
Abstract
Excessive myocardial collagen deposition and cross-linking (CCL), a process regulated by lysyl oxidase (LOX), determines left ventricular (LV) stiffness and dysfunction. The angiotensin II antagonist losartan, metabolized to the EXP3179 and EXP3174 metabolites, reduces myocardial fibrosis and LV stiffness in hypertensive patients. Our aim was to investigate the differential influence of losartan metabolites on myocardial LOX and CCL in an experimental model of hypertension with myocardial fibrosis, and whether EXP3179 and EXP3174 modify LOX expression and activity in fibroblasts. In rats treated with NG-nitro-L-arginine methyl ester (L-NAME), administration of EXP3179 fully prevented LOX, CCL and connective tissue growth factor (CTGF) increase, as well as fibrosis, without normalization of blood pressure (BP). In contrast, administration of EXP3174 normalized BP and attenuated fibrosis but did not modify LOX, CCL and CTGF. In TGF-β1-stimulated fibroblasts, EXP3179 inhibited CTGF and LOX expression and activity with lower IC50 values than EXP3174. Our results indicate that, despite a lower antihypertensive effect, EXP3179 shows higher anti-fibrotic efficacy than EXP3174, likely through its ability to prevent the excess of LOX and CCL. It is suggested that the anti-fibrotic effect of EXP3179 may be partially mediated by the blockade of CTGF-induced LOX in fibroblasts.
Similar content being viewed by others

Introduction
Myocardial fibrosis is involved in the development of left ventricular (LV) dysfunction and clinically overt heart failure (HF) in hypertensive patients1,2,3. Diverse experimental studies indicate that collagen-dependent LV chamber stiffness is influenced not only by the amount of collagen fibers but also by the degree of collagen cross-linking (CCL) within the fibers, a process whereby collagen fibrils are covalently linked to one another by the enzyme lysyl oxidase (LOX), providing stiffness and resistance to degradation of the resulting fibers4,5,6,7. Of notice, recent clinical studies point to an excess of CCL as a major determinant of LV dysfunction and clinical outcomes in patients with HF of hypertensive etiology1,8,9,10. In this context, the reduction of myocardial LOX expression and CCL has been demonstrated to be associated with diminution of LV chamber stiffness in hypertensive patients with HF2.
The angiotensin II type 1 receptor antagonist losartan is a prodrug metabolized by the cytochrome-P450 pathway in the liver producing two metabolites: EXP3174 and EXP3179. EXP3174 is the final metabolite and the pharmacological blocker of the AT1 receptor by which losartan exerts its antihypertensive actions11. EXP3179 is an intermediate metabolite that has no AT1 receptor blocking properties and mediates a variety of AT1 receptor-independent, non-hemodynamic actions12,13,14,15. It is known that losartan reduces LV stiffness in hypertensive patients, an effect associated with the reduction of the content of myocardial collagen fibers16. However, there is no information on the effects of this drug on myocardial LOX and CCL in hypertension. Therefore, we have investigated the effects of EXP3179 and EXP3174 on fibrosis, and LOX and CCL in the myocardium of rats with arterial hypertension induced by the NG-nitro-L-arginine methyl ester (L-NAME), an experimental model characterized by myocardial transforming growth factor-β1 (TGF-β1) overexpression and fibrosis17,18,19. In addition, we aimed to explore whether losartan metabolites influence LOX expression and activity in fibroblasts stimulated with TGF-β1.
Results
In Vivo Findings
Effects of Losartan Metabolites on Blood Pressure (BP)
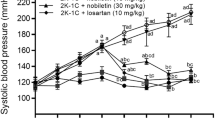
From the third week until the end of treatment, systolic and diastolic blood pressure (SBP and DBP) were elevated in L-NAME rats compared to control normotensive rats (Fig. 1). L-NAME + EXP3179 rats exhibited reduced SBP and DBP as compared with L-NAME rats, although BP values remained significantly increased as compared to control normotensive rats (Fig. 1A,B). On the other hand, L-NAME + EXP3174 rats showed SBP and DBP values similar to control normotensive rats throughout the entire 10-week treatment period (Fig. 1A,B). Treatment with either EXP3179 or EXP3174 in the absence of L-NAME did not influence either SBP or DBP (data not shown).

