
Abstract
Modelling Down syndrome (DS) in mouse has been crucial for the understanding of the disease and the evaluation of therapeutic targets. Nevertheless, the modelling so far has been limited to the mouse and, even in this model, generating duplication of genomic regions has been labour intensive and time consuming. We developed the CRISpr MEdiated REarrangement (CRISMERE) strategy, which takes advantage of the CRISPR/Cas9 system, to generate most of the desired rearrangements from a single experiment at much lower expenses and in less than 9 months. Deletions, duplications, and inversions of genomic regions as large as 24.4 Mb in rat and mouse founders were observed and germ line transmission was confirmed for fragment as large as 3.6 Mb. Interestingly we have been able to recover duplicated regions from founders in which we only detected deletions. CRISMERE is even more powerful than anticipated it allows the scientific community to manipulate the rodent and probably other genomes in a fast and efficient manner which was not possible before.
Similar content being viewed by others


Introduction
Mouse models have been key elements to better understand the physiopathology of Down syndrome (DS)1,2,3,4,5,6. Nevertheless new models in rat may offer opportunities for exploring behaviour, cognition, memory and social interaction. In addition, being separated from mouse for more than 11 million years the rat represent an alternate model of choice. Mouse, rat and human share many homologies at the genomic7,8,9,10 and physiological levels so that conclusions drawn in two rodent species would be stronger for translational approach in human. More than 200 protein coding genes have been identified on human chromosome 21 (HSA21), for which 188 orthologs have been found on rat chromosomes 11 and 20 (www.ensembl.org/biomart; see Fig. 1). For example, a large fragment ranging from Lipi to Zfp295 encompassing 24.4 Mb and 113 protein-coding genes is located on rat chromosome 11 (RNO11; RNO for Rattus norvegicus). It is similar to the region located on the mouse chromosome 16. The most telomeric part of HSA21 long arm is located on a unique segment on the RNO20, with 74 genes found between Umodl1 and Prmt2. By contrast, the mouse orthologs for these genes are present in segments of two chromosomes in the mouse11. On this basis, we focused on developing rat models to decode the cellular and molecular mechanisms linked to the neuropathological, cognitive, physiological, and morphological alterations found in human DS patients.

The relative position of the mouse homologous regions on MMU16, −17, −10 and rat homologous region on RN011 and −20 to HSA21 are shown with the genes located at the borders of the genetic interval. In blue and grey, the position of the DS models (rat or mouse respectively) attempted in this paper.
So far the lack of embryonic stem cell was a limiting step for generating DS rat model. Indeed modelling segmental duplication has been achieved so far by using the Cre-lox technology12,13,14,15 in combination with ES cells or alternative strategies16,17,18 only in mouse. Briefly, this strategy requires the generation of two mouse lines with loxP site inserted in the same orientation at the border of the region of interest, followed by 3-step breeding to introduce a Cre driver expressed in the germ line15,14. For inversions, new experimental design and mouse lines had to be generated19 with inverted loxP sites. The overall strategy necessitates at least 3 years to generate a model but demonstrated the power of a mapping panel of duplication to better understand the contribution of HSA21 regions to DS phenotypes3. Recently, CRISPR/Cas9 was used to manipulate large genomic region in the mouse genome. The Wu lab20 demonstrated that DNA elements of up to 29 kb can be manipulated directly in mice using the CRISPR/Cas9 technology. Zheng and Li’s lab showed that deletion of up to 95 kb can be generated in mice21,22. Similarly, gene clusters of up to 800 kb can be deleted, duplicated, and inversed in HEK293T cells23. CRISPR/Cas9 was used for the generation of up to 1 Mb structural deletion and inversion around the Tyrosinase locus24 in mouse zygotes but duplications appeared less frequently and did not pass the germ line.
In this paper, we investigated how mouse or rat segmental duplications, and other large scale rearrangements, can be easily generated using the CRISPR/cas9 technology in zygotes either for a single candidate gene or for larger to very large genomic regions. These models and their analyses will allow us to better understand the complexity of the genetic interactions that are involved in DS cognitive phenotypes and could challenge the hypothesis regarding the multigenic nature of DS in a new species.
Results
Successful segmental inversion, deletion and duplications of two rat genes by CRISMERE
We defined the CRISMERE approach by using two pairs of sgRNAs, one pair located upstream and the other pair downstream of the region of interest (called here break site), for all the experiments described in this paper. All sgRNAs microinjected were selected and validated in vitro (see Methods) on a PCR fragment containing the target sequence, but we (like others) observed that the in vivo efficiency can vary from one sgRNA to another. For our first attempt, the pairs of sgRNA were used in combination with the D10A nickase Cas9 mutant but no chromosomal rearrangement was observed. When the same guides were used with wild type (wt) Streptococcus pyogenes Cas9 the occurrence of rearrangement increased substantially. We decided to keep this configuration for our subsequent experiments. The two sgRNAs of a pair recognized close sequences (distance between two guides was always lower than 150 bps) to increase the probability to obtain a double strand break (DSB) (see Supplementary Data).
