
Abstract
In the aconitase superfamily, which includes the archetypical aconitase, homoaconitase, and isopropylmalate isomerase, only aconitase X is not functionally annotated. The corresponding gene (LhpI) was often located within the bacterial gene cluster involved in L-hydroxyproline metabolism. Screening of a library of (hydroxy)proline analogues revealed that this protein catalyzes the dehydration of cis-3-hydroxy-L-proline to Δ1-pyrroline-2-carboxylate. Furthermore, electron paramagnetic resonance and site-directed mutagenic analyses suggests the presence of a mononuclear Fe(III) center, which may be coordinated with one glutamate and two cysteine residues. These properties were significantly different from those of other aconitase members, which catalyze the isomerization of α- to β-hydroxy acids, and have a [4Fe-4S] cluster-binding site composed of three cysteine residues. Bacteria with the LhpI gene could degrade cis-3-hydroxy-L-proline as the sole carbon source, and LhpI transcription was up-regulated not only by cis-3-hydroxy-L-proline, but also by several isomeric 3- and 4-hydroxyprolines.
Similar content being viewed by others
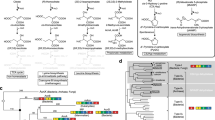
Introduction
L-Hydroxyproline has been found in certain proteins, particularly in collagen, and in some peptide antibiotics. In mammalian systems, the L-proline residue is post-translationally hydroxylated to trans-4-hydroxy-L-proline [(2S,4R)-4-hydroxyproline] or trans-3-hydroxy-L-proline [(2S,3S)-3-hydroxyproline] by a diverse set of prolyl 4-hydroxylase (EC 1.14.11.2) and prolyl 3-hydroxylase (EC 1.14.11.7) with different protein substrates, respectively1. A number of bacterial and fungal enzymes are known to directly hydroxylate free L-proline to trans-4-hydroxy-L-proline2, trans-3-hydroxy-L-proline3,4, cis-4-hydroxy-L-proline [(2S,4S)-4-hydroxyproline]5, or cis-3-hydroxy-L-proline [(2S,3R)-3-hydroxyproline]6, which are also found as components of several peptide antibiotics produced by these microorganisms7,8,9,10,11.
Among the several stereoisomers of L-hydroxyproline, trans-4-hydroxy-L-proline is the most common in nature, and is converted to pyruvate and glyoxylate via three intermediates in mammals (Table S1)1. On the other hand, a number of bacteria, including Pseudomonas aeruginosa PAO1 and Pseudomonas putida KT2440, metabolize trans-4-hydroxy-L-proline to α-ketoglutarate by four enzymes (encoded by LhpA~F genes) (Fig. 1a and Table S1)12,13. In contrast to trans-4-hydroxy-L-proline, the metabolic pathways of trans-3-hydroxy-L-proline in bacteria and mammalians may be similar, and consist of a dehydratase (LhpG) and a reductase (LhpH or LhpK), by which trans-3-hydroxy-L-proline is converted to L-proline via Δ1-pyrroline-2-carboxylate (Fig. 1b and Table S1)14,15. It is likely that the reductase is also involved in cis-3-hydroxy-L-proline metabolism in bacteria, in which Δ1-pyrroline-2-carboxylate is produced by a different dehydratase than the LhpH protein (LhpJ) (Fig. 1c and Table S1)16. These LhpA~K genes often cluster together with the putative L-hydroxyproline (ABC-type) transporter genes, transcriptional regulator gene, aconitase-like gene (LhpI) and proline racemase-like gene (LhpL) on bacterial genomes (Fig. 1d; referred to as the “L-hydroxyproline gene cluster”)12,13,15,16. Among these hypothetical genes, it is likely that the LhpI gene (protein) in particular is not necessary for the known pathway of L-hydroxyproline metabolism due to its putative function, based on amino acid sequence similarity.

L-Hydroxyproline metabolism in bacteria.
(a) trans-4-Hydroxy-L-proline, (b) trans-3-hydroxy-L-proline, and (c) cis-3-hydroxy-L-proline pathways. (d) Schematic gene clusters related to the metabolism of L-hydroxyproline. Enzyme names and EC numbers are listed in Table S1. Homologous genes are indicated in the same color and correspond to Fig. 1(a,b,c). A characteristic pair of catalytic amino acid residues of proline racemase superfamily enzymes are shown in the bottom of the LhpA, LhpG, and LhpL genes (see “Supplementary Discussion”). Gray and TR genes are putative L-hydroxyproline transporters and transcriptional regulators, respectively. Genes with up arrows from P. aeruginosa PAO1 and S. meliloti 2011 are induced by trans-4-hydroxy-L-proline28,29. Mutant strains of the underlined genes cannot grow on trans-4-hydroxy-L-proline as a sole carbon source.
The aconitase superfamily contains four functional hydro-lyase enzymes: aconitase (EC 4.2.1.3; Acn), 2-methylcitrate dehydratase (EC 4.2.1.79; AcnD), homoaconitase (EC 4.2.1.114; HACN), and isopropylmalate isomerase (EC 4.2.1.33; IPMI), which have been classified into eight phylogenetic subfamilies: (1) AcnA of bacteria17; (2) AcnB of bacteria18; (3) mitochondrial Acn (mAcn)19; (4) cytoplasmic Acn and iron regulatory protein (IRP) of mammalians20; (5) AcnD of bacteria21; (6) HACN of bacteria and archaea22,23; (7) IPMI of bacteria, archaea, and fungi24,25,26; (8) function unknown aconitase X (AcnX) (Fig. 2). All of these subfamilies (referred to as “Acn enzymes”), except for the last one, catalyze the homologous stereospecific isomerization of α- to β-hydroxyl acids by sequential dehydration and hydration (anti-elimination/addition), and their active centers contain a [4Fe-4S] iron-sulfur cluster. By contrast, AcnX (subfamily) was initially discovered by a comparative analysis of archaeal genomes in 200327. Although the secondary structural elements of Acn enzymes may also be conserved in this subfamily, the sequence similarities between them are insignificant. The AcnX subfamily has been further classified into “AcnXType I”, consisting of a single polypeptide from bacteria, and “AcnXType II” found in a number of bacteria (AcnXType IIa) and archaea (AcnXType IIb), which consists of (fragmented) small and large polypeptide chains. The small polypeptide (subunit) of AcnXType II is homologous to the so-called “fourth domain” of Acn enzymes as well as the IPMI and HACN of bacteria and archaea (Fig. 2)22,23,24,25. Among the three AcnX subfamilies, LhpI protein corresponds to AcnXType I.

