
Abstract
Angiogenesis is the growth of new blood vessels from pre-existing microvessels. Peripheral arterial disease (PAD) is caused by atherosclerosis that results in ischemia mostly in the lower extremities. Clinical trials including VEGF-A administration for therapeutic angiogenesis have not been successful. The existence of anti-angiogenic isoform (VEGF165b) in PAD muscle tissues is a potential cause for the failure of therapeutic angiogenesis. Experimental measurements show that in PAD human muscle biopsies the VEGF165b isoform is at least as abundant if not greater than the VEGF165a isoform. We constructed three-compartment models describing VEGF isoforms and receptors, in human and mouse, to make predictions on the secretion rate of VEGF165b and the distribution of various isoforms throughout the body based on the experimental data. The computational results are consistent with the data showing that in PAD calf muscles secrete mostly VEGF165b over total VEGF. In the PAD calf compartment of human and mouse models, most VEGF165a and VEGF165b are bound to the extracellular matrix. VEGF receptors VEGFR1, VEGFR2 and Neuropilin-1 (NRP1) are mostly in ‘Free State’. This study provides a computational model of VEGF165b in PAD supported by experimental measurements of VEGF165b in human and mouse, which gives insight of VEGF165b in therapeutic angiogenesis and VEGF distribution in human and mouse PAD model.
Similar content being viewed by others


Introduction
Angiogenesis is the process of new blood vessel formation from the pre-existing microvessels. Members of vascular endothelial growth factor (VEGF) superfamily critically but differentially regulate angiogenesis in normal physiological and pathophysiological conditions including exercise, ischemic cardiovascular diseases, and cancer1. The VEGF family includes five ligands VEGF-A, VEGF-B, VEGF-C, VEGF-D and PlGF (Placental growth factor), and five receptors VEGFR1, VEGFR2, VEGFR3, NRP1 (neuropilin-1) and NRP2 (neuropilin-2). Among the members of VEGF family, VEGF-A and VEGFR2 are considered to be potent pro-angiogenic molecules. However, recent identification of VEGFxxxb isoforms has changed the classical paradigm of VEGF-A:VEGFR2 function in regulation of angiogenesis2.
Alternate splicing in the 8th exon of VEGF-A results in the formation of sister families: pro-angiogenic VEGFxxxa (VEGF165a, in human) isoform (xxx denotes number of amino acids) containing an amino acid sequence ‘CDKPRR’ and anti-angiogenic VEGF165b isoform containing an amino acid sequence ‘PLTGKD’ in their C-terminus, respectively. The positively charged cysteine and arginine residues (CDKPRR) in pro-angiogenic VEGF-A isoform facilitate the binding of VEGF165a to VEGFR2 and NRP1 to induce a conformational change and internal rotation of intracellular domain and maximal activation of VEGFR. However, replacement of cysteine and arginine residues with neutral lysine and aspartic acid in VEGFxxxb isoform was predicted to result in partial VEGFR2 activation that cannot induce torsional rotation required for autophosphorylation and downstream signaling. Hence, the balance between VEGF165a and VEGF165b levels may play a crucial role in promoting angiogenesis especially in ischemic cardiovascular diseases such as peripheral arterial disease (PAD) or coronary artery disease (CAD).
PAD is caused by atherosclerosis, which results in ischemia most frequently in the lower extremities. Clinical trials including exogenous VEGF-A administration to activate VEGFR2 dependent therapeutic angiogenesis were not successful. While suboptimal delivery or dosage might be the contributing factors, induction of VEGF165b in ischemic muscle could compete with pro-angiogenic VEGF165a isoform for binding sites on VEGFR2 to decrease VEGFR2 activation. The mechanism of VEGF165b binding to VEGFR2 suggests the potential reason for the failure of therapeutic angiogenesis in VEGF-A clinical trials. Currently, the balance between VEGF165b and VEGF165a isoforms that can modulate VEGFR2 activation and angiogenic signaling in the ischemic skeletal muscle of PAD patients is not fully understood.