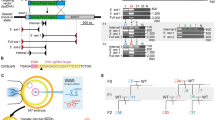
First we selected the locus of the cystathionine beta synthase gene, Cbs, located in a region contributing to DS phenotypes in mouse models25,26. In human, CBS recessive mutation deficiency leads to homocystinuria (OMIM 236200). Cbs homozygous knock-out in mouse are severely growth retarded and die within 5 weeks of birth while heterozygotes survive27. In rat, Cbs (ENSRNOG00000029528) is located on RNO20 in the syntenic HSA21 interval (Fig. 1). The generation of Cbs structural variant models was tested by CRISMERE. We designed two pairs of sgRNAs targeting both extremities of the 37.2 kb region containing the Cbs gene (for sequence details see Supplementary Table 1 and Supplementary Data S1). Injection conditions and genotyping results are synthetized in Table 1. Given that DNA duplications, deletions, and inversions have two junctions, we designed two pairs of specific PCR primers near the cleavage sites of the Cas9 to identify upstream and downstream DSB (Fig. 2B). We used different combinations to identify inversion, deletion, and duplication junctions to make sure we would detect any event (Fig. 2A and B). Twenty-four F0 founder rats were born and screened by PCR at 4 weeks for the whole variety of potential alleles. We detected 12 F0 founders (50%; 12 out of 24) with a deletion of the whole Cbs gene (for details see Fig. 2C and Supplementary Data S1). Interestingly, five F0 founders showed two deletion on two alleles, of distinct sizes and intensity, which were confirmed by Sanger sequencing. For instance, rat founder F0-278 had two different deleted alleles (close and large arrows on Fig. 1B; see also Supplementary Data S1) that went germline. The deletion annotated by the faint arrow occurred frequently (12/25 F1 pups) while the other deletion was only detected in one F1 pups (1/25). Inversion events were less frequent (7 F0 rats; 29%) but still abundant. Only one F0 founder had detectable tandem duplication (4.2%) (F0-264; Fig. 2D). This duplication was transmitted to the F1 (3/14 pups). The inverted allele observed in the same founder (Fig. 2D and Supplementary Data S1) was also transmitted to the germ line at lower frequency (3/21 pups).

(A) Variety of alleles detected in founders F0 and F1 when two pairs of sgRNAs for two sites flanking a region of interest are used. In blue the genomic region 5′ of the region of interest, in yellow the region of interest and in orange the genomic region 3′ of the region of interest. (B) Diagram for rat Cbs and Dyrk1a genes. Shown are the position of the primers used for genotyping, and position of the sgRNAs (C,D) concern Cbs. (C) Shown are deletion junction chromatograms of 3 different founders. Only one deleted allele was obtained in F0-274 (one band detected by PCR) whereas 2 deleted alleles were detected for founders F0-268 and -278 (2 bands observed by PCR and confirmed by a close analyze of the chromatograms). For founder F0-278, a bold arrow faint arrow shows the two distinct deletion alleles (see also Supplementary data S1). Both alleles passed germ line. (D) Shown are duplication and inversion junctions for Cbs amplified by PCR from ear clipping rat founder F0-264 with specific primers pairs. An example of sequence chromatograms of the inversion and duplication junctions is shown. The same inversion and duplication events were found independently on F1 rats (E,F) concern Dyrk1a (E). The duplication junctions amplified by PCR from ear clipping rat founders F0-384 and F0-386 with specific primers pairs are shown. Inversion junctions of F0-355 founder is shown as an example (see also Supplementary data S2) (F) Detection of rats with one copy of Dyrk1a. Appearance of a small founder F0 (yellow arrow) compared to its wt littermate. Monosomic rats for Dyrk1a were first detected by droplet digital PCR. Two ddPCRs (in the first and last exon) were performed in Dyrk1a gene and the loss of one complete copy was confirmed for F0-356, -368, -378 and F0-385. The junction for F0-378 is detected only with primers located farther from the sgRNAs and shows a more important deletion than expected (see Supplementary Fig. S1).
We next investigated a second locus for the Dual-specificity tyrosine phosphorylation-regulated kinase 1a (Dyrk1a). In the last decade, DYRK1a has become one of the top candidate gene in DS for therapeutic intervention28,29. It is mutated in the Mental Retardation Autosomal Dominant 7 (MRD7, OMIM 614104)30,31,32. This gene is located within the DS-critical region on HSA21 (ENSG00000157540; see Fig. 1). Transgenic Dyrk1a mouse models exist and have already provided key information on the role of this gene33,34. The homozygous Dyrk1a-deleted (Dyrk1a−/−) mice present a general embryonic growth delay and die during mid gestation. Heterozygous Dyrk1a+/− mice display reduced postnatal survival, postnatal growth retardation, microcephaly, behavioural and motor deficits, and altered neocortical pyramidal cell morphology35,36,37,38. As no rat models exist, the availability of Dyrk1a trisomic or monosomic rat models would be valuable tools for understanding this gene’s function in DS and MRD7. The rat Dyrk1a (ENSRNOG00000001662) is located on RNO11 (Fig. 1). We designed two pairs of sgRNAs in order to generate structural variants of rat Dyrk1a gene (see Fig. 2B and Supplementary Table 1). The size of the whole targeted region was 121.7 kb. Injection conditions are described in Table 1. We observed 7.4% (2 out of 28) of the pups showing the 121.7 kb duplication (Fig. 2E) and three of them (10.7%; 3 out of 28 pups) showed a clear inversion (Fig. 2E). We were not able to detect the deletion of the region by standard PCR using six combinations of primers (F1 + R3 or R4; F3 + R3 or R4; F5 + R3 or R4, see Supplementary Table 1). This was most surprising as we clearly observed that seven of the potential founder pups were significantly smaller than wild-type rat at early post-natal time (2–3 weeks; Fig. 2F) reminding us the phenotype observed in Dyrk1a+/− mice35,36,37,38. We suspected a failure in the amplification of the junction PCR detecting the deletion. We, therefore, decided to use droplet digital PCR (ddPCR) to quantify Dyrk1a allele copy number. From this, we were able to confirm one of the duplication events (F0-384) but not the other (F0-386) (Fig. 2F). Germline transmission was confirmed for founder F0-384 using both ddPCR and junction sequencing. In founder F0-386, allele counting demonstrated that Dyrk1a remained at two copies, suggesting that one allele of Dyrk1a is deleted whereas the second allele is duplicated in most of the F0 founder cells. Most striking was the confirmation of the presence of a single copy of Dyrk1a in four out of the seven small rats (Fig. 2F). We were not able to detect any modification in the three remaining small rats by ddPCR or junction sequencing. Again, this might be due to mosaicism rendering copy counting difficult or other undetectable chromosomic rearrangement. As expected, inversions were not detected by ddPCR, the number of copy remains stable when an inversion occurs. Founders were weighted at ten-eleven weeks. We observed a clear reduction in weight (average of 368 grams) for the 4 founders with one Dyrk1a copy (ddPCR detection) compared to the other founders (average of 464 grams) when two Dyrk1a copies detected by ddPCR (Supplementary Fig. 1A). Despite its low weight, the male F0-378 (1 copy detected by ddPCR) fertilized a female before undergoing euthanasia (very bad general state) at 10 weeks of age. We collected tissue samples, purified genomic DNA and performed long range PCR in order to assess the precise junction of the deletion. We selected new primers farther to the expected break sites and were able to amplify and sequence the junction (Supplementary Fig. 1B and D and Supplementary Data). The size of the region deleted was indeed larger than expected and explained why we were not able to detect it with the standard primers used initially. Three F1 pups (out of 8) transmitted the deletion as confirmed by ddPCR and junction PCR (Supplementary Fig. 1C and D). As expected, these rats are 40% smaller than their wt littermates (Supplementary Fig. 1E).