Schematic reactions of four members of the Acn superfamily.
A linear representation of the sequential domain arrangement of six phylogenetic subfamilies is also included. “c” is the connector domain, and “heat” is a protein-protein interaction domain found only in AcnB18. In propionate metabolism in bacteria21,51, (2S,3S)-2-methylcitrate is converted to 2-methyl-cis-acotinate through two routes: (1) sequential reactions by AcnD and PrpF via 2-methyl-trans-acotinate; (2) reaction by a single protein, PrpD (gray-colored routes). 2-Methyl-cis-acotinate is subsequently metabolized to (2S,3R)-2-methylisocitrate by AcnB. The AcnX subfamily is further classified into three groups, among which AcnXType I, consisting of a single polypeptide from bacteria, was functionally characterized in this study.
We report here that LhpI protein catalyzes the dehydration of cis-3-hydroxy-L-proline to Δ1-pyrroline-2-carboxylate, by a reaction that was not homologous with those of known Acn enzymes, and contains mononuclear Fe(III), but not the [4Fe-4S] cluster. Bacterial growth and transcriptional analysis suggested that this enzyme is surely involved in cis-3-hydroxy-L-proline metabolism.
Results and Discussion
Identification of substrates and functions
A genetic analysis of the L-hydroxyproline gene cluster (PA1252-1269) from Pseudomonas aeruginosa PAO1 was recently reported in detail (Fig. 1d)28. Among the 18 components, 10 genes including PA1259 (LhpI) were significantly induced by trans-4-hydroxy-L-proline. Furthermore, the LhpI mutant, constructed by transposon insertion, as well as mutants of the known trans-4-hydroxy-L-proline metabolic gene lost the ability to grow on trans-4-hydroxy-L-proline. However, this phenotype may be due to a polar effect on downstream genes28. The non-polar LhpI deletion mutant (SMb20269; Fig. 1d) of Sinorhizobium meliloti 1021 was previously reported to grow normally on trans-4-hydroxy-L-proline29. Therefore, there has been no evidence to show that the LhpI gene plays a role in the metabolism of L-hydroxyproline.
The recombinant (His)6-tagged PaLhpI protein was successfully expressed in P. putida cells, and purified to homogeneity using a nickel-chelating affinity column (Fig. 3a,b). The apparent molecular masses, estimated by SDS-PAGE and analytic gel filtration, were 60 and 67 kDa, indicating a monomeric structure. Initially, no hydro-lyase activity toward citrate and/or cis-acotinate was observed in this protein. Therefore, in the next approach for functional analysis, eight proline derivatives (10 mM; Fig. S1) were tested as substrates in Tris-HCl buffer (pH 8.0) without additives. Among them, only cis-3-hydroxy-L-proline was consumed in a time-dependent manner (specific activity of 39.5 μmol·min−1·mg−1) (Fig. 4a), and the reaction product was identified as Δ1-pyrroline-2-carboxylate by 1H NMR (Fig. 4g). This result confirmed that when the reaction was performed in the co-presence of Δ1-pyrroline-2-carboxylate reductase15, L-proline was produced in a time-dependent manner (Fig. 4b). Collectively, these results suggest that the PaLhpI protein catalyzes (only) the irreversible dehydration reaction of cis-3-hydroxy-L-proline to Δ1-pyrroline-2-carboxylate via a putative Δ2-pyrroline-2-carboxylate intermediate (Fig. 1c). Since cis-3-hydroxy-L-proline possesses the hydroxyl group and proton to be eliminated at the same “anti” positions as Acn enzymes, it is likely that trans-3-hydroxy-L-proline (“syn” positions) is not a substrate for this protein.

Properties of purified LhpI proteins.
(a) SDS-PAGE analysis of recombinant proteins (5 μg in a 10% (w/v) gel). The theoretical molecular masses of PaLhpI, AtLhpI, and TrLhpI are 63354.77, 60389.43, and 65215.77, respectively. The bottom panel is a photograph of LhpI proteins (~30 mg/ml). (b) Elution profile of PaLhpI during gel filtration. The molecular mass, 67 kDa, was determined by using the following equation: y = 80855e−0.082x (R2 = 0.9784). (c) Effects of pH on the activity of PaLhpI. Reactions were buffered with 50 mM acetic acid-NaOH (pH 4.0–6.0) (red), 50 mM potassium phosphate (pH 6.0–8.5) (orange), 50 mM Tris-HCl (pH 7.0–9.0) (blue), and 50 mM glycine-NaOH (pH 9.0–11.0) (green), instead of 50 mM Tris-HCl (pH 8.0) used under standard assay conditions. Values are the average ± SD, n = 3.

Characterization as a cis-3-hydroxy-L-proline dehydratase.
Analysis of reaction products of cis-3-hydroxy-L-proline (a,b,e,f) and 2,3-cis-3,4-cis-dihydroxy-L-proline (c,d) by PaLhpI (a,c), PaLhpI + PaLhpH + NADPH (b,d), TrLhpI (e), or TrLhpI + PaLhpH + NADPH (f) using an amino acid analyzer. Concentrations are expressed as the percentage of the amount initially present. Three independent experiments were performed, and the typical result is shown. (g) 1H NMR spectra of cis-3-hydroxy-L-proline, the reaction product of cis-3-hydroxy-L-proline by PaLhpI, Δ1-pyrroline-2-carboxylate, and trans-3-hydroxy-L-proline (identical to trans-3-hydroxy-D-proline). Asterisks are peaks derived from an internal standard. Left panels show the assignments of protons in D2O. (h) Lineweaver-Burk plots and kinetic parameters of PaLhpI (red), AtLhpI (blue), and TrLhpI (yellow) toward cis-3-hydroxy-L-proline. Values are the average ± SD, n = 3.
In order to investigate substrate specificity in more detail, we prepared a unique 2,3-cis-3,4-cis-3,4-dihydroxy-L-proline [(2 S,3 S,4 R)-3,4-dihydroxyproline] from cis-3-hydroxy-L-proline by L-proline cis-4-hydroxylase from S. meliloti30. This proline derivative was an additional substrate for PaLhpI, whereas specific activity (0.130 μmol·min−1·mg−1), estimated by HPLC analysis, was ~300-fold lower than that of cis-3-hydroxy-L-proline (Fig. 4c). Although the 1H NMR analysis did not identify the enzyme product directly (Fig. S2), cis-4-hydroxy-L-proline was produced by coupling with Δ1-pyrroline-2-carboxylate reductase (Fig. 4d); the reaction product of 2,3-cis-3,4-cis-3,4-dihydroxy-L-proline must be a Δ1-pyrroline-4S-hydroxy-2-carboxylate. Furthermore, no dehydration (degradation) was observed in L-serine, in spite of carbons 1~3 as well as the hydroxyl and amino groups being identical to those of cis-3-hydroxy-L-proline (Fig. S3). These results indicate that PaLhpI recognizes not only the framework and chiral selectivity of 3-hydroxyproline, but also the 4′-functional group of cis-3-hydroxy-L-proline.