Time-course changes in systolic blood pressure (SBP) (panel A) and diastolic blood pressure (DBP) (panel B) in normotensive controls rats (□), L-NAME-treated rats (●), L-NAME + EXP3179-treated rats (x) and L-NAME + EXP3174-treated rats (○). Data points represent mean ± SEM (n = 5). *P < 0.01 vs Control and L-NAME + EXP3174, #P < 0.05 vs L-NAME and Control.
Effects of losartan metabolites on LV morphology and function
As shown in Table 1, compared to control normotensive rats, L-NAME rats exhibited LV hypertrophy, as indicated by the increased relative wall thickness (RWT) and LV mass index (LVMI). The LVMI was normalized in L-NAME + EXP3179 rats but remained increased in L-NAME + EXP3174 rats (Table 1).
In addition, Table 1 shows that L-NAME rats exhibited reduced LV systolic (e.g., reduced ejection fraction and fractional shortening) and diastolic (e.g., reduced E/A ratio) function compared to control normotensive rats. Co-treatment of L-NAME rats with either EXP3179 or EXP3174 prevented these alterations (Table 1). None of the parameters assessing LV morphology and function were modified in rats treated with either metabolite alone (data not shown).
Effects of Losartan Metabolites on Myocardial TGF-β1 and CTGF Expression
Compared to control normotensive rats, L-NAME rats exhibited increased expression of myocardial TGF-β1 mRNA, which was fully prevented in L-NAME + EXP3179 and in L-NAME + EXP3174 rats (Fig. 2A). In addition, L-NAME rats showed increased expression of CTGF mRNA (Fig. 2B) and protein (Fig. 2C) compared to control normotensive rats. Co-treatment of L-NAME rats with EXP3179, but not with EXP3174, significantly decreased CTGF mRNA and protein expression induced by L-NAME (Fig. 2B,C, respectively). Treatment with either EXP3179 or EXP3174 in the absence of L-NAME did not exert any effect on these parameters (Fig. 2).

Histograms represent the expression of cardiac TGF-β1 (panel A) and CTGF (panel B) mRNA and CTGF protein (panel C) in the myocardium of Vehicle-, EXP3179-, EXP3174-, L-NAME + Vehicle-, L-NAME + EXP3179- and L-NAME + EXP3174-treated rats. Representative Western blot autoradiograms for CTGF are presented at the bottom of panel C. Bars represent mean + SEM (n = 10). *P < 0.05 vs Vehicle, EXP3179 and EXP3174, †P < 0.05 vs L-NAME + Vehicle, #P < 0.05 vs Vehicle and L-NAME + Vehicle.
Effects of Losartan Metabolites on Myocardial Collagen Synthesis and Accumulation
Collagen volume fraction (CVF) was increased in L-NAME rats compared to control normotensive rats, being normal in L-NAME + EXP3179 rats but still abnormally increased in L-NAME + EXP3174 rats (Fig. 3A,B). Compared to control normotensive rats, L-NAME rats exhibited increased procollagen type I expression in cardiac tissue, which was fully prevented in L-NAME + EXP3179 rats and only partially prevented in L-NAME + EXP3174 rats (Fig. 3C). None of these parameters were modified in rats treated with either EXP3179 or EXP3174 in the absence of L-NAME (Fig. 3).

Histograms represent the percentage of collagen volume fraction (panel A) in the myocardium of Vehicle-, EXP3179-, EXP3174-, L-NAME + Vehicle-, L-NAME + EXP3179- and L-NAME + EXP3174-treated rats. Representative images of myocardial tissue from one rat of each group are shown in panel B. Sections were stained with picrosirius red and collagen fibers were identified in red. Expression of procollagen type I mRNA is shown in panel C. Bars represent mean + SEM (n = 10). *P < 0.05 vs Vehicle, EXP3179 and EXP3174, †P < 0.05 vs L-NAME + Vehicle, #P < 0.05 vs Vehicle and L-NAME + Vehicle.
In addition, the myocardium of L-NAME rats exhibited increased LOX mRNA (Fig. 4A) and protein (Fig. 4B) expression, as well as an excess of CCL (Fig. 4C). These alterations were fully prevented in L-NAME + EXP3179 rats but not in L-NAME + EXP3174 rats. Treatment with either EXP3179 or EXP3174 in the absence of L-NAME did not exert any effect on these parameters (Fig. 4).