Generation of large regions structural variant ranging from 3.6 Mb to 24.4 Mb in rat
We wanted to assess if larger chromosomic rearrangements are also possible. We targeted the rat 3.6 Mb region spanning from Umodl1 (ENSRNOG00000001157) to Prmt2 (ENSRNOG00000001297) located on RNO20 and encompassing the most telomeric part of the HSA21. This region is the second larger syntenic region to HSA21 (see Fig. 1). Two sgRNA pairs targeting the 5′ region of Umodl1 at the most telomeric part (sgRNA95 and sgRNA91) and the 3′ region of Prmt2 (sgRNA86 and sgRNA61) were synthesized (Fig. 3A; Supplementary Table 1 and Supplementary Data S3A). Injection conditions are described in Table 1. We observed 5% (2 out of 40 pups) pups showing a 3.6 Mb deletion (Fig. 2B). The experiment showed that manipulation of large genomic sequence is feasible. No germ line transmission was achieved. Another set of experiments with two different sgRNA pairs was performed (Fig. 3C; Supplementary Table 1 and Supplementary Data S3C). Rat pups born after microinjection were screened by ddPCR (Fig. 3D) and junction PCR. One copy of Cbs (gene located in the region) was clearly detected in F0-782 whereas 1.7 and 1.8 copies were counted in F0-801 and F0-802 (Fig. 3D, left panel). No PCR products were obtained with junction PCRs. These 3 founders were bred in order to analyse their offspring. Germ line transmission of both the duplication and deletion was obtained from F0-802 (Fig. 3D, right panel). Both junctions were determined by junction PCR and sequencing (Fig. 3F and E and Supplementary Data S3). Nine different ddPCRs were performed on different gene present on the 3.6 Mb fragment, all confirmed that the whole region was indeed duplicated or deleted (Fig. 3F).

(A) Diagrams show both regions of interest (in orange, surrounded by 5′ (blue) and 3′ (brown) regions). Two pairs of sgRNAs flank both regions of interest. Shown are the position of the primers used for genotyping and position of the sgRNAs. (B) Targeted deletion of Umodl2-Prmt2 region (3.6 Mb) in founders. Shown the deletion junction chromatograms obtained after PCR amplification with specific primers pairs (F4 + R1) in rat founders F0-334 and F0-335. (C) Scheme of the 3.6 Mb genomic region with zoom at both extremities. Positions of the sgRNAs and PCR primers are illustrated. ddPCRs were performed on 9 genes (Abcg1, green; Pde9a, black; Cbs, blue; Sik1, brown; Trpm2, purple; Itgb2, pink; Slc19a1, dark blue; Lss, orange and S100b, red) distributed over the 3.6 Mb region. The region highlighted with a large light green arrow is found at the junction of the duplication. Oligonucleotides used (F and R) are shown (D) Droplet digital PCR results. Left panel of the graph shows some F0 results: a single copy of Cbs was detected in F0-782 whereas 1.8 copies of Cbs were detected in F0-802. On the right panal are F1 results: 1 or 3 copies of Cbs were clearly detected on the F0-802 first litter showing that both deletion and duplication went germ line. (E) Confirmation of the tandem duplication and deletion by junction sequencing. A 590 bp inversed fragment corresponding to the region 5′ of the first pair of sgRNA is found at the duplication junction. A small (70 bp) inversed sequence is found at the deletion junction (see Supplementary data S3D and E for details) (F) ddPCR results with 9 primer pairs on 2 F1 pups (291-DEL and 294-DUP) confirmed the deletion and duplication of the whole genomic region; a WT rat was taken as control (2 copies) (G) Targeted deletion and duplication of >24 Mb region in a stillborn rat F0. Shown are duplication and deletion junctions amplified by PCR on a stillborn rat F0 (Supplementary nts in gray). The gel pictures of both duplication and deletion junctions are presented.