Labrenzia aggregata IAM 12614 possesses the L-hydroxyproline gene cluster, which contains the LhpJ gene (SIAM614_RS19325) instead of the LhpI gene (Fig. 1d)16. The LhpJ protein belongs to the muconate lactonizing enzyme subclass of the enolase superfamily, distinct from the Acn superfamily, and shows the bifunctional activity of (1) the reversible 2-epimerization of cis-3-hydroxy-L-proline to trans-3-hydroxy-D-proline and (2) the irreversible dehydration of cis-3-hydroxy-L-proline to Δ1-pyrroline-2-carboxylate (Fig. 1c); the latter is homologous with the activities of hydroxyproline betaine 2-epimerase31 and proline betaine racemase32, other members in this protein superfamily. On the other hand, epimerization activity was not found in PaLhpI (Fig. 4g), possibly due to the different catalytic mechanisms of the proteins, as described below.
Catalytic properties
In the absence of substrate under oxidative conditions, one of the iron ions, so-called “Feα” (Fig. 5a), is easily lost from the [4Fe-4S] cluster in Acn enzymes, leading to the catalytically inactive [3Fe-4S] state; therefore, reconstruction (reactivation) is typically necessary under reducing conditions33. Purified PaLhpI was brown in color, indicating the presence of cofactor(s) (Fig. 3a), whereas inactivation was not detected after three months at −35 °C. The pH dependence of dehydration activity with cis-3-hydroxy-L-proline was estimated by a colorimetric method based on the reaction of 2-aminobenzaldehyde with Δ1-pyrroline-2-carboxylate14,15: optimum pH of 8.0–9.5 (Fig. 3c). Moreover, we developed a more conventional spectrophotometric assay method using NADPH-dependent Δ1-pyrroline-2-carboxylate reductase as a coupling enzyme (see Fig. 4b). Kinetic parameters with cis-3-hydroxy-L-proline are shown in Fig. 4h. The specific activity value, 41.3 μmol·min−1·mg−1, was similar to that obtained by HPLC.

Identification of active sites.
(a) Active site models of mAcn from bovine (PDB ID 1ACO)19 and ([Fe-S] cluster unbound) HACN of M. jannaschii (4KP2)42. So-called “Feα” is not ligated to a cysteine residue. (b) Site-directed mutagenic study of PaLhpI. Numbers in parentheses correspond to the sites in Fig. 7. Relative activity values (average ± SD, n = 3) are expressed as percentages of the values obtained in the WT enzyme. Proteins with asterisks were not expressed in P. putida cells. Inset. Stoichiometric analysis of the iron atom. Values are the average ± SD, n = 3. (c) EPR spectra of Na2S2O4-reduced AtLhpI at 10 K.
Among the various divalent metal ions tested, only Zn2+, Cd2+, and Hg2+ inhibited activity (Fig. S4a), and their IC50 values were 9.75 × 10−3, 2.05 × 10−3, and 2.47 × 10−2 mM, respectively (Fig. 6a). The pattern was competitive, and the Ki value for Zn2+ was 1.07 × 10−3 mM (Fig. S4b). In the case of Acn enzymes, only mitochondrial Acn from mammals was inhibited by Zn2+ (Ki = 2.00 × 10−3 mM)34. Furthermore, pyrrole-2-carboxylate (and its derivative 2-thiophenecarboxylate) inhibited the activity of PaLhpI (Fig. 6a), whereas several characteristic inhibitors for Acn enzymes including trans-aconitate, fluorocitrate, and oxalomalate did not. Pyrrole-2-carboxylate is structurally analogous to the (putative) transition state of cis-3-hydroxy-L-proline, Δ2-pyrroline-2-carboxylate (see Fig. 1c), and its IC50 value is markedly higher than those of divalent metal ions: 440-fold that of Cd2+. Collectively, these results strongly indicate that the substrate binding pocket of the PaLhpI protein is not similar to Acn enzymes, and that binding sites for divalent metal ions and pyrrole-2-carboxylate are spatially distinct.

Inhibition study on cis-3-hydroxy-L-proline dehydratase activity.
PaLhpI (a), AtLhpI (b), and TrLhpI (c). Relative specific activity values (average ± SD, n = 3) are expressed as percentages of the values obtained in the absence of an inhibitor. IC50 values (in parentheses) were calculated by curve fitting using ImageJ software (http://rsb.info.nih.gov/ij/).
Characterization of other AcnXType I enzymes
Although the bacterial LhpI gene is often located within the L-hydroxyproline gene cluster, there are several combinations of trans-4-hydroxy-L-proline and/or trans-3-hydroxy-L-proline metabolic genes. For example, there are two types of cis-4-hydroxy-D-proline dehydrogenases13: P. aeruginosa PAO1 etc., α4β4γ4-type enzyme encoded by LhpB (encoding to β-subunit), LhpC (α-subunit) and LhpD genes (γ-subunit); Agrobacterium tumefaciens C58 etc., homomeric-type enzyme encoded by LhpB gene (Fig. 1d). Furthermore, there is no sequence similarity between LhpH and LhpK proteins encoding to Δ1-pyrroline-2-carboxylate reductase. Although the AcnX protein was originally considered to only be present in archaea and bacteria27, a homology search using the Protein-BLAST program revealed that a large number of fungi possess the LhpI homologous gene; the ANI_1_578044 gene from Aspergillus niger CBS 513.88 is closely located to the putative trans-3-hydroxy-L-proline dehydratase gene (Fig. 1d)14.