Histograms represent the fold change in the expression of LOX mRNA (panel A), LOX protein (panel B) and collagen cross-linking (panel C) in the myocardium of Vehicle-, EXP3179-, EXP3174-, L-NAME + Vehicle-, L-NAME + EXP3179- and L-NAME + EXP3174-treated rats. Representative Western blot autoradiograms for LOX protein expression are presented at the bottom of panel B. Bars represent mean + SEM (n = 10). *P < 0.05 vs Vehicle, EXP3179 and EXP3174, †P < 0.05 vs L-NAME + Vehicle.
In Vitro Findings
Effects of Losartan Metabolites on Procollagen Type I Expression, and LOX Expression and Activity in TGF-β1-stimulated adult human dermal fibroblasts (HDFa)
As it has been previously published for cardiac fibroblast20, incubation of HDFa with TGF-β1 for 24 h induced a dose-dependent increase in both procollagen type I mRNA (see Supplementary Fig. S1A) and LOX (see Supplementary Fig. S1B) expression. TGF-β1 10−4 μg/mL was selected as the lowest concentration with submaximal effects on these parameters (see Supplementary Fig. S1).
HDFa fibroblasts were incubated with 10−4 μg/mL TGF-β1 in the presence of EXP3179 and EXP3174 at increasing concentrations. TGF-β1-induced procollagen type I expression was significantly inhibited by EXP3179 (Fig. 5A) and it tended to be reduced by EXP3174 (Fig. 5B).

Histograms represent the fold change in procollagen type I mRNA expression in HDFa fibroblasts stimulated without and with 10−4 μg/mL TGF-β1 for 24 hours, in the absence or the presence of 1, 2, 5, 10, 20 and 50 μM of EXP3179 (panel A) and EXP3174 (panel B), or incubated with the two compounds alone at 20 and 50 μM, as compared with controls cells. Bars represent mean + SEM (n = 5 to 8 experiments). *P < 0.05 vs Control, †P < 0.05 vs TGF-β1.
In addition, EXP3179 inhibited TGF-β1 effects on LOX mRNA (Fig. 6A) and protein (Fig. 6B) expression, with a clear tendency to inhibit extracellular LOX activity (Fig. 6C). Moreover, EXP3174 inhibited TGF-β1-induced LOX mRNA expression (Fig. 6D) and tended to reduce TGF-β1-induced LOX protein (Fig. 6E) and activity (Fig. 6F) only at high doses.

Histograms depict the fold change in LOX mRNA, protein and activity in HDFa fibroblasts stimulated without and with 10−4 μg/mL TGF-β1 for 24 hours, in the absence or the presence of 1, 2, 5, 10, 20 and 50 μM of EXP3179 (panels A–C) and EXP3174 (panels D–F), or incubated with the two compounds alone at 20 and 50 μM, as compared to control cells. Line graphs show percentage of inhibition in LOX mRNA (panel G), protein (panel H), and activity (panel I), in HDFa stimulated with 10−4 μg/mL TGF-β1 in combination with EXP3174 (□) or EXP3179 (∆) at the above mentioned concentrations, compared to cells incubated with TGF-β1 alone. Bars represent mean +SEM (n = 5 to 8 experiments) and curves show means ± SEM (n = 5 to 8). *P < 0.05 vs Control, †P < 0.05 vs TGF-β1, ††P < 0.01 vs TGF-β1 and $P < 0.05 vs EXP3174.
For all LOX parameters, EXP3179 was identified as a more potent inhibitor as compared to EXP3174 (Fig. 6G–I), with lower IC50 values than EXP3174 (LOX mRNA: 1.6 ± 0.1 vs 29.4 ± 1.5 μM, P < 0.001; LOX protein: 8.9 ± 1.0 vs 38.5 ± 5.0 μM, P < 0.05; LOX activity: 7.4 ± 0.3 vs 37.3 ± 2.9 μM, P < 0.05).
Involvement of Intracellular Pathways on EXP3179-mediated Inhibition of LOX Expression
We investigated whether classical intracellular pathways involved in the actions described for EXP3179 in other experimental settings12,13,14,15 were also involved in its inhibitory effects on TGF-β1-induced LOX upregulation in HDFa fibroblasts. However, none of the compounds tested (i.e. the inhibitors of PPAR-γ [G3335] and PI3K [LY294002], and the activators of PKC [PMA] and COX [LPS]) were able to counteract the actions of EXP3179 on LOX mRNA expression (see Supplementary Fig. S2).