A larger 24.4 Mb chromosomal rearrangement was then attempted to recapitulate the human largest syntenic region to HSA21 (spanning from Lipi to Zfp295, see Fig. 1). We designed two pairs of sgRNAs for upstream and downstream sites flanking the rat 24.4 Mb DNA region located on RNO11 (Fig. 3A; Table 1; Supplementary Data S4 and Supplementary Table 1). Different combinations of primers were used to identify inversion, deletion, and duplication junctions (Fig. 3A and Supplementary data S4A). The monosomy of this 24.4 Mb region induces lethality in mice but the trisomy is viable both in mice39 and human (Down syndrome patients). We took care to genotype the stillborn rat pups. On the unique stillborn founder F0, we were able to detect both the deletion and tandem duplication of the 24.4 Mb region by junction PCR and Sanger sequencing (Fig. 3G and Supplementary Data S4). In addition, small deletions at both break points were also detected in the same stillborn rat (Supplementary Data S4B). A deletion of 51 bps was found near Lipi between the two DSBs induced by the Cas9 3 bps upstream of the PAM sites. A deletion of 35 bps was found between the two DSBs accompanied with a short insertion of 10 nts in near Zfp295. This shows the high efficiency of the sgRNAs at both extremities. Eight out of the 9 founders had indels at a least one or the other break points. Subsequently, we performed injections in order to obtain the duplication but have not been able to obtain a surviving founder F0 with the duplication only. These experiments highlight the limit of our approach, namely the inherent viability problems of the variant generated in the rodent. We are currently assessing if duplication can occur in the germ line cells of founder F0s by breeding a few founders in which we detected DSBs at both 5′ and 3′ break points.
We show with these four examples that any kind of chromosomal rearrangement of any size can be fairly easily achieved in rat using CRISPR/Cas9. The only limitation we find is the lethality of the variant generated. Indeed, our evidence suggests that the larger the genomic fragment, the higher the risk to cause lethality.
Generation of structural variants of various size in mouse
We have demonstrated that the CRISMERE strategy in rat can be easily used to manipulate large genomic regions. Thus, we decided to test the strategy in mouse to generate a panel of DS models.
First, we attempted the deletion and duplication of the high mobility group nucleosomal binding domain 1 (Hmgn1; ENSMUSG00000040681), a 16.8 kb locus (Fig. 1). In human, overexpression of HMGN1 (ENSG00000205581), a nucleosome remodelling protein encoded on chromosome 21q2240, suppresses histone 3 lysine 27 trimethylation (H3K27me3) and promotes both B cell proliferation in vitro and B cell acute lymphoblastic leukemia in vivo41. Two pairs of sgRNAs surrounding the murine Hmgn1 sequence were synthesized (see Fig. 4A, Supplementary Data S5 and Supplementary Table 1). Injection conditions are described in Table 1. We were able to detect the deleted allele in 4 out of 8 founders (50%). Two founders were bred with wild type animals and both gave germline transmission (founder F0-3: 13 F1 out of 49 (26%); founder F0-8: 4 F1 out of 20 (20%) (Fig. 4A). These mice are currently breeding to establish phenotyping cohorts.

Diagrams are as in Fig. 3. Shown are junctions amplified by PCR from mouse tail with specific primers. Examples of sequence chromatograms of the different junctions are shown. (A) Generation of monosomic Hmgn1 mouse line. The chromatogram of the deletion junction observed in F0-3 is shown (details in supplementary data S6). Germ line transmission was confirmed in 7 out of 15 offspring. (B,C) Deletion and duplication of mouse Tiam1 (226 kb). (B) Shown the chromatogram of the duplication junction observed in F0-14, the gel picture of the PCR products from F0-14 and a F1-30. Graph showing ddPCR results on both F0-14, 7 F1 offspring and a WT animal. F0-14 mosaicism is shown as 2.3 Tiam1 copies are detected with 2 distinct ddPCRs (5′ and 3′ of Tiam1, red and blue dots). Germ line transmission was confirmed by ddPCRs (3 copies detected in F1-30). (C) Shown the chromatogram of the deletion junction (primers F2 + R4) of F0-41 and gel picture with the PCR for F0-38, F0-40 and F0-41. Germ line transmission for the deletion was confirmed by ddPCR (F1-30 to F1-34; one copy detected) whereas the founder (F0-41) had a mosaic profile (1.7 copies of Tiam1). Note also a duplicated allele (3 copies of Tiam1) is also detected in F0-41 offspring (F1-28). A single copy of Tiam1 was detected in F0-38 by ddPCR (next graph) suggesting absence of mosaicism. The same ddPCR profile was observed for 2 of the offspring (F1-15 and -13). (D) Targeted deletion and inversion of the mouse genomic region ranging from Runx1 to Cbr1 (1 Mb). Shown the chromatogram of the deletion junction amplified by PCR from F0-32 tail (F1 + R3) with accompanying gel picture. Inversion of the whole region is confirmed by PCR amplification and Sanger sequencing of the 5′ junction on F0-7 (F1 + F4). Droplet digital PCRs on 3 genes distributed over the 1 Mb region (Runx1, blue; Gm28003, red and Cbr1, green) detected 3 copies in F0-138 as well as some of its offspring (F1-3, -6 and -7). A WT control (2 copies) and a deletion (1 copy) are included.