Therefore, in order to elucidate the catalytic functions of other AcnXType I proteins, we enzymatically investigated Atu4683 from A. tumefaciens C58 (AtLhpI) and TRIREDRAFT_59073 from Trichoderma reesei QM6a (TrLhpI), and found 46.2% and 51.7% sequence identity with PaLhpI, respectively. The (His)6-tagged AtLhpI and TrLhpI proteins were expressed in Escherichia coli, purified, and characterized (Fig. 3a). As observed for PaLhpI, among eight proline derivatives (Fig. S1), only cis-3-hydroxy-L-proline was consumed in a time-dependent manner by both proteins, and similar inhibition by Zn2+ was observed (Fig. 6b,c). By contrast, the kcat/Km value of TrLhpI was 316- and 83.4-fold lower than those of PaLhpI and AtLhpI, respectively (Fig. 4h). Although analysis by an amino acid analyzer suggested the reaction product of TrLhpI is Δ1-pyrroline-2-carboxylate (specific activity of 3.75 μmol·min−1·mg−1), the consumption rate of cis-3-hydroxy-L-proline by coupling assay with Δ1-pyrroline-2-carboxylate reductase decreased significantly (Fig. 4e,f). Furthermore, no effect of pyrrole-2-carboxylate (and 2-thiophenecarboxylate) was found on the activity (Fig. 6c). These results suggest that the fungal TrLhpI protein may well have an entirely different main (physiological) substrate from cis-3-hydroxy-L-proline.
Putative active sites
There are ~23 amino acid residues that are completely (or highly) conserved between Acn enzymes (yellow- and orange-shadowed letters in Fig. 7), implying similar structural organization and catalytic mechanisms35. However, most of these residues had no readily detectable counterpart in AcnXType I proteins. On the other hand, several amino acid residues were completely (or highly) conserved within the AcnX subfamily only (Fig. 7 and Fig. S5), and some of them may be located around the active sites of Acn enzymes. Based on these insights, we selected thirty amino acid residues at sites 1~4, 9, 11~14, 17, and 21~23 for a site-directed mutagenic study, and constructed each alanine mutant of PaLhpI (Fig. 5b). Among them, only D35A (136%), C207A (80.5%), and N457A (55.9%) mutants showed similar activity to the wild-type (WT) enzyme, whereas C275A, W292A, H404A, C516A, and Y542A mutants were not expressed in host cells, S66A, S70A, E294A, S295A, S303A, T309A, C518A, and K538A mutants were inactive, and C68A (0.4%), H459A (0.4%), D523A (14.4%) and S545A (7.3%) mutants decreased the activity significantly (the values in the parentheses are the expression of specific activity relative to the WT enzyme), suggesting their potential role(s) in catalysis and/or structure folding. Among them, Ser66 at site 2 may play a role in abstracting a proton from the Cα of cis-3-hydroxy-L-proline as a catalytic base, as in Acn enzymes33. However, in contrast to Cys275 of PaLhpI, mutation of histidine of Acn enzymes at site 8 does not markedly change activity levels from those of the WT enzyme36, probably caused by the lack of a direct ligand for [4Fe-4S] cluster, as described below.

Partial multiple amino acid sequence alignments of AcnX and other Acn subfamilies.
PaAcnX and AcnX from P. aeruginosa PAO1; AtAcnX, A. tumefaciens C58; TrAcnX, AcnX from T. reesei QM6a; BxAcnX, Burkholderia xenovorans LB400; ApAcnX, Aeropyrum pernix K1; AcnB from E. coli18; mAcn from bovine19; AcnD from Shewanella oneidensis MR-121; IRP from human20; IPMI from Saccharomyces cerevisiae26; HACN1 and HACN2 from M. jannaschii23,42; HACN3 from Pyrococcus horikoshii OT343. “S” and “L” indicate small and large subunits, respectively. Red- and black-bold letters indicate amino acids highly conserved in the AcnX subfamily and Acn enzymes, respectively. The complete alignment of the AcnX subfamily is shown in Fig. S5.
Putative iron binding sites
Inhibition by the Hg2+ and Cd2+ of LhpI protein(s) is observed in many proteins that possess a cysteine residue(s) as the active site: furthermore, an incubation with Hg2+ results in irreversible inactivation (Fig. S4c). In the case of Acn enzymes, three iron ions of the [4Fe-4S] cluster were coordinated by three conserved cysteine residues (orange-shadowed letters at sites 15, 18, and 20 in Fig. 5a and Fig. 7)33, among which PaLhpI possessed only one equivalent residue to site 18 (Cys516). Therefore, as other potential ligand(s), we selected Cys207, Cys275, Glu294, Asn457, and Cys518 at sites 5, 8, 10, 15, and 19, the corresponding residues of the Acn enzymes that were located around the [4Fe-4S] cluster, and measured the iron content in each alanine mutant by a colorimetric assay. As results, WT, C207A, E294A, N457A, and C518A contained 0.84, 0.76, 0.01>, 1.63, and 0.52 iron equivalents per (monomeric) protein (Fig. 5b): Cys275A and C516A mutants were not expressed in host cells, as described above. These findings indicate that PaLhpI contains an iron ion (but not [4Fe-4S] cluster) that is necessary for catalysis, and that Cys275, Glu294, and Cys516 at sites 8, 10, and 18 may be important for substrate binding (pink-shadowed letters). Complete conservation at these sites in the AcnX subfamily appears to support this hypothesis (Fig. S5). It is noteworthy that in the catalytic mechanism of the enolase type of cis-3-hydroxy-L-proline dehydratase (LhpJ), an enolate anion intermediate is stabilized by an essential Mg2+ ligated by two tightly conserved aspartate residues and one glutamate residue, and two lysine residues function as a basic and/or acidic catalyst16. Therefore, although AcnXType I has no evolutionary relationship with the enolase type enzyme, the catalytic mechanism including metal ion binding may be more similar than those of Acn enzymes.
Non-heme iron analysis
In order to examine the nature of non-heme iron in more detail, electron paramagnetic resonance (EPR) analysis was performed using AtLhpI. AtLhpI treated with dithionite (Na2S2O4) exhibited EPR signals at g = 5.40, 4.65, and 2.00 (Fig. 5c), whereas reduced rubredoxin, a typical mononuclear iron-sulfur protein, was EPR-silent. This EPR spectrum is markedly different from those of other reduced iron-sulfur clusters, such as [2Fe-2S] ferredoxin, the [2Fe-2S] Rieske center, [3Fe-4S] ferredoxin, [3Fe-4S] Acn enzymes, and [4Fe-4S] ferredoxin, the EPR signals of which were observed in the g ≈ 2 region37. Furthermore, it is interesting to note that the observed g-values of AtLhp1 were not a typical high-spin (S = 5/2) electronic ground state. Similar EPR spectra have been reported for cytochrome c’ from some photosynthetic bacteria, the electronic ground states of which are interpreted as quantum mechanical admixtures of an intermediate-spin state (S = 3/2) and high-spin state (S = 5/2)38,39. The S = 3/2 and S = 5/2 components at the ground state of the iron center in AtLhp1 are estimated to be 49 and 51%, respectively, from observed g-values (5.40 and 4.65). Although the electronic ground state of iron is still ambiguous, these results suggest that the iron site in AtLhpI is an unprecedented mononuclear Fe(III) center that has a low redox potential that is resistant to the reduction of dithionite, but not an iron-sulfur cluster. In rubredoxin, Fe(III) is coordinated to four cysteinyl sulfurs occurring on two Cys-X2-Cys segments or one Cys-X2-Cys and one Cys-Cys40. By contrast, in order to coordinate the Fe(II) center, “cupin superfamily” proteins have a conserved motif, in which no cysteine is contained as a ligand: His-X2-[His/Asp]-X4-Glu-Xn-His41. These specific motifs are not found in the sequences of AcnX enzymes (Fig. S5).