In addition, we analysed in HDFa fibroblasts the involvement of diverse fibrosis-related genes in the EXP3179-induced LOX inhibition, by using the RT2 ProfilerTM PCR Array Human Fibrosis. As shown in table S1, 84 genes were examined, confirming the TGF-β1-induced increment in LOX expression and its inhibition in the presence of EXP3179, both exceeding a 1.5 fold change. Therefore, this threshold was chosen to select those genes overexpressed by TGF-β1 that were inhibited by EXP3179. According to the array results and following the proposed criteria, CTGF and thrombospondin-1 (THBS1) were selected as genes that were potentially involved in the anti-fibrotic actions of EXP3179 (Supplemental Table S1). The inhibitory actions of EXP3179 on these genes were confirmed by real-time RT-PCR (data not shown). Therefore, CTGF and THBS1 genes were selected for further analyses.
By silencing CTGF expression with siRNA in HDFa, TGF-β1-induced LOX expression was inhibited. On the contrary, siRNA-mediated THBS1 inhibition did not exert any influence on this pathway (Supplementary Fig. S3A). In addition, we observed that, as in the case of LOX, EXP3179 inhibited CTGF mRNA expression in a dose-dependent manner (see Supplementary Fig. S3B).
Discussion
The main findings of this study are the following: 1) Chronic inhibition of NO synthesis with L-NAME in Wistar rats resulted in a hypertensive model of myocardial fibrosis with increased expression of CTGF and LOX, as well as enhanced CCL; 2) Administration of EXP3179 in L-NAME rats exerted a partial anti-hypertensive effect, whereas EXP3174 fully prevented the increase in BP; 3) Whereas the administration of EXP3179 in L-NAME rats fully prevented myocardial CTGF and LOX overexpression, and excessive CCL and fibrosis, EXP3174 administration was not able to prevent LOX overexpression and CCL, and only partially reduced myocardial fibrosis; and 4) EXP3179 inhibited the TGF-β1-induced upregulation of LOX expression and activity in fibroblasts, probably through CTGF regulation, in a more effective manner than EXP3174.
The evidence supports that L-NAME-induced hypertension is a well-established experimental model, representative of the left ventricular remodeling, and specifically of the myocardial fibrosis, occurring in hypertensive heart disease17,18,19,21,22,23,24,25. In addition, Tsukamoto et al. further characterized this model demonstrating that the lack of NO is associated with impairment of LV systolic and diastolic function22. In this regard, we confirm these findings and expand the characterization of myocardial fibrosis in this model with novel data demonstrating that reduced NO bioavailability is associated with increased myocardial LOX expression and enhanced cross-linking of collagen fibrils.
Although several clinical16,26,27 and experimental studies28,29,30 have demonstrated that losartan prevents and/or regresses the myocardial fibrosis associated with arterial hypertension, the role of losartan metabolites in this anti-fibrotic effect has not been characterized. In this regard, we report here that administration of EXP3179 completely prevented the excess of CTGF, LOX and CCL, and fully abrogated myocardial fibrosis, yet without normalization of BP. In contrast, we found that although administration of EXP3174 normalized BP, it failed to prevent myocardial CTGF and LOX overexpression, as well as the excess of CCL, and only partially reduced myocardial fibrosis. Of notice, both treatments prevented LV systolic and diastolic dysfunction. Therefore, we may speculate that EXP3179 may improve LV function by inhibiting CTGF-dependent pro-fibrotic mechanisms in cardiac fibroblasts, with a mild anti-hypertensive effect, whereas EXP3174 may improve LV function through normalization of BP with a concomitant, mild inhibitory effect on myocardial fibrosis.