Next we focused on the T-cell lymphoma invasion and metastasis 1 gene (TIAM1; ENSG00000156299). Indeed TIAM1 is located on HSA21 and has been shown to be a critical regulator of different aspects of Ras-induced tumour formation42. We aimed to generate the deletion and duplication of the whole murine Tiam1 gene (ENSMUSG00000002489) a region spanning 226 kb. We were mainly interested in the trisomic version of this gene as the monosomic model (i.e. heterozygous knock-out mice) has already been studied42. We thus decided to duplicate the Tiam1 gene using the double sgRNA pair strategy. Microinjection conditions can be found in Table 1. Forty one mouse pups were born and PCR primer pairs were designed to detect all the combination of alleles (see Fig. 4B). We observed 19.5% (8 out of 41) of the pups showing a 226 kb deletion and one of them showed a clear duplication (Fig. 4B) (2.4%; 1 out of 41 pups) (Table 1). No inversions were detected by junction PCR. Three founders with a deleted allele were bred with wild type C57BL/6 N mice and germ line transmission was obtained for each of them (Fig. 4C). Junction sequencing of F1 mice confirmed the deletion observed in founders. Seventeen F1 pups were born from founder F0-38, seven (41%) had the deletion. The Tiam1 deletion was also confirmed by droplet digital PCR (Fig. 4C) with two different pairs of primers specific from both extremities of the Tiam1 gene. Interestingly, ddPCR confirmed that founder F0-38 was carrying mostly heterozygous cells as only one copy was detected. This mutant allele also demonstrated a high Mendelian transmission rate observed (41%, close to the 50% rate expected). Thirty-four mice were born after breeding the founder F0-41 and only eight F1 (23%) had the deleted allele. ddPCR copy counting (1.7 Tiam1 copy number, Fig. 4C) confirmed the mosaic nature of this animal. Interestingly, we were able to detect 3 Tiam1 copy number in one F1 mouse (out of 7 F1 pups analysed by ddPCR). Thus, a duplication event can be transmitted to the germ line that is not necessarily detected in a founder F0 (see Fig. 4C, mouse 28). Considering that we were not able to detect this duplication by standard junction genotyping, this highlights the complementarity of both genotyping techniques. Finally, the F0-14 founder for which we detected a duplication by Sanger sequencing also showed a mosaic pattern by ddPCR (2.3 copies detected), we obtained only one single F1 pups with the same duplication (out of 33 genotyped) showing that the founder was highly mosaic. We demonstrate here that ddPCR allows us to detect deletions and duplications in F1 or later generations and eliminates the possibility of PCR failures (as the PCR is not performed at the junctions but inside the copy number variant). However ddPCR fails to detect inversions and rare/mosaic rearrangement and some mosaic founders (del/dup in G1, see discussion). Regardless of the means of detection, we have demonstrated that DNA region deletion or duplication by CRISPR with two pairs of sgRNAs can be transmitted through germline in mice.
We next focused on the 1 Mb long mouse genomic region comprising three genes (Runx1, Setd4 and Cbr1) (see Fig. 1). Two lincRNAs are also located in this interval (1810053B23Rik and 1700029J03Rik). Runx1 homozygote knockouts die by embryonic day 12.5 from a complete lack of definitive hematopoiesis and lethal central nervous system (CNS) hemorrhage43 whereas heterozygotes have reduced erythroid and myeloid progenitor numbers. Two pairs of sgRNAs surrounding this region were chosen (Fig. 4D and Supplementary Data S6). Microinjection conditions are described in Table 1. We were able to genotype one founder with an inversion of the 1.1 Mb region (3% of genotyped pups) as confirmed by Sanger sequencing of the 5′ junction (Fig. 4D). Another F0 founder (F0-32) showed a confirmed deletion (3%). This F0 pup died after 3 weeks. We performed another round of microinjection with the same sgRNAs and obtained founders with one copy of the region and copy numbers ranging from 1 to 2.6 (data not shown). We obtained germ line transmission for the deletion (1 copy by ddPCR; 3 genes spread across the 1.1 Mb region- Runx1, Gm28003 and Cbr1) and the duplication (3 copies by ddPCR, Fig. 4D). Altogether, these data definitively demonstrated for the first time that segmental duplication, deletion, or inversion of any size can be efficiently induced in rodents by CRISMERE with two pairs of sgRNAs (see Table 1 for summary).
Discussion
In this study we have shown that any kind of chromosomal rearrangements can be generated in both rat and mouse using CRISMERE. The rearrangements ranged in size from a few kilobases to more than 24 megabases and were achieved with relative ease in vivo for rat and mice. Our method for generating DNA fragment inversions, deletions and duplications is simple and efficient. The efficiency of these rearrangements mediated by CRISPR/Cas9 is a significant improvement over methods through recombinases and nucleases15,20,44,45,46 paving the way for efficient DNA fragment inversion, deletion, and duplication in any species in vivo. Indeed, editing large DNA fragments of several hundred kb can be used to model DNA segmental duplications, which are common in mammalian genomes47. Previously, the generation of such a model was possible only in a very narrow range of species, very time consuming, expensive, and limited in the variety of rearrangements one sought to mimic among the many observed in human disease. Now with the CRISMERE strategy described above such chromosomal modifications should be straightforward.
According to our results there are two possibilities to obtain genomic DNA region rearrangements for two concomitant DSBs (here generated by pairs of sgRNAs): intra-chromosomal recombination between two DSBs on a single chromosome (Fig. 5A) and trans-allelic recombination between two DSBs each on one of the two chromosomes (Fig. 5B). These events must happen in mitotic phase G1 before the chromosome replication. The range of events that can be obtained with CRISMERE is even broader than what was first expected. Indeed, three or four DSBs can also occur in mitotic phase G1 and could lead to different combination of duplication (head to head or tail to tail) (Fig. 5C). Chromosomic recombination via CRISMERE can also occur in a cis configuration in G2 leading directly to monosomic and trisomic daughter cells after mitosis (Fig. 5D). The mechanism would be similar to targeted asymmetric sister chromatid event of recombination TASCER48,49. Moreover, in rat and mouse founders, the variety of alleles is high and the size of the genomic region to rearrange does not seem to lead to consequences. Noteworthy, the orientation of the sgRNAs does not seem to favour one or the other rearrangement (data not shown).