AcnXType IIb proteins possess two homologous cysteine residues, not only at site 18 but also site 15, with Acn enzymes (green-shadowed cysteine in Fig. 7 and Fig. S5). On the other hand, in the HACN subfamily, only enzymes from methanogenic archaea, including Methanococcus jannaschii, have the “fourth cysteine” at site 7 (light-green shadowed cysteine), which may be located near a sulfur atom in the [4Fe-4S] cluster; however, its role is currently unclear (Fig. 5a)42,43. Indeed, we have already found that purified AcnXType IIb protein (from Aeropyrum pernix K1) also contains iron ion(s), although functional annotation is in progress (unpublished). Therefore, the EPR analysis of AcnXType IIb protein would be useful for further understanding of the unique binding mode of Fe(III) of AcnX subfamily, and identifying which of the [4Fe-4S] cluster, or Fe(III), common ancestors of the Acn superfamily possessed.
Degradation pathway of L-hydroxyproline(s) in P. aeruginosa PAO1
To the best of our knowledge, cis-3-hydroxy-L-proline has only been found as one of the building blocks of telomycin, a peptide antibiotic produced by Streptomyces canus C1597,8, and these bacteria may also hydroxylate free L-proline to cis-3-hydroxy-L-proline enzymatically6. Since the amount of cis-3-hydroxy-L-proline in nature is markedly less than that of trans-4-hydroxy-L-proline, the possibility of catabolism by (micro)organisms had not previously been considered. In order to estimate the potential metabolism of L-hydroxyproline, P. aeruginosa PAO1 was cultivated in minimal medium supplemented with L-proline, trans-4-hydroxy-L-proline, trans-3-hydroxy-L-proline, or cis-3-hydroxy-L-proline as the sole carbon source. Measurements of concentration by an amino acid analyzer revealed that cis-3-hydroxy-L-proline (but not trans-3-hydroxy-L-proline) was consumed in a time-dependent manner: 72% degradation after 20 days (Fig. 8a,b). Furthermore, in comparisons with L-proline in a quantitative real-time PCR (qRT-PCR) analysis, the PaLhpI gene was induced not only by trans-4-hydroxy-L-proline and cis-4-hydroxy-D-proline, but also by cis-3-hydroxy-L-proline, whereas the PaLhpA and PaLhpH genes were only induced by trans-4-hydroxy-L-proline and cis-4-hydroxy-D-proline (Fig. 8e). Since the co-presence of trans-4-hydroxy-L-proline clearly enhanced the metabolism of cis-3-hydroxy-L-proline (Fig. 8c), slow degradation may be due to the lack of significant induction of the PaLhpH gene. Similarly, Starkeya novella DSM 506, which possesses the LhpJ gene (Fig. 1d), may utilize cis-3-hydroxy-L-proline as a carbon source, and transcription is up-regulated by cis-3-hydroxy-L-proline (and trans-4-hydroxy-L-proline)16. Although the co-presence of trans-4-hydroxy-L-proline also resulted in the degradation of trans-3-hydroxy-L-proline, half of the trans-3-hydroxy-L-proline consumed accumulated as a cis-3-hydroxy-D-proline (Fig. 8d), indicating the presence of trans-3-hydroxy-L-proline metabolic enzyme(s). We herein demonstrated that PaLhpL gene (PA1255; Fig. 1d) in the L-hydroxyproline gene cluster encodes a bifunctional dehydratase and 2-epimerase toward trans-3-hydroxy-L-proline as the leading candidate (Supplementary Discussion and Fig. S6).

L-Hydroxyproline metabolism in P. aeruginosa PAO1.
L-Hydroxyproline degradation when P. aeruginosa PAO1 was cultivated on cis-3-hydroxy-L-proline (a), trans-3-hydroxy-L-proline (b), cis-3-hydroxy-L-proline + trans-4-hydroxy-L-proline (c), and trans-3-hydroxy-L-proline + trans-4-hydroxy-L-proline (d) as a sole carbon source(s). Concentrations of L-hydroxyproline in medium were measured using an amino acid analyzer. Three independent experiments were performed, and the typical results obtained are shown. (e) Transcriptional analysis. Expression profiles in cells grown with cis-3-hydroxy-L-proline, trans-4-hydroxy-L-proline, and cis-4-hydroxy-D-proline compared to those in cells grown with L-proline as a sole carbon source. Numbers indicate relative expression levels that were measured by qRT-PCR.
Overall, the L-hydroxyproline gene cluster from microorganisms is often related not only to the metabolism of trans-4-hydroxy-L-proline, but also to that of cis-3-hydroxy-L-proline and trans-3-hydroxy-L-proline, suggesting that the origin of these free L-hydroxyproline compounds in nature is the degradation of several peptide antibiotics, such as telomycin (containing cis-3-hydroxy-L-proline and trans-3-hydroxy-L-proline)7,8, microcolin A (cis-4-hydroxy-L-proline)9, pneumocandins (trans-4-hydroxy-L-proline and trans-3-hydroxy-L-proline)10, and etamycin (cis-4-hydroxy-D-proline)11, rather than collagen.
Conclusion
We herein showed that LhpI protein catalyzes the dehydration of cis-3-hydroxy-L-proline to Δ1-pyrroline-2-carboxylate: over 10 years after the discovery of AcnX (subfamily) in Acn superfamily27, the functional characterization can now be done successfully. The gene context in the bacterial genome was of considerable help in estimating the potential substrate, rather than the amino acid sequence similarity. Although cis-3-hydroxy-L-proline is believed to hardly exist in nature, our findings may indicate the significant physiological role in (micro)organisms, similar to trans-3-hydroxy-L-proline14,15. Until now, the Acn superfamily (enzymes) was a typical example suitable for a recruitment hypothesis of enzyme evolution proposed by Jensen (1976)44; gene duplication of multi-specific enzymes containing a [4Fe-4S] cluster, followed by narrowing of substrate specificity. Moreover, this functional annotation of a Fe(III)-containing AcnX protein must promote interest in common ancestors of the Acn superfamily, including the nature of the iron-sulfur cluster.