In order to further evaluate the potential molecular mechanisms involved in the differential anti-fibrotic effects of losartan metabolites, in vitro experiments were performed. In accordance with previous studies20,31, we observed an upregulation of procollagen type I and LOX expression and activity in fibroblasts stimulated with TGF-β1. In addition, we observed that CTGF mediates TGF-β1-induced LOX expression, confirming previous studies in which such an association was suggested32. Moreover, we report that EXP3179 showed a higher efficacy than EXP3174 in reducing LOX expression in HDFa, with significant effects at doses similar to those present in blood from patients chronically treated with losartan (~2μM)33. In this regard, examination of a large number of genes in the fibrosis pathway and the in vitro experiments with specific siRNAs revealed that EXP3179 effects on LOX are likely mediated through downregulation of CTGF. Our results provide mechanistic support to previous findings demonstrating that losartan is able to interfere with the profibrotic activity of TGF-β1, including downregulation of CTGF34,35,36. In this regard, since CTGF may be a critical mediator of the EXP3179 actions on LOX expression, evaluation of different intracellular pathways participating in the regulation of CTGF (e.g., the c-Jun N-terminal kinase [JNK] pathway)37,38 could be considered for future studies. This is of particular interest taking into account that we observed that rats co-treated with L-NAME and EXP3174 exhibited CTGF overexpression despite TGF-β1 being normalized, with a mild anti-fibrotic effect. In this regard, the TGF-β1-independent upregulation of CTGF and collagen synthesis induced by angiotensin II has been reported in different in vivo models of renal damage39, and in atrial fibrillation40. In addition, other mechanisms besides TGF-β1, and related to the renin-angiotensin system, hemodynamic stress41 or inflammation/oxidative stress17, have been reported as inductors of myocardial fibrosis in L-NAME-treated rats. On the other hand, the mild antifibrotic effect shown by EXP3174 may be due to abrogation of chronic hemodynamic stress, independently of CTGF. This notion is supported by previous studies demonstrating that treatment with losartan or hydrochlorothiazide showed similar anti-fibrotic and anti-hypertensive effects in the myocardium of SHR rats receiving high salt diet, being losartan more effective in decreasing CTGF expression42.
Some limitations of the present study must be acknowledged. First, a losartan group was not included in the study. Nevertheless, the aim of the study was to examine the effects of both metabolites on myocardial fibrosis present in L-NAME rats, in order to determine their individual anti-fibrotic actions on the hypertensive myocardium. Second, few studies have applied EXP3179 as an individual potential drug and therefore a dose-dependent pharmacological profile of EXP3179 has not been established yet. Third, further experiments are needed to analyse the myocardial distribution of extracellular matrix/profibrotic proteins in the L-NAME-induced hypertension model. Fourth, in the in vitro study, the potential contribution of the remaining differentially expressed genes in the array was not analysed in depth. In addition, further studies are required to confirm these findings in human cardiac fibroblasts. Finally, it is unknown whether the anti-fibrotic effects of EXP3179 were solely due to prevention of collagen synthesis and deposition or if the metabolite also influences the enzymes that control collagen degradation.
We conclude that despite a lower hypertensive efficacy, the losartan metabolite EXP3179 is more effective improving myocardial fibrosis than EXP3174 in L-NAME rats. In particular, we found that EXP3179 is more efficient than EXP3174 downregulating LOX expression, with these effects resulting in a normalization of CCL. Of interest, these molecular and anti-fibrotic effects of EXP3179 in L-NAME rats may be mediated by the inhibition of the TGF-β1-CTGF pathway that is activated in the myocardium of these rats.
Methods
For detailed description, see Methods in the online Data Supplement.
In Vivo Procedure
Forty five 10-week-old male Wistar rats were treated for 10 weeks, once-daily, with 30 mg/kg of L-NAME by oral administration, a dose sufficient to induce arterial hypertension43 and 45 untreated rats were considered as controls. Fifteen animals in each group were co-treated, once-daily, with either vehicle, EXP3179 or EXP3174 by oral administration. EXP3174 was administered at 5 mg/kg/day, a dose resulting in circulating metabolite levels similar to those found in patients chronically treated with losartan33,44. EXP3179 was administered at the same dose. Blood pressure (BP) was assessed in five animals from each group by radiotelemetry monitoring.
Echocardiographic Studies
Echocardiography was performed using a Vevo 770 ultrasound system. The heart rate (HR) of the animals was recorded immediately before the echocardiographic study.
Collagen Cross-linking Analysis
The evaluation of the degree of CCL was performed using Fast Green-Sirius Red and Sircol-based colorimetric assays in myocardial tissue.
Assessment of Collagen Volume Fraction
CVF was determined in heart sections of rats as a percentage of total myocardial area occupied by collagen tissue.