We have previously reported experimental evidence that VEGF165b levels are significantly higher in biopsies of PAD patients3. Kikuchi et al. measured the ratios of VEGF165b versus VEGF165a in serum (by western blot analysis) and peripheral blood mononuclear cells (PBMCs, by mRNA) in PAD patients, which are 4:1 and 8:1, respectively4. Currently there is no study measuring the ratio of VEGF165b and VEGF165a in the calf muscles to correlate with VEGFR2 activation in PAD patients. Besides, the understanding of the secretion rates of VEGF165b in different compartments such as diseased calf muscles, blood and normal tissues (all tissues and organs except the diseased calf) is currently lacking. In this study, we expand the previously developed three-compartment models5,6 to predict the concentration of VEGF165b and VEGF165a in PAD patients. We also report the experimental measurements of the expression level of VEGF165b and VEGF164a in experimental PAD non-ischemic and ischemic muscle samples in mice with hindlimb ischemia to support our computational models.
Results
In this study, we use both experimental and computational approaches to predict the secretion rate of VEGF165b and receptor occupancy of VEGFR1 and VEGFR2 in PAD. In practice, it is not possible to measure the secretion rates of VEGF in different tissues in vivo. These important physiological parameters could be only estimated or calculated from the model. This is the first computational model to investigate and account for the experimental ratios of VEGF165b and total VEGF165 in human PAD muscle biopsies. The predominance of VEGF165b in human muscles provides the potential mechanism why the clinical trials of VEGF therapeutic angiogenesis have failed. Based on the measurement of ratio of VEGF165b and total VEGF165 in muscle biopsies, we use the three-compartment model to predict the secretion rates of VEGF165b in different tissues. This model enables prediction of receptor occupancy with VEGF165b and other isoforms in human and mouse.
Specificity and sensitivity of VEGF-A and VEGF165b antibodies
VEGF-A ELISA detected both recombinant VEGF165a and VEGF165b with equal sensitivity and specificity. This result indicates that the actual measurement of pro-angiogenic VEGF-A by VEGF-A antibodies have been overestimated due to its cross reactivity with anti-angiogenic VEGF165b isoforms. However, VEGF165b antibody raised against the unique 6 amino acid sequence (SLTRKD) used in ELISA specifically detected only recombinant VEGF165b but not recombinant VEGF165a at any concentrations indicating that a ratio of VEGF165b:total VEGF165 measurements will be needed to determine the actual amount of pro-angiogenic VEGF-A isoforms in biological samples (Fig. 1).

ELISA showing the specificity and sensitivity of VEGF165a (VEGFA) and VEGF165b antibodies.
Three-compartmental models of PAD: motivation, assumptions and simulations
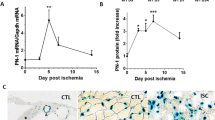
In our western analysis we observed a significant increase in total VEGF165 levels in ischemic muscle compared to non-ischemic at day 3 and day 7 post hindlimb ischemia (HLI). However, the levels of VEGF165b were also significantly higher at day 3 and day 7 post HLI. The ratio VEGF165b:VEGF-A (from the densitometry values of western blot analysis) showed a ~4 fold induction in the fraction of VEGF165b in total VEGF-A in ischemic muscle at day 7 post HLI compared to non-ischemic (i.e. VEGF165b isoforms constitute ~80% of total VEGF-A) in experimental PAD muscle compared to control (Fig. 2).

Western blot analysis of VEGF165b, VEGF-A and Actin in non-ischemic (NGA) and ischemic gastrocnemius muscle (IGA) at day 3 and day 7 post hindlimb ischemia (HLI).
(n = 4. One-way ANOVA with Dunnetts post-test. p < 0.05 considered significant).
We used the above experimental results in our model to predict the concentrations of VEGF165b and VEGF165a in the calf muscles. Kikuchi et al. measured the ratios of VEGF165b versus VEGF165a in serum (protein) and peripheral blood mononuclear cells (PBMCs, mRNA) in PAD patients, which were reported as 4:1 and 8:1, respectively4. Hoier et al. measured the interstitial total VEGF protein concentration (i.e. including both VEGF165a and VEGF165b) in the thigh skeletal muscle of PAD patients as 69 ± 21 and 190 ± 78 pg/ml in rest and active exercise, respectively. We convert these numbers to molar concentrations as 69 pg/ml · 1000 ml/l · 1 mole/46000 g = 1.5 pM and 190 pg/ml = 4.1 pM, respectively, based on the molecular weight 46 kDa for VEGF homodimers7. We have constrained the model so that the predictions of steady-state concentration of VEGF165b and total VEGF165 are within an experimentally observed range (e.g. 0–10 pM) in any of the three compartments to be consistent with the experimental data.