Chromosomes are represented by a thick gray line, the centromeres by a gray dot and the region of interest in yellow. (A–C) Standard chromosomic recombination when Cas9 edits the genome in G1. After mitosis, two alleles distinct from the initial WT allele will be obtained (A) Intra-chromosomal recombination between two DSBs on a single chromosome (B) Trans-allelic recombination between two DSBs each on one of the two chromosome. (C) Trans-allelic recombination between (three or) four DSBs on the two chromosomes ending with head to head, tail to tail duplication (D) Schematic of the event that should take place in the eggs were Cas9 edits the genome in Cis configuration in G2 leading to monosomic and trisomic daughter cells after mitosis. Trans-allelic recombination between two DSBs on the two chromatids in G2 (similar to targeted asymmetric sister chromatid event of recombination TASCER).
CRISMERE is a powerful tool to generate duplication, deletion and inversion of different sizes, in different species and different genes or locus. All of the structural variants described in these paper will be valuable tools for the study of gene dosage in the context of Down syndrome. We anticipate that the only limit for CRISMERE will be the nature of the genomic region, i.e. the viability of its deletion, inversion, or duplication. Owning to the high efficiency of generating blunt end by Cas9, we showed that inversion, deletion and duplication of DNA fragments with defined length ranging in size from tens to thousands of kilobases can be easily achieved. Applying this technology to the editing of small DNA fragments such as millions of regulatory DNA elements50 is possible now. Also, generating mutations involving large DNA fragments of several hundred kilobases can be used to study complex gene clusters, the organization of chromosome in its genomic milieu, and copy number variants common in mammalian genomes47.
Materials and Methods
All experiments were carried out in accordance with” relevant guidelines and regulations. Animal experiments were approved by the Com’Eth N°17 and accredited by the French Ministry for Superior Education and Research (“MESR”, MESR 00130 & MESR 01639), and were supervised by M.C.B. and YH who are qualified in compliance with the European Community guidelines for laboratory animal care and use (2010/63/UE) in our animal facility (Agreement C67-218-40).
In vitro transcription of Cas9 mRNA and sgRNAs
We used the CRISPOR software (http://tefor.net/crispor/crispor.cgi), developed by the French TEFOR infrastructure, to select sgRNAs. Each sgRNA name refers to the specificity score given by the software. The Cas9 vector (T7-Cas9 wt cloned in pUC57) with T7 promoter was first linearized with AccI for use as a template for in vitro transcription with T7 polymerase. Cas9 mRNA was transcribed using mMESSAGEmMACHINE T7 Ultra Kit according to the manufacturer’s manual (Life Technologies). sgRNAs templates with T7 promoter were obtained by PCR amplification with primers (Supplementary Table S1). The MEGAshortscript Kit (Life Technologies) was used to transcribe sgRNAs from the PCR product template. Cas9 mRNA and sgRNAs were purified with the MEGAclear Kit (Life Technologies) and eluted in TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, InVitrogen) for microinjections.
In vitro cleavage assay to validate sgRNA’s functionality
Before injecting sgRNAs in eggs, all the guides were tested in vitro on the targeted DNA PCR product in order to validate their efficiency. PCR surrounding the targets were performed as described in Mouse and rat genotyping for detecting DNA fragment inversions, duplications, and deletions section below. Approximately 200 ng of target PCR product were mixed with 300 ng of Cas9 protein (PNA Bio Inc), 150 ng of sgRNA, 10X NEB buffer 3, 10X BSA and nuclease water qsp 10 μl. As control the Cas9 protein was omitted. The reactions were incubated at 37 °C for 1 h. One μl STOP solution (30% glycerol, 1.2% SDS, 250 mM EDTA, pH8.0) was added and the mix was incubated for 15 mins at 37 °C. The reaction was loaded on a 3% agarose gel. The correct sizes after Cas9 cleavage allow to validate the functionality of the tested guide. Only sgRNAs showing a cut (even partial) were injected in eggs.
One-cell embryos injection
All mice were housed at 21 °C on a 14/12 h light-dark cycle (5:00am–07:00 pm) in the SPF facilities. Sexually immature female C57BL/6 N mice (4–5 weeks olds) were superovulated by intraperitoneal injection of 5 IU eCG followed by 2.5 IU hCG at an interval of 48 h and mated overnight with C57BL/6NCrl male mice that were >10 weeks old. Zygotes were collected after 20 h of hCG injection by oviductal flashing, and pronuclei-formed zygotes were put into the M2 medium (Sigma M-7167). Microinjection was performed using a microinjector (Eppendorf Femtojet 4i) equipped microscope. RNA solution was injected into the cytoplasm and the pronucleus of each zygote using continuous pneumatic pressure. After injection, embryos were in vitro cultured in the M16 medium (Sigma M-7292) at 37 °C in a 5% CO2 incubator. The survivors of the injected embryos were implanted into the oviducts of pseudo-pregnant CD1 mice.
All rats were housed at 21 °C on a 14/12 h light-dark cycle (5:00am–07:00 pm) in the SPF facilities. Sexually immature female Sprague Dawley rat (4–5 weeks olds) were superovulated by intraperitoneal injection of 20 IU eCG followed by 30 IU hCG at an interval of 48 h and mated overnight with Sprague Dawley male mice that were >10 weeks old. Zygotes were collected after 20 h of hCG injection by oviductal flashing, and pronuclei-formed zygotes were put into the M2 medium. Microinjection was performed using a microinjector equipped microscope. RNA solution was injected into the cytoplasm and the pronucleus of each zygote using continuous pneumatic pressure. After injection, embryos were in vitro cultured in the M16 medium at 37 °C in a 5% CO2 incubator. The survivors of the injected embryos were implanted into the oviducts of pseudo-pregnant CD1 mice.