In Vitro Procedure
After carrying out dose-response curves to human recombinant TGF-β1 (RαD systems), adult human dermal fibroblasts (HDFa line; GIBCO) were incubated for 24 hours with or without 10−4 μg/mL, in the absence or presence of the metabolites EXP3174 and EXP3179 (provided by Merck & Co, Inc.) at a range of concentrations. In addition, HDFa cells were co-incubated with EXP3179 in the absence or presence of the following compounds: the inhibitors of PPAR-γ (G3335) and phosphatidylinositol 3-kinase (PI3K) pathway (LY294002) and the activators of protein kinase-C (PKC) (Phorbol 12-myristate 13-acetate [PMA]) and cyclo-oxygenase-2-pathway (lipopolysaccharide [LPS]).
Human fibrosis array
Samples from cells incubated in control conditions or with TGF-β1, in the absence or presence of EXP3179 (20μM) were examined by using the Human Fibrosis RT2 Profiler PCR Array (SABiosciences Corp.) consisting of a panel of 84 key genes involved in fibrosis.
Quantitative Real-time PCR and Gene Silencing
Gene expression was analysed by quantitative real-time PCR by using specific TaqMan fluorescent probes. Expression of connective tissue growth factor (CTGF) and thrombospondin 1 (THBS1) was silenced by small interfering RNAs in HDFa.
Assessment of Protein Expression and Evaluation of LOX Activity
The expression of several proteins was analysed by Western Blot. LOX activity was assessed by a commercially available fluorimetric assay.
Statistical Analysis
Variables are expressed as means ± SEM. Differences among different conditions were tested by one-way ANOVA followed by the Fisher’s least significant difference method for post-hoc comparisons once normality was checked (Shapiro-Wilks test); otherwise, the nonparametric Kruskal-Wallis test followed by a Mann-Whitney U test (adjusting the α-level by Bonferroni inequality) was used. Statistical significance was defined as two-sided P < 0.05. The analyses were performed using the programs SPSS (15.0 version) and STATA (12.1 version).
Ethical standards
All authors in this work gave their informed consent prior to their participation in the study. The manuscript does not contain clinical studies or patient data. The research conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No 85–23, revised 1996), and was approved by the Ethical Committee for Animal Experimentation of the University of Navarra (036/08).
Additional Information
How to cite this article: Miguel-Carrasco, J. L. et al. Mechanisms underlying the cardiac antifibrotic effects of losartan metabolites. Sci. Rep. 7, 41865; doi: 10.1038/srep41865 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Zile, M. R. et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 131, 1247–1259 (2015).
López, B., Querejeta, R., González, A., Beaumont, J., Larman, M. & Díez, J. Impact of treatment on myocardial lysyl oxidase expression and collagen cross-linking in patients with heart failure. Hypertension 53, 236–242 (2009).
Dai, Z., Aoki, T., Fukumoto, Y. & Shimokawa, H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol. 60, 416–421 (2012).
Norton, G. R., Tsotetsi, J., Trifunovic, B., Hartford, C., Candy, G. P. & Woodiwiss, A. J. Myocardial stiffness is attributed to alterations in cross-linked collagen rather than total collagen or phenotypes in spontaneously hypertensive rats. Circulation 96, 1991–1998 (1997).
Badenhorst, D. et al. Cross-linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovasc Res. 57, 632–641 (2003).
Shoulders, M. D. & Raines, R. T. Collagen structure and stability. Annu Rev Biochem. 78, 929–958 (2009).
Brower, G. L. et al. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg. 30, 604–610 (2006).
Kasner, M. et al. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol 57, 977–985 (2011).
López, B., Querejeta, R., González, A., Larman, M. & Díez, J. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension 60, 677–683 (2012).
López, B. et al. Myocardial collagen cross-linking is associated with hospitalization for heart failure in patients with hypertensive heart failure. J Am Col Cardiol 67, 251–260 (2016).
Lo, M. W., Goldberg, M. R., McCrea, J. B., Lu, H., Furtek, C. I. & Bjornsson, T. D. Pharmacokinetics of losartan, an angiotensin II receptor antagonist, and its active metabolite EXP3174 in humans. Clin Pharmacol Ther 58, 641–649 (1995).
Krämer, C. et al. Angiotensin II receptor-independent antiinflammatory and antiaggregatory properties of losartan: role of the active metabolite EXP3179. Circ Res 90, 770–776 (2002).