Methods
Plasmid construction for the expression of recombinant proteins
The primer sequences used in this study are shown in Table S2. PaLhpI, TrLhpI, and AtLhpI genes were amplified by PCR using primers containing appropriate restriction enzyme sites at the 5′- and 3′-ends and the genome DNA of P. aeruginosa PAO1, T. reesei QM6a, or A. tumefaciens C58 as a template. Each amplified DNA fragment was introduced into BamHI-HindIII sites in pQE-80L (Qiagen), a plasmid vector for conferring an N-terminal (His)6 tag on the proteins expressed, in order to obtain pQE/PaLhpI, pQE/TrLhpI, and pQE/AtLhpI, respectively. Regarding the expression of the PaLhpI gene in P. putida cells, a DNA fragment of the (His)6-PaLhpI-t0 terminator was amplified by PCR using pQE/PaLhpI as a template, and introduced into the SalI-EcoRI sites in pUCP26KmAhpCp45 in order to obtain pUCP/PaLhpI. A site-directed mutation was introduced into the PaLhpI gene by sequential steps of PCR using sense and antisense primers and pUCP/PaLhpI as a template.
Expression and purification of the recombinant protein
P. putida KT2442-oxyR146 harboring pUCP/PaLhpI was grown at 30 °C overnight in LB medium containing kanamycin (50 mg/liter), whereas E. coli strain DH5α harboring pQE/TrLhpI or pQE/AtLhpI was grown at 37 °C to a turbidity of 0.6 at 600 nm in Super broth medium containing ampicillin (50 mg/liter). After the addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG), the culture was grown at 18 °C for a further 18 h in order to induce the expression of the (His)6-tagged protein. Grown cells were harvested, suspended in Buffer A (50 mM sodium phosphate buffer (pH 8.0) containing 300 mM NaCl and 10 mM imidazole), disrupted by sonication, and then centrifuged. The supernatant was loaded onto a Ni-NTA Superflow column (Qiagen) equilibrated with Buffer A linked to the BioAssist eZ system (TOSOH). The column was washed with Buffer B (50 mM sodium phosphate buffer (pH 8.0) containing 300 mM NaCl, 10% (v/v) glycerol, and 50 mM imidazole). The enzymes were then eluted with Buffer C (pH 8.0, as Buffer B but containing 250 mM instead of 50 mM imidazole), concentrated by ultrafiltration, dialyzed against 50 mM Tris-HCl buffer (pH 8.0) containing 50% (v/v) glycerol, and stored at –35 °C until use. The native molecular mass of the recombinant protein was estimated by gel filtration using a HiLoad 16/60 Superdex 200 pg column (GE Healthcare) equilibrated with 50 mM Tris-HCl buffer (pH 8.0). A high-molecular-weight gel filtration calibration kit (GE Healthcare) was used for molecular markers. Protein concentrations were measured by the method of Lowry et al.47 with bovine serum albumin as the standard.
Screening of the proline derivative library for the LhpI protein
The purified PaLhpI protein (5 μg) was added to 50 mM Tris-HCl buffer (pH 8.0) (1 ml) containing 10 mM substrate (Fig. S1), and incubated at 30 °C. After varying the incubation times, the enzyme reaction was stopped by briefly incubating at −80 °C. Substrate consumption was estimated by HPLC using an Ultron ES-CD chiral separation column (150 × 2.0 mm, Shinwa Chemical Industries)48 and/or Hitachi L-8900 PH Amino Acid Analyzer (Hitachi, Tokyo, Japan).
Enzyme assay
Unless noted otherwise, cis-3-hydroxy-L-proline dehydratase was assayed spectrophotometrically in the coupling system with Δ1-pyrroline-2-carboxylate reductase (LhpH protein from P. aeruginosa PAO1)15. The reaction mixture consisted of 50 mM Tris-HCl (pH 8.0), 0.15 mM NADPH, and purified Δ1-pyrroline-2-carboxylate reductase (10 μg). The reaction was initiated by the addition of 100 mM cis-3-hydroxy-L-proline (100 μl) with a final reaction volume of 1 ml. One unit of enzyme activity refers to 1 μmol NADPH consumed/min. Km and kcat values were calculated by a Lineweaver-Burk plot. The enzyme was alternatively assayed by a colorimetric method based on the reaction of 2-aminobenzaldehyde with Δ1-pyrroline-2-carboxylate, which yields a yellow reaction product13,14. This method was used to clarify the optimum pH for enzyme activity. Potential Acn activity in PaLhpI protein was measured spectrophotometrically by monitoring the change of absorption at 240 nm derived from unsaturated cis-aconitate49.
Identification of reaction products
In order to remove glycerol in the storage buffer, purified PaLhpI was dialyzed at 4 °C overnight in 50 mM K2HPO4/KH2PO4 buffer (pH 7.0), lyophilized, and solvated in D2O (600 μl) containing 20 mM cis-3-hydroxy-L-proline or 2,3-cis-3,4-cis-3,4-dihydroxy-L-proline. NMR spectra were recorded at 25 °C on a JEOL JNM-EC400 NMR spectrometer (JEOL Ltd., Tokyo, Japan) operating at 400 MHz. 2,2-Dimethyl-2-silapentane-5-sulfonate was used as an internal standard. 2,3-cis-3,4-cis-3,4-Dihydroxy-L-proline was enzymatically synthesized from cis-3-hydroxy-L-proline by L-proline cis-4-hydroxylase from S. meliloti, as described previously30.
Non-heme iron analysis
The content of non-heme iron was assessed using a Metallo Assay kit (AKJ Global Technology, Japan). The (potential) iron-sulfur cluster was analyzed by electron paramagnetic resonance (EPR) using a JEOL TE-300 X-band spectrometer operating with a 100-kHz field modulation. A temperature-dependent analysis was performed in the range of 10 to 40 K using a LTR-3 liquid helium cryostat (Air Products). The purified AtLhpI protein (~40 mg/ml) was prepared in 50 mM Tris-HCl (pH8.0) containing 50% (v/v) glycerol for this purpose, and treated with a 10-fold excess amount of Na2S2O4 under a nitrogen atmosphere. EPR spectra were recorded using the following representative conditions; microwave frequency; 8.9995 GHz, microwave power; 5.0 mW, 100 kHz modulation magnitude; 0.63 mT, center field; 280 ± 250 mT, sweep time; 4.0 min, time constant; 0.1 sec, and receiver amplitude; 500. In the present study, g-values were evaluated based on the g-value of the Li-salt of tetracyanoquinodimethane (2.0025) as an external standard. The magnetic field strength of EPR spectra was calibrated using the hyperfine coupling constant (8.69 mT) of the Mn(II) ion doped in MgO powder.