However, because of the sparseness of experimental data it is not possible to identify all the unknown parameters from the available data. Thus, we choose an initial set of secretion rates based on our previous analyses and perform the sensitivity analysis to determine a range of secretion rates constrained with available experimental data, both our own and others. Note that future experiments may provide additional data that could allow further constraining the solutions; presently, the analysis is heuristic aiming at stimulating further experiments.
Prediction of secretion rates of VEGF165b and secretion ratio of VEGF165b over total VEGF in three compartments
The values of secretion rate of VEGF in normal and blood compartments are adopted from the tumor angiogenesis model8. The secretion rate in PAD calf compartment is set up the same as the secretion rate in the normal compartment. The initial set of secretion rates of total VEGF including VEGF165a, VEGF165b and VEGF121 is assumed as 0.02, 0.031 and 0.02 molecules/cell/s, respectively, in all the three compartments (normal, blood and PAD calf). The secretion ratio of VEGF121 over total VEGF (VEGF165a + VEGF165b + VEGF121) is assumed to be 10%, i.e. 0.002, 0.0031 and 0.002 in normal, blood and disease compartments, respectively. We scan the ratio of secretion rate of VEGF165b over total VEGF165 (i.e. the sum of VEGF165a and VEGF165b) in the three compartments from 0 and 1 in 0.1 increments. When the ratio of VEGF165b over total VEGF165 is 0.5 in all three compartments (i.e. secretion rates of VEGF165a and VEGF165b have equal values 0.009, 0.01395 and 0.009 in normal and disease compartments, respectively), the concentration of VEGF165b in the normal compartment reaches 31 pM. When the ratio of VEGF165b over total VEGF165 in all three compartments is 10%, the concentration of VEGF165b in the normal compartment drops to 6.5 pM, which is within the experimentally observed range (<10 pM).
We can also change the secretion ratio of VEGF165b to total VEGF165 in each of the three compartments individually. We plot the predictions for steady-state concentrations of VEGF165b and VEGF165a in Fig. 3. We first change the secretion ratio of VEGF165b/total VEGF165 from 0 to 1 in 0.1 increments in the normal compartment while keeping the total secretion rate of VEGF165 (VEGF165a + VEGF165b) constant at 0.018 molecules/cell/s in the normal and disease compartments and fixing the secretion ratios in blood and disease compartments at 0.1 (Fig. 3A). We follow the same strategy in Fig. 3B,C, except we vary the secretion ratio of VEGF165b/total VEGF165 in the blood compartment for B and the disease compartment for C, while the total secretion rates are fixed and the ratios are fixed at 0.1 in the other compartments. The model predicts that the concentration of VEGF165b remains at approximately 6.5 pM, when the VEGF165b/total VEGF165 secretion ratio is 0.1 in normal and blood, across the full range of secretion ratios in the disease compartment (Fig. 3, left column). Additionally, the steady state concentration of VEGF165b is consistently predicted to be approximately 30% higher than VEGF165a in the disease compartment. This result is qualitatively consistent with our experimental data. This prediction gives us an important biological insight that the PAD disease calf muscles comprise mostly VEGF165b over other total VEGF isoforms. This finding could be further extended for the development of VEGF165b antibody treatment in PAD.

Concentrations of VEGF165a and VEGF165b plotted against the secretion ratio of VEGF165b to total VEGF165 isoforms in normal (left column), blood (middle column) and disease compartments (right column).
Each row represents the variation of secretion ratio of VEGF165b to total VEGF165 in (A) normal, (B) blood, and (C) disease compartment.