Watanabe, T., Suzuki, J., Yamawaki, H., Sharma, V. K., Sheu, S. S. & Berk, B. C. Losartan metabolite EXP3179 activates Akt and endothelial nitric oxide synthase via vascular endothelial growth factor receptor-2 in endothelial cells: angiotensin II type 1 receptor-independent effects of EXP3179. Circulation 112, 1798–1805 (2005).
Fortuño, A. et al. Losartan metabolite EXP3179 blocks NADPH oxidase-mediated superoxide production by inhibiting protein kinase C. Potential clinical implications in hypertension. Hypertension 54, 744–750 (2009).
Schupp, M. et al. Regulation of peroxisome proliferator-activated receptor gamma activity by losartan metabolites. Hypertension 47, 586–589 (2006).
Díez, J., Querejeta, R., López, B., González, A., Larman, M. & Martínez Ubago, J. L. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 105, 2512–2517 (2002).
Ndisang, J. F., Chibbar, R. & Lane, N. Heme oxygenase suppresses markers of heart failure and ameliorates cardiomyopathy in L-NAME-induced hypertension. Eur J Pharmacol 734, 23–34 (2014).
Zambrano, S. et al. L-Carnitine protects against arterial hypertension-related cardiac fibrosis through modulation of PPAR-γ expression. Biochem Pharmacol 85, 937–944 (2013).
Silambarasan, T., Manivannan, J., Krishna Priya, M., Suganya, N., Chatterjee, S. & Raja, B. Sinapic acid prevents hypertension and cardiovascular remodeling in pharmacological model of nitric oxide inhibited rats. PLoS One 9, e115682 (2014).
Voloshenyuk, T. G., Landesman, E. S., Khoutorova, E., Hart, A. D. & Gardner, J. D. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine 55, 90–97 (2011).
Bunbupha, S., Prachaney, P., Kukongviriyapan, U., Kukongviriyapan, V., Welbat, J. U. & Pakdeechote, P. Asiatic acid alleviates cardiovascular remodelling in rats with L-NAME-induced hypertension. Clin Exp Pharmacol Physiol 42, 1189–1197 (2015).
Tsukamoto, O. et al. The antagonism of aldosterone receptor prevents the development of hypertensive heart failure induced by chronic inhibition of nitric oxide synthesis in rats. Cardiovasc Drug Ther 20, 93–102 (2006).
Yamashita, T. et al. Apoptosis signal-regulating kinase-1 is involved in vascular endothelial and cardiac remodeling caused by nitric oxide deficiency. Hypertension 50, 519–524 (2007).
Ferreira-Melo, S. E. et al. Sildenafil reduces cardiovascular remodeling associated with hypertensive cardiomyopathy in NOS inhibitor-treated rats. Eur J Pharmacol 542, 141–147 (2006).
Tomita, H. et al. Early induction of transforming growth factor-beta via angiotensin II type 1 receptors contributes to cardiac fibrosis induced by long-term blockade of nitric oxide synthesis in rats. Hypertension 32, 273–279 (1998).
Shimada, Y. J. et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol HF 1, 480–487 (2013).
López, B. et al. Usefulness of serum carboxy-terminal propeptide of procollagen type I in assessment of the cardioreparative ability of antihypertensive treatment in hypertensive patients. Circulation 104, 286–291 (2001).
Varo, N. et al. Losartan inhibits the post-transcriptional synthesis of collagen type I and reverses left ventricular fibrosis in spontaneously hypertensive rats. J Hypertens 17, 107–114 (1999).
Boffa, J. J., Lu, Y., Placier, S., Stefanski, A., Dussaule, J. C. & Chatziantoniou C. Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol 14, 1132–1144 (2003).
Varo, N., Iraburu, M. J., Varela, M., López, B., Etayo, J. C. & Díez, J. Chronic AT(1) blockade stimulates extracellular collagen type I degradation and reverses myocardial fibrosis in spontaneously hypertensive rats. Hypertension 35, 1197–1202 (2000).
Roy, R., Polgar, P., Wang, Y., Goldstein, R. H., Taylor, L. & Kagan, H. M. Regulation of lysyl oxidase and cyclooxygenase expression in human lung fibroblasts: interactions among TGF-beta, IL-1 beta, and prostaglandin E. J Cell Biochem 62, 411–417 (1996).
