the MICINN-FEDER, Project CGL2011-30079, and from the Project “icyMARS”, funded by the European Research Council, Starting Grant no 307496. One of the authors (C.G.L.) also acknowledges the financial support by the Spanish Ministry of Science and Innovation under FPU Grant AP2008-03712. The authors would like to thank Dr. José Benito Rodríguez and Dr. Eugenio Solla for their inestimable help in the preparation of FIB samples and HREM analyses and Dra. Serra Rodríguez for the valuable help with XPS analysis.
Author information
Authors and Affiliations
Contributions
All persons who meet authorship criteria are listed as authors, and all authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript. Furthermore, each author certifies that this material or similar material has not been and will not be submitted to or published in any other publication before its appearance in the Scientific Reports journal. Conception and design of study: Carolina Gil Lozano, Luis Gago Duport. Acquisition of data: Carolina Gil Lozano. Analysis and/or interpretation of data: Carolina Gil Lozano, Alfonso F. Davila, Elisabeth Losa. Adams, Alberto G. Fairén, Luis Gago Duport. Drafting the manuscript: Carolina Gil Lozano, Luis Gago Duport. Revising the manuscript critically for important intellectual content: Carolina Gil Lozano, Alfonso Davila, Elisabeth Losa Adams, Alberto G. Fairén, Luis Gago Duport. Approval of the version of the manuscript to be published: Carolina Gil Lozano, Alfonso. Davila, Elisabeth Losa Adams, Alberto G. Fairén, Luis Gago Duport.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Gil-Lozano, C., Davila, A., Losa-Adams, E. et al. Quantifying Fenton reaction pathways driven by self-generated H2O2 on pyrite surfaces. Sci Rep 7, 43703 (2017). https://doi.org/10.1038/srep43703
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43703
This article is cited by
-
Magnetically modified-mitoxantrone mesoporous organosilica drugs: an emergent multimodal nanochemotherapy for breast cancer
Journal of Nanobiotechnology (2024)
-
Electrochemical deposition of nickel from aqueous electrolytic baths prepared by dissolution of metallic powder
Journal of Solid State Electrochemistry (2022)
-
Sunlight Irradiated Pyrite-Fenton System for Advanced Oxidative Treatment of Textile Dyes Mixture
Water, Air, & Soil Pollution (2022)
-
Dynamic interactions at the mineral–organic matter interface
Nature Reviews Earth & Environment (2021)
-
Pyrite-induced uv-photocatalytic abiotic nitrogen fixation: implications for early atmospheres and Life
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