Sensitivity analysis of VEGF165a, VEGF165b and sVEGFR1
We use SimBiology to perform a local sensitivity analysis, which quantifies how changes in a given parameter value influence predicted concentrations of interest. We calculate the normalized sensitivity coefficient  based on the algorithm9, where Y is the species VEGF165a, VEGF165b and VEGF121, and X is the three VEGF receptors VEGFR1, VEGFR2 and NRP1 in the disease compartments; [X] and [Y] denote the molar concentrations. Figure 4 shows that VEGF165a in the disease compartment and VEGF165b in the blood compartment are more sensitive to VEGFR2 than VEGFR1, whereas VEGF121 in the disease and blood compartment is sensitive to both VEGFR1 and VEGFR2. VEGF165a, VEGF165b, VEGF121 are not sensitive to NRP1. The sensitivity analysis in Fig. 4 demonstrates the importance of VEGF165b-VEGFR2 binding.
based on the algorithm9, where Y is the species VEGF165a, VEGF165b and VEGF121, and X is the three VEGF receptors VEGFR1, VEGFR2 and NRP1 in the disease compartments; [X] and [Y] denote the molar concentrations. Figure 4 shows that VEGF165a in the disease compartment and VEGF165b in the blood compartment are more sensitive to VEGFR2 than VEGFR1, whereas VEGF121 in the disease and blood compartment is sensitive to both VEGFR1 and VEGFR2. VEGF165a, VEGF165b, VEGF121 are not sensitive to NRP1. The sensitivity analysis in Fig. 4 demonstrates the importance of VEGF165b-VEGFR2 binding.

Local sensitivity analysis for VEGF165a, VEGF165b and VEGF121.
The results show that VEGF165a in the disease compartment and VEGF165b in the blood compartment are more sensitive to VEGFR2 than VEGFR1, whereas VEGF121 in the disease and blood compartment is sensitive to both VEGFR1 and VEGFR2. VEGF165a, VEGF165b, VEGF121 are not sensitive to NRP1. The sensitivity analysis demonstrates the importance of VEGF165b-VEGFR2 binding.
Tissue VEGF distribution and VEGFR occupancy in human three compartment model
We show the steady-state distribution of VEGF ligands and their receptors for each tissue in Fig. 5A,B, respectively. The y-axis represents the percentage of each species in x-axis in each VEGF ligand (VEGF165a, VEGF165b and VEGF121) in Fig. 5A and each receptor (VEGFR1, VEGFR2, NRP1, and sVEGFR1) in Fig. 5B. In PAD calf, most VEGF165a is bound to the ECM and parenchymal basement membrane (PBM) (55% and 17%, respectively); most VEGF165b is also bound to ECM and PBM (62% and 20%, respectively). Here ECM represents the total extracellular matrix minus the endothelial and parenchymal basement membranes. Most VEGF121 isoform is bound to VEGFR1 and NRP1 as the VEGF121:VEGFR1:NRP1 complex in the disease (48%) and normal (51%) compartment. Regarding receptor occupancy, in the normal compartment, the three receptors VEGFR1, VEGFR2 and NRP1 are in the free states, except the VEGF121:VEGFR1:NRP1 (33%) and VEGF165a:VEGFR2:NRP1 (49%). In the PAD calf compartment, most receptors are in the free states, except the complexes VEGF165a:VEGFR2:NRP1 (20%) and VEGFR1:NRP1 (18%). Most sVEGFR1 are bound to ECM, endothelial basement membrane (EBM) and PBM (62%, 15% and 19%, respectively). Our model in PAD resembles the similar distribution of VEGF and VEGFR occupancy in tumor10.

(A) VEGF distribution and (B) VEGFR occupancy in human. The bars in y-axis represent the percentage of each species for each VEGF ligand VEGF165a, VEGF165b and VEGF121 in (A) and each receptor VEGFR1, VEGFR2, NRP1, and sVEGFR1 in (B).