Mouse and rat genotyping for detecting DNA fragment inversions, duplications, and deletions
PCR was used to identify inversions, duplications, and deletions in mice with appropriate primer pairs (Supplementary Table S1). Those PCRs are performed with Phusion Taq HS (Fermentas) in a final volume of 20 μl. The PCR conditions are: predenaturing at 94 °C for 30 sec; followed by 35 cycles of 94 °C denaturation for 8 sec, 60 °C annealing for 10 sec, and 72 °C extension for 1 min; followed by a final extension at 72 °C for 5 min. Ten μl of these PCR products were denaturated, re-annealed and digested with T7 endonuclease in order to detect small deletions. Two size markers were used (GeneRuler 50 bp DNA ladder SM0372 and Generuler DNA ladder Mix SM0333).
Droplet digital PCR validation and Copy number Variation
To confirm the deletion or the duplication in mice or rats, several ddPCR were performed using the QX200 droplet reader (Bio-Rad). Each ddPCR reaction contained duplex TaqMan assay reagents for the target and reference genes. The ddPCR reaction mixture consisted of 10 μl of a 2 × ddPCR Mastermix (Bio-Rad), 1 μl of target 20x primer/probe mix (IDT), 1 μl of reference 20× primer/probe mix (IDT), and 2 μl of DNA in a final volume of 20 μl. All primers and probes sequences are listed in Supplementary Table 2. The ratio between primer and probe was kept at 3:1. Primers and probes were designed with Prime time assays from Integrated DNA technologies or with Universal ProbeLibrary website from Roche Diagnostics. Each assembled ddPCR reaction mixture was then loaded into the sample well of an eight-channel disposable droplet generator cartridge (Bio-Rad). A volume of 70 μL of droplet generation oil (Bio-Rad) was loaded into the oil well for each channel. The cartridge was placed into the droplet generator (Bio-Rad). The cartridge was removed from the droplet generator, where the droplets that collected in the droplet well were then manually transferred with a multichannel pipet to a 96-well PCR plate. The plate was heat-sealed with a foil seal and then placed on a conventional thermal cycler and amplified to the end-point as follow: 95 °C for 10 min, followed by 40 cycles of 95 °C for 20 s and 58.2 °C for 60 s, 1 cycle of 98 °C for 10 min and ending at 12 °C. After PCR, the 96-well PCR plate was loaded on the droplet reader (Bio-Rad), which automatically reads the droplets from each well of the plate (32 wells/h). Analysis of the ddPCR data and CNV were performed with QuantaSoft analysis software (Bio-Rad) that accompanied the droplet reader.
Additional Information
How to cite this article: Birling, M.-C. et al. Efficient and rapid generation of large genomic variants in rats and mice using CRISMERE. Sci. Rep. 7, 43331; doi: 10.1038/srep43331 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Dierssen, M., Herault, Y. & Estivill, X. Aneuploidy: from a physiological mechanism of variance to Down syndrome. Physiol Rev 89, 887–920 (2009).
Choong, X. Y., Tosh, J. L., Pulford, L. J. & Fisher, E. M. Dissecting Alzheimer disease in Down syndrome using mouse models. Front Behav Neurosci 9, 268 (2015).
Lana-Elola, E. et al. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. Elife 5 (2016).
Brault, V. et al. Opposite phenotypes of muscle strength and locomotor function in mouse models of partial trisomy and monosomy 21 for the proximal Hspa13-App region. PLoS Genet 11, e1005062 (2015).
Jiang, X. et al. Genetic dissection of the Down syndrome critical region. Hum Mol Genet (2015).
Olson, L. E., Richtsmeier, J. T., Leszl, J. & Reeves, R. H. A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science 306, 687–690 (2004).
Brudno, M. et al. Automated whole-genome multiple alignment of rat, mouse, and human. Genome Res 14, 685–692 (2004).
Zhao, S. et al. Human, mouse, and rat genome large-scale rearrangements: stability versus speciation. Genome Res 14, 1851–1860 (2004).
Mullins, L. J. & Mullins, J. J. Insights from the rat genome sequence. Genome Biol 5, 221 (2004).
Nigam, R. et al. Rat Genome Database: a unique resource for rat, human, and mouse quantitative trait locus data. Physiol Genomics 45, 809–816 (2013).
Zhang, L. et al. Human chromosome 21 orthologous region on mouse chromosome 17 is a major determinant of Down syndrome-related developmental cognitive deficits. Hum Mol Genet 23, 578–589 (2014).
Brault, V., Pereira, P., Duchon, A. & Herault, Y. Modeling chromosomes in mouse to explore the function of genes, genomic disorders, and chromosomal organization. PLoS Genet 2, e86 (2006).
Brault, V., Besson, V., Magnol, L., Duchon, A. & Herault, Y. Cre/loxP-mediated chromosome engineering of the mouse genome. Handb Exp Pharmacol 178, 29–48 (2007).
Herault, Y., Rassoulzadegan, M., Cuzin, F. & Duboule, D. Engineering chromosomes in mice through targeted meiotic recombination (TAMERE). Nat Genet 20, 381–384 (1998).
Ramirez-Solis, R., Liu, P. & Bradley, A. Chromosome engineering in mice. Nature 378, 720–724 (1995).
Zheng, B., Mills, A. A. & Bradley, A. A system for rapid generation of coat color-tagged knockouts and defined chromosomal rearrangements in mice. Nucleic Acids Res 27, 2354–2360 (1999).
Adams, D. J. et al. Mutagenic insertion and chromosome engineering resource (MICER). Nat Genet 36, 867–871 (2004).
Ruf, S. et al. Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nature Genetics 43, 379–386 (2011).
Spitz, F., Herkenne, C., Morris, M. A. & Duboule, D. Inversion-induced disruption of the Hoxd cluster leads to the partition of regulatory landscapes. Nat Genet 37, 889–893 (2005).