Growth and gene expression analysis of P. aeruginosa PAO1
P. aeruginosa PAO1 was cultured aerobically with vigorous shaking at 30 °C in 5 ml of minimal medium15 supplemented with a 30 mM carbon source(s). The concentration of L-hydroxyproline in medium after varying cultivation times was analyzed using an amino acid analyzer (L-8900; Hitachi, Tokyo, Japan). The preparation of RNA samples and one-step real-time RT-PCR were performed as described previously50. The primers used for RT-PCR are listed in Table S2. The PA0393 gene encoding pyrroline-5-carboxylate reductase was used as an internal control.
Sequence comparison
Protein sequences were analyzed using the Protein-BLAST and Clustal W programs distributed by DDBJ (DNA Data Bank of Japan).
Additional Information
How to cite this article: Watanabe, S. et al. Functional characterization of aconitase X as a cis-3-hydroxy-L-proline dehydratase. Sci. Rep. 6, 38720; doi: 10.1038/srep38720 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Adams, E. & Frank, L. Metabolism of proline and the hydroxyprolines. Annu. Rev. Biochem. 49, 1005–1061 (1980).
Shibasaki, T., Mori, H., Chiba, S. & Ozaki, A. Microbial proline 4-hydroxylase screening and gene cloning. Appl. Environ. Microbiol. 65, 4028–4031 (1999).
Petersen, L., Olewinski, R., Salmon, P. & Connors, N. Novel proline hydroxylase activities in the pneumocandin-producing fungus Glarea lozoyensis responsible for the formation of trans 3- and trans 4-hydroxyproline. Appl. Microbiol. Biotechnol. 62, 263–267 (2003).
Houwaart, S., Youssar, L. & Hüttel, W. Pneumocandin biosynthesis: involvement of a trans-selective proline hydroxylase. Chembiochem. 15, 2365–2369 (2014).
Hara, R. & Kino, K. Characterization of novel 2-oxoglutarate dependent dioxygenases converting L-proline to cis-4-hydroxy-L-proline. Biochem. Biophys. Res. Commun. 379, 882–886 (2009).
Mori, H., Shibasaki, T., Uozaki, Y., Ochiai, K. & Ozaki, A. Detection of novel proline 3-hydroxylase activities in Streptomyces and Bacillus spp. by regio- and stereospecific hydroxylation of L-proline. Appl. Environ. Microbiol. 62, 1903–1907 (1996).
Sheehan, J. C., Mania, D., Nakamura, S., Stock, J. A. & Maeda, K. The structure of telomycin. J. Am. Chem. Soc. 90, 462–470 (1968).
Fu, C. et al. Biosynthetic studies of telomycin reveal new lipopeptides with enhanced activity. J. Am. Chem. Soc. 137, 7692–7705 (2015).
Koehn, F. E., Longley, R. E. & Reed, J. K. Microcolins A and B, new immunosuppressive peptides from the blue-green alga Lyngbya majuscula. J. Nat. Prod. 55, 613–619 (1992).
Schwartz, R. E. et al. Pneumocandins from Zalerion arboricola. I. Discovery and isolation. J. Antibiot. 45, 1853–1866 (1992).
Haste, N. M. et al. Activity of the streptogramin antibiotic etamycin against methicillin-resistant Staphylococcus aureus. J. Antibiot. 63, 219–224 (2010).
Watanabe, S., Yamada, M., Ohtsu, I. & Makino, K. α-Ketoglutaric semialdehyde dehydrogenase isozymes involved in metabolic pathways of D-glucarate, D-galactarate and hydroxy-L-proline: molecular and metabolic convergent evolution. J. Biol. Chem. 282, 6685–6695 (2007).
Watanabe, S. et al. Identification and characterization of D-hydroxyproline dehydrogenase and Δ1-pyrroline-4-hydroxy-2-carboxylate deaminase involved in novel L-hydroxyproline metabolism of bacteria: metabolic convergent evolution. J. Biol. Chem. 287, 32674–32688 (2012).
Visser, W. F., Verhoeven-Duif, N. M. & de Koning, T. J. Identification of a human trans-3-hydroxy-L-proline dehydratase, the first characterized member of a novel family of proline racemase-like enzymes. J. Biol. Chem. 287, 21654–21662 (2012).
Watanabe, S. et al. Identification and characterization of trans-3-hydroxy-L-proline dehydratase and Δ1-pyrroline-2-carboxylate reductase involved in trans-3-hydroxy-L-proline metabolism of bacteria. FEBS Open Bio 4, 240–250 (2014).
Zhang, X. et al. A unique cis-3-hydroxy-L-proline dehydratase in the enolase superfamily. J. Am. Chem. Soc. 137, 1388–1391 (2015).
Gruer, M. J. & Guest, J. R. Two genetically-distinct and differentially-regulated aconitases (AcnA and AcnB) in Escherichia coli. Microbiology 140, 2531–2541 (1994).
Williams, C. H. et al. E. coli aconitase B structure reveals a HEAT-like domain with implications for protein-protein recognition. Nat. Struct. Biol. 9, 447–452 (2002).
Robbins, A. H. & Stout, C. D. Structure of activated aconitase: formation of the [4Fe-4S] cluster in the crystal. Proc. Natl. Acad. Sci. USA 86, 3639–3643 (1989).
Dupuy, J. et al. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure 14, 129–139 (2006).
Grimek, T. L. & Escalante-Semerena, J. C. The acnD genes of Shewenella oneidensis and Vibrio cholerae encode a new Fe/S-dependent 2-methylcitrate dehydratase enzyme that requires prpF function in vivo. J. Bacteriol. 186, 454–62 (2004).
Jia, Y., Tomita, T., Yamauchi, K., Nishiyama, M. & Palmer, D. R. Kinetics and product analysis of the reaction catalysed by recombinant homoaconitase from Thermus thermophilus. Biochem. J. 396, 479–485 (2006).