VEGF distribution and VEGFR occupancy in murine three compartment model
Constructing a computational model of mouse VEGF distribution is necessary to make use of the experimental measurements in hindlimb ischemia (HLI) models in mice where numerous studies have been conducted3,11,12. In the mouse, VEGF isoforms are shorter by one amino acid, e.g., VEGF164a, and VEGF120, but VEGF165b. Construction of the three-compartment model in mouse can help identify the relative concentration of VEGF164a and VEGF165b in the different compartments, and predict VEGF receptor occupancy. We replace the geometric parameters in the three-compartment model from human to mouse. These geometric parameters of mouse are described in the tumor xenograft model6 as well as in the mouse two-compartment model13. The results of VEGF distribution and VEGFR occupancy are shown in Fig. 6(A,B), respectively. Figures 5(A) and 6(A) show that VEGF165b (human and mouse) mostly bind to the ECM and rarely exist as free ligands in both human and mouse. VEGF164a and VEGF165b bind to PBM with higher percentage in mouse than the corresponding isoforms in human. The VEGF receptor occupancies in Figs 5(B) and 6(B) show the similar trend between mouse and human. However, there is lower percentage of free VEGFR1 and VEGFR2 in human (Fig. 5B) than in mouse (Fig. 6B). This result is due to the higher percentage of VEGF121:VEGFR1:NRP1 and VEGF165a:VEGFR2:NRP1 in human than the corresponding complexes in mouse.

(A) VEGF distribution and (B) VEGFR occupancy in mouse. The bars represent the percentage of each species for each VEGF ligand in (A) and each receptor in (B), respectively, same as Fig. 5.
Discussion
We previously developed the three-compartment models of VEGF in tumor6,8,10 and in PAD14. The anti-angiogenic form of VEGF165b has not been included in any of the previous computational models; this is the first study to include VEGF165b in our human PAD model. This is also the first study that compares human PAD and mouse hindlimb ischemia (HLI) models. By changing the geometric parameters in human PAD calf muscles and adding the kinetics equations of VEGF165b, we predict the VEGF165b level in the PAD calf, blood and normal tissue compartments. We summarize the predictions in Table 1. Our computational model shows that the ratio of VEGF165b/VEGF165a in PAD is higher than 1 (1.70, 2.10 and 1.41 in disease, blood and normal compartments, respectively). This prediction is consistent with the experimental measurements of VEGF165b as the predominant isoform of VEGF in PAD4. The ratio of VEGF165b/total VEGF165 in the disease compartment of our model is slightly lower than the experiment in human biopsies; however, it should be noted that our predictions are for VEGF concentration in the interstitial fluid in the normal and diseased tissues, whereas the measurements represent the bulk tissue including intracellular components.
We do not include intrinsic heterogeneity that arises from the stochastic nature of biochemical reactions because we consider relatively large concentrations (pM). It may be more relevant to consider extrinsic heterogeneity (i.e., due to variations in protein concentrations)15. Others have included heterogeneity in the receptor distributions16 and found that variability in receptor expression can influence the response to anti-VEGF cancer treatment. The focus of the present work is to investigate and quantify how VEGF165b molecular interactions influence overall VEGF distribution in human and mouse. This is the first model to incorporate the VEGF165b isoform and we focus on the issues related to the expression of this isoform. Future work can incorporate variability in protein levels.
This study is also important for computational modeling of pharmacokinetics and pharmacodynamics (PK/PD) of administering VEGF165b-antibody as a potential pro-angiogenic therapeutic. Current VEGF antibodies including bevacizumab bind both total VEGF (VEGF165a, VEGF165b and VEGF121). We propose to introduce an antibody to VEGF165b as a PAD therapeutic to stimulate angiogenesis. This strategy will be explored in upcoming computational and experimental studies.
Conclusions
We applied the three-compartment model of VEGF distribution to predict the concentration of VEGF isoforms, including VEGF165b, in the body in peripheral arterial disease. The experimental data show that the expression level of VEGF165b is higher than VEGF165a in the human biopsies and the computational model results are consistent with these measurements. The secretion ratio of VEGF165b to total VEGF165 is estimated as 1 in the disease compartment, and 0.1 in the blood and normal compartments. Our predictions support the importance of the VEGF165b in PAD, and provide a foundation for therapeutic inhibition of VEGF165b in PAD in the future. This multiscale model can also provide a basis for simulating the pharmacokinetics of VEGF165b antibody in PAD in human and mouse models.
Methods
ELISA to measure VEGF165b antibody sensitivity
VEGF-A and VEGF165b (R&D) Dual sandwich ELISAs were used to determine the sensitivity and specificity of VEGF-A and VEGF165b antibodies in differentiating recombinant pro-angiogenic VEGF-A (VEGF165a) and anti-angiogenic VEGF-A (VEGF165b) isoforms. Recombinant VEGF165a and VEGF165b isoforms were serially diluted at 1000, 500, 250, 125 pg/ml concentrations and standard VEGF-A and VEGF165b ELISA were performed according to manufacturer instructions on all the samples to determine the sensitivity and specificity of VEGF-A and VEGF165b Mice.