Hong, H. H., Uzel, M. I., Duan, C., Sheff, M. C. & Trackman, P. C. Regulation of lysyl oxidase, collagen, and connective tissue growth factor by TGF-beta1 and detection in human gingiva. Lab Invest 79, 1655–1667 (1999).
Kappert, K. et al. Chronic treatment with losartan results in sufficient serum levels of the metabolite EXP3179 for PPARgamma activation. Hypertension 54, 738–743 (2009).
Cohn, R. D. et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med 13, 204–210 (2007).
Geirsson, A. et al. Modulation of transforming growth factor-β signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation 126, S189–197 (2012).
Friedberg, M. K. et al. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased transforming growth factor-β1 signaling and is abrogated by angiotensin receptor blockade. Am J Respir Cell Mol Biol 49, 1019–1028 (2013).
Shi, L., Chang, Y., Yang, Y., Zhang, Y., Yu, F. S. & Wu, X. Activation of JNK signalling mediates connective tissue growth factor expression and scar formation in corneal wound healing. PLoS One 7, e32128 (2012).
Weng, C. M., Yu, C. C., Kuo, M. L., Chen, B. C. & Lin, C. H. Endothelin-1 induces connective tissue growth factor expression in human lung fibroblasts by ETAR-dependent JNK/AP-1 pathway. Biochem Pharmacol 88, 402–411 (2014).
Yang, F., Chung, A. C., Huang, X. R. & Lan, H. Y. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 54, 877–884 (2009).
Kiryu, M. et al. Angiotensin II-mediated up-regulation of connective tissue growth factor promotes atrial tissue fibrosis in the canine atrial fibrillation model. Europace 14, 1206–1214 (2012).
Sadek, S. A., Rashed, L. A., Bassam, A. M. & Said, E. S. Effect of aliskiren, telmisartan and torsemide on cardiac dysfunction in l-nitro arginine methyl ester (l-NAME) induced hypertension in rats. J Adv Res 6, 967–974 (2015).
Gröholm, T. et al. Cardioprotective effects of vasopeptidase inhibition vs. angiotensin type 1-receptor blockade in spontaneously hypertensive rats on a high salt diet. Hypertens Res 27, 609–618 (2004).
Morán, A., de Urbina, A. V., Martín, M. L., Rodríguez-Barbero, A. & Román, L. S. Characterization of the contractile 5-hydroxytryptamine receptor in the autoperfused kidney of L-NAME hypertensive rats. Eur J Pharmacol 620, 90–96 (2009).
Yan, Y. D. et al. The physicochemical properties, in vitro metabolism and pharmacokinetics of a novel ester prodrug of EXP3174. Mol Pharm 7, 2132–2140 (2010).
Acknowledgements
The authors thank Ana Montoya, Idoia Rodríguez, Ana Igea, Sonia Martínez and María J. González for their valuable technical assistance. This study was supported by the Ministry of Economy and Competitiveness, Madrid, Spain (SAF2011-29610, SAF2013-49088-R, Instituto de Salud Carlos III grant RD12/0042/0009 and CIBER-CV CB16/11/00483), and the European Commission FP7 Programme, Brussels, Belgium (MEDIA project grant HEALTH-2010-261409, and FIBRO-TARGETS project grant FP7-HEALTH-2013-602904).
Author information
Authors and Affiliations
Contributions
J.L.M-C., J.B. and G.S.J., co-designed the study, conducted animal studies and revised the manuscript critically for important intellectual content. M.U.M., B.L. and A.G., co-designed the study, performed experiments and revised the manuscript critically for important intellectual content. G.Z., A.F., S.R. and J.D., co-designed the study, wrote and edited the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Miguel-Carrasco, J., Beaumont, J., San José, G. et al. Mechanisms underlying the cardiac antifibrotic effects of losartan metabolites. Sci Rep 7, 41865 (2017). https://doi.org/10.1038/srep41865
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41865
This article is cited by
-
Irbesartan, an angiotensin II type 1 receptor blocker, inhibits colitis-associated tumourigenesis by blocking the MCP-1/CCR2 pathway
Scientific Reports (2021)
-
Nebivolol is more effective than atenolol for blood pressure variability attenuation and target organ damage prevention in L-NAME hypertensive rats
Hypertension Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