Drevland, R. M., Jia, Y., Palmer, D. R. & Graham, D. E. Methanogen homoaconitase catalyzes both hydrolyase reactions in coenzyme B biosynthesis. J. Biol. Chem. 283, 28888–28896 (2008).
Fultz, P. N. & Kemper, J. Wild-type isopropylmalate isomerase in Salmonella typhimurium is composed of two different subunits. J. Bacteriol. 148, 210–219 (1981).
Yasutake, Y., Yao, M., Sakai, N., Kirita, T. & Tanaka, I. Crystal structure of the Pyrococcus horikoshii isopropylmalate isomerase small subunit provides insight into the dual substrate specificity of the enzyme. J. Mol. Biol. 344, 325–333 (2004).
Skala, J., Capieaux, E., Balzi, E., Chen, W. N. & Goffeau, A. Complete sequence of the Saccharomyces cerevisiae LEU1 gene encoding isopropylmalate isomerase. Yeast 7, 281–285 (1991).
Makarova, K. S. & Koonin, E. V. Filling a gap in the central metabolism of archaea: prediction of a novel aconitase by comparative-genomic analysis. FEMS Microbiol. Lett. 227, 17–23 (2003).
Li, G. & Lu, C. D. Molecular characterization of LhpR in control of hydroxyproline catabolism and transport in Pseudomonas aeruginosa PAO1. Microbiology 162, 1232–1242 (2016).
White, C. E., Gavina, J. M., Morton, R., Britz-McKibbin, P. & Finan, T. M. Control of hydroxyproline catabolism in Sinorhizobium meliloti. Mol. MicroBiol. 85, 1133–1147 (2012).
Hara, R., Uchiumi, N., Okamoto, N. & Kino, K. Regio- and stereoselective oxygenation of proline derivatives by using microbial 2-oxoglutarate-dependent dioxygenases. Biosci. Biotechnol. Biochem. 78, 1384–1388 (2014).
Zhao S., et al. Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature 502, 698–702 (2013).
Kumar, R. et al. Prediction and biochemical demonstration of a catabolic pathway for the osmoprotectant proline betaine. MBio 5, e00933–13 (2014).
Beinert, H., Kennedy, M. C. & Stout, C. D. Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 96, 2335–2374 (1996).
Costello, L. C., Liu, Y., Franklin, R. B. & Kennedy, M. C. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J. Biol. Chem. 272, 28875–28881 (1997).
Frishman, D. & Hentze, M. W. Conservation of aconitase residues revealed by multiple sequence analysis: implications for structure/function relationships. Eur. J. Biochem. 239, 197–200 (1996).
Zheng, L., Kennedy, M. C., Beinert, H. & Zalkin, H. Mutational analysis of active site residues in pig heart aconitase. J. Biol. Chem. 267, 7895–7903 (1992).
Messerschmidt, A., Huber, R., Poulos, T. & Wieghardt, K. Handbook of Metalloproteins, Vol. I, pp.505–609 Eds. John Wiley & Sons, Ltd., West Sussex, England.
Maltempo, M. M., Moss, T. H. & Cusanovich, M. Magnetic studies on the changes in the iron environment in Chromatium ferricytochrome c’. Biochim. Biophys. Acta 342, 290–305 (1974).
Fujii, S. et al. Electron paramagnetic resonance studies of ferric cytochrome c’ from photosynthetic bacteria. Biochim. Biophys. Acta 1251, 161–169 (1995).
Meyer, J. Iron-sulfur protein folds, iron-sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 13, 157–170 (2008).
Buongiorno, D. & Straganz, G. D. Structure and function of atypically coordinated enzymatic mononuclear non-heme-Fe(II) centers. Coord. Chem. Rev. 257, 541–563 (2013).
Lee, E. H., Lee, K. & Hwang, K. Y. Structural characterization and comparison of the large subunits of IPM isomerase and homoaconitase from Methanococcus jannaschii. Acta Crystallogr. D 70, 922–931 (2014).
Lombo, T. et al. Functional analysis of the small subunit of the putative homoaconitase from Pyrococcus horikoshii in the Thermus lysine biosynthetic pathway. FEMS Microbiol. Lett. 233, 315–324 (2004).
Jensen, R. A. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 30, 409–425.
Kobayashi, Y., Ohtsu, I., Fujimura, M. & Fukumori, F. A mutation in dnaK causes stabilization of the heat shock sigma factor σ32, accumulation of heat shock proteins and increase in toluene-resistance in Pseudomonas putida. Environ. MicroBiol. 13, 2007–2017 (2011).
Hishinuma, S., Yuki, M., Fujimura, M. & Fukumori, F. OxyR regulated the expression of two major catalases, KatA and KatB, along with peroxiredoxin, AhpC in Pseudomonas putida. Environ. Microbiol. 8, 2115–2124 (2006).
Lowry, O. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275 (1951).
Watanabe, S., Tanimoto, Y., Nishiwaki, H. & Watanabe, Y. Identification and characterization of bifunctional proline racemase/hydroxyproline epimerase from archaea: discrimination of substrates and molecular evolution. PLoS One 10, e0120349 (2015).
Doi, Y. & Takaya, N. A novel A3 group aconitase tolerates oxidation and nitric oxide. J. Biol. Chem. 290, 1412–1421 (2015).
Ito, F., Tamiya., T., Ohtsu, I., Fujimura, M. & Fukumori, F. Genetic and phenotypic characterization of the heat shock response in Pseudomonas putida. MicrobiologyOpen 3, 922–936 (2014).
Garvey, G. S., Rocco, C. J., Escalante-Semerena, J. C. & Rayment, I. The three-dimensional crystal structure of the PrpF protein of Shewanella oneidensis complexed with trans-aconitate: insights into its biological function. Protein Sci. 16, 1274–1284 (2007).
Acknowledgements
This work was partially supported by JSPS KAKENHI Grants 25440049 and 16K07297 (to S.W.).
Author information
Authors and Affiliations
Contributions
S.W. conceived, designed, and performed the experiments, analyzed the data, and wrote the manuscript. K.T. and S.F. carried out EPR measurements, analyzed the data obtained, and wrote some of the manuscript. F.F. contributed to qRT-PCR experiments. R.H. and K.K. synthesized a unique dihydroxyproline compound. R.F. and M.M. prepared the recombinant mutated LhpI proteins. Y.W. contributed to improve the article.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Watanabe, S., Tajima, K., Fujii, S. et al. Functional characterization of aconitase X as a cis-3-hydroxy-L-proline dehydratase. Sci Rep 6, 38720 (2016). https://doi.org/10.1038/srep38720
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38720
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