Murine Model of Hindlimb Ischemia
Hindlimb ischemia (HLI) induced by femoral artery ligation and resection was used as an experimental model of human PAD. All animal experiments were approved by the University of Virginia Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. HLI was performed on 8- to 12-week-old age and sex matched C57Bl/6 mice as described previously11,12. Briefly, mice were anesthetized by a combination of ketamine and Xylazine (ketamine 90 mg/kg and xylazine 10 mg/kg) and femoral artery was ligated and resected from just above the inguinal ligament to its bifurcation at the origin of saphenous and popliteal arteries. The inferior epigastric, lateral circumflex, and superficial epigastric artery branches were also ligated.
Antibodies
VEGF165b antibody was purchased from Millipore (Clone 56/1, Cat No: MABC595), VEGF-A antibody was purchased from Santa Cruz Biotech (Cat No: SC-7269) and β-Actin was purchased from Sigma (Cat No: A2103).
Western blotting, Densitometry and Statistics
Mice were sacrificed with an overdose of anesthesia and non-ischemic and ischemic gastrocnemius muscle samples were collected at day 3 and day 7 post HLI. Tissue was homogenized in RIPA with protease inhibitor cocktail. Equal amounts of protein were resolved by Sodium Dodecyl Sulphate-Poly Acrylamide Gel Electrophoresis, transferred onto nitro cell cellulose membranes and western blotted against VEGF165b, VEGF-A and Actin by chemiluminescent method. Bands on the film were scanned and quantified by densitometric analysis of the band intensity by NIH image-J (1.6) software. Densitometric values were plotted on Graph PAD Prism 6 or 7 and analyzed for statistical significance. One-way ANOVA with Dunnetts post–test was used to check statistical significance. p < 0.05 considered statistically significant.
Three-compartment models for human and mouse
The model is comprised of three components: normal tissue (representing all tissues and organs except the diseased calf and blood), blood, and diseased calf of PAD (Fig. 7). For clarity, we use the notation VEGF165a to denote the pro-angiogenic isoform; we refer to the combination of VEGF165a and VEGF165b as total VEGF165. VEGF165a, VEGF165b and VEGF121 are secreted by myocytes and possibly stromal cells in the normal tissues and by calf muscles in PAD, respectively, and also by endothelial cells in all compartments. VEGF receptors (VEGFR1 and VEGFR2) and co-receptor neuropilin-1 (NRP1) are localized on the surfaces of endothelial and parenchymal cells. We include soluble VEGFR1 and glycosaminoglycan (GAG) chains in the interstitial space of normal and calf compartments, and alpha-2-macroglobulin (α2M) in the blood. These soluble factors bind VEGF and can be present in high concentrations; therefore, it is important to include them in the model. Geometric parameters are used to characterize the compartment and enable conversion of the concentrations from units used in the model (moles/cm3 tissue) to more standard units (molarity). The geometric parameters in the normal and blood human compartments are described in ref. 17 where applications to cancer are considered. The geometric parameters in the diseased calf compartment are described in ref. 14. The geometric parameters for mouse compartments are described in refs 6 and 13. We list the details of geometric parameters used in our model in Table 2.

Three-compartment model of VEGF in peripheral arterial disease.
121: VEGF121. 165a: VEGF165a. 165b: VEGF165b. N: neuropilin-1. 1: soluble and membrane-bound VEGFR1. 2: VEGFR2. GAG: glycosaminoglycan. α2M: alpha-2 macroglobulin.
Binding of VEGF165a, VEGF165b and VEGF121 to VEGFR1 and VEGFR2
The molecular interactions between VEGF isoforms and their receptors are illustrated in Fig. 8. VEGFR1 and VEGFR2 are the transmembrane receptors that bind to VEGF165a, VEGF165b and VEGF121. VEGF165b has the equivalent binding to VEGFR2 as VEGF165a18, and functions as a competitive inhibitor of the major downstream effects of VEGF165a. VEGF165a and VEGF165b are the two glycoproteins with heparin binding domain that can bind the extracellular matrix. VEGF121 is a freely diffusible protein lacking a heparin-binding domain. VEGF165b could bind to VEGFR1 and VEGFR2 but cannot bind the co-receptor NRP1 because it lacks exon 8a19. The densities of cell receptors VEGFR1, VEGFR2 and NRP1 are listed in Table 3 based on available in vivo and in vitro experimental data. The kinetic parameters are listed in Table 4. The model is described in terms of 80 ordinary differential equations (ODE) and is presented in the Supplementary File.