Li, J. et al. Efficient inversions and duplications of mammalian regulatory DNA elements and gene clusters by CRISPR/Cas9. J Mol Cell Biol 7, 284–298 (2015).
Wang, L. et al. Large genomic fragment deletion and functional gene cassette knock-in via Cas9 protein mediated genome editing in one-cell rodent embryos. Scientific Reports 5, 17517 (2015).
Zhang, L. et al. Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS One 10, e0120396 (2015).
Kraft, K. et al. Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell Rep 10, 833–339 (2015).
Boroviak, K., Doe, B., Banerjee, R., Yang, F. & Bradley, A. Chromosome engineering in zygotes with CRISPR/Cas9. Genesis 54, 78–85 (2016).
Lopes Pereira, P. et al. A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum Mol Genet 18, 4756–4769 (2009).
Yu, T. et al. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Research 1366, 162–171 (2010).
Watanabe, M. et al. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci USA 92, 1585–1589 (1995).
De la Torre, R. et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol Nutr Food Res 58, 278–288 (2014).
Luco, S. M. et al. Case report of novel DYRK1A mutations in 2 individuals with syndromic intellectual disability and a review of the literature. BMC Med Genet 17, 15 (2016).
Bronicki, L. M. et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. Eur J Hum Genet 23, 1482–1487 (2015).
van Bon, B. W. et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiatry 21, 26–32 (2016).
Møller, R. S. et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am J Hum Genet 82, 1165–1170 (2008).
Guedj, F. et al. DYRK1A: a master regulatory protein controlling brain growth. Neurobiol Dis 46, 190–203 (2012).
Altafaj, X. et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum Mol Genet 10, 1915–1923 (2001).
Fotaki, V. et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol 22, 6636–6647 (2002).
Benavides-Piccione, R. et al. Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/− mouse. Neurobiol Dis 20, 115–122 (2005).
Fotaki, V., Martinez De Lagran, M., Estivill, X., Arbones, M. & Dierssen, M. Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav Neurosci 118, 815–821 (2004).
Arque, G. et al. Impaired spatial learning strategies and novel object recognition in mice haploinsufficient for the dual specificity tyrosine-regulated kinase-1A (Dyrk1A). PLoS One 3, e2575 (2008).
Li, Z. et al. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet 16, 1359–1366 (2007).
Deng, T. et al. HMGN1 modulates nucleosome occupancy and DNase I hypersensitivity at the CpG island promoters of embryonic stem cells. Mol Cell Biol 33, 3377–3389 (2013).
Lane, A. A. et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet 46, 618–623 (2014).
Malliri, A. et al. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 417, 867–871 (2002).
Growney, J. D. et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 106, 494–504 (2005).
Wu, S., Ying, G., Wu, Q. & Capecchi, M. R. Toward simpler and faster genome-wide mutagenesis in mice. Nat Genet 39, 922–930 (2007).
Lee, H. J., Kweon, J., Kim, E., Kim, S. & Kim, J. S. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome Res 22, 539–548 (2012).
Gupta, A. et al. Targeted chromosomal deletions and inversions in zebrafish. Genome Res 23, 1008–1017 (2013).
Pan, D. & Zhang, L. Tandemly arrayed genes in vertebrate genomes. Comp Funct Genomics, 545269 (2008).
Duchon, A., Besson, V., Pereira, P. L., Magnol, L. & Herault, Y. Inducing segmental aneuploid mosaicism in the mouse through targeted asymmetric sister chromatid event of recombination. Genetics 180, 51–59 (2008).
Herault, Y. et al. Controlled somatic and germline copy number variation in the mouse model. Curr Genomics 11, 470–480 (2010).
Tai, D. J. et al. Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci 17 517–522 (2016).
Acknowledgements
This work was supported by the National Centre for Scientific Research (CNRS), the French National Institute of Health and Medical Research (INSERM), the University of Strasbourg (UDS), the “Centre Européen de Recherche en Biologie et en Médecine”. This study also received support from French state funds through the “Agence Nationale de la Recherche” under the frame programme Investissements d’Avenir labelled ANR-10-IDEX-0002-02, ANR-10-LABX-0030-INRT, ANR-10-INBS-07 PHENOMIN, and the Fondation Jérôme Lejeune (grant #1166) and the Fondation Bettencourt-Schueller to Y.H. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We are grateful to G. Prevost for caring our rats, J-V Figerolle and C Kujath for mouse injection, D Ali-Hadji and her team for caring for the animals, and P Reilly for critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
L.S. performed most of the molecular biology experiments and did all PCR analyses. P.A. performed all the rat microinjection experiments. L.L. performed ddPCR experiments and M.-C.B. conceived the experimental strategy, analyzed the data. D.M., A.A. and T.S. supervise the mouse and rat breeding work and M.-C.B., G.P. and Y.H. designed the study and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Birling, MC., Schaeffer, L., André, P. et al. Efficient and rapid generation of large genomic variants in rats and mice using CRISMERE. Sci Rep 7, 43331 (2017). https://doi.org/10.1038/srep43331
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43331
This article is cited by
-
Exon skipping induced by CRISPR-directed gene editing regulates the response to chemotherapy in non-small cell lung carcinoma cells
Gene Therapy (2022)
-
Cas9-induced large deletions and small indels are controlled in a convergent fashion
Nature Communications (2022)
-
Importing genetically altered animals: ensuring quality
Mammalian Genome (2022)
-
Progress towards completing the mutant mouse null resource
Mammalian Genome (2022)
-
Targeting the RHOA pathway improves learning and memory in adult Kctd13 and 16p11.2 deletion mouse models
Molecular Autism (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