Molecular Interactions of VEGF165a, VEGF165b and VEGF121.
We assume that the total secretion rate of VEGF isoforms to be 0.02 molecules/cells/s in the disease and normal compartment, and 0.031 in the blood compartment (see Results for the details). The model equations were implemented in MATLAB R2014b (MathWorks, Natick, MA) using the SimBiology toolbox and were solved with the Sundials solver.
Transport parameters
Molecular species are transported between compartments via microvascular permeability (kp) and lymphatic drainage (kL) as shown in Fig. 7. All isoforms of unbound VEGF and soluble VEGFR1 (sVEGFR1) in the tissue compartments are subject to proteolytic degradation (kdeg) and are removed from the blood via plasma clearance (cv). We list the transport parameters in Table 5.
Sensitivity analysis
The sensitivity analysis is implemented using Matlab SimBiology toolbox. The time-dependent sensitivities of the species states are calculated with respect to species initial conditions and parameter values in the model. The sensitivity is calculated using  , which determines how changes in parameter X influences the output Y by taking the partial derivatives.
, which determines how changes in parameter X influences the output Y by taking the partial derivatives.
Additional Information
How to cite this article: Chu, L.-H. et al. A multiscale computational model predicts distribution of anti-angiogenic isoform VEGF165b in peripheral arterial disease in human and mouse. Sci. Rep. 6, 37030; doi: 10.1038/srep37030 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Mac Gabhann, F. & Popel, A. S. Systems biology of vascular endothelial growth factors. Microcirculation 15, 715–738 (2008).
Woolard, J., Bevan, H. S., Harper, S. J. & Bates, D. O. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation 16, 572–592 (2009).
Jones, W. S. et al. Alteration in angiogenic and anti-angiogenic forms of vascular endothelial growth factor-A in skeletal muscle of patients with intermittent claudication following exercise training. Vasc Med 17, 94–100 (2012).
Kikuchi, R. et al. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat Med 20, 1464–1471 (2014).
Finley, S. D., Angelikopoulos, P., Koumoutsakos, P. & Popel, A. S. Pharmacokinetics of Anti-VEGF Agent Aflibercept in Cancer Predicted by Data-Driven, Molecular-Detailed Model. CPT Pharmacometrics Syst Pharmacol 4, 641–649 (2015).
Finley, S. D., Dhar, M. & Popel, A. S. Compartment model predicts VEGF secretion and investigates the effects of VEGF trap in tumor-bearing mice. Frontiers in oncology 3, 196 (2013).
Hoier, B. et al. Angiogenic response to passive movement and active exercise in individuals with peripheral arterial disease. J Appl Physiol (1985) 115, 1777–1787 (2013).
Finley, S. D. & Popel, A. S. Effect of tumor microenvironment on tumor VEGF during anti-VEGF treatment: systems biology predictions. J Natl Cancer Inst 105, 802–811 (2013).
Ingalls, B. P. & Sauro, H. M. Sensitivity analysis of stoichiometric networks: an extension of metabolic control analysis to non-steady state trajectories. J Theor Biol 222, 23–36 (2003).
Stefanini, M. O., Wu, F. T., Mac Gabhann, F. & Popel, A. S. The presence of VEGF receptors on the luminal surface of endothelial cells affects VEGF distribution and VEGF signaling. Plos Comput Biol 5, e1000622 (2009).
Hazarika, S. et al. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res 101, 948–956 (2007).
Hazarika, S. et al. MicroRNA-93 controls perfusion recovery after hindlimb ischemia by modulating expression of multiple genes in the cell cycle pathway. Circulation 127, 1818–1828 (2013).
Yen, P., Finley, S. D., Engel-Stefanini, M. O. & Popel, A. S. A two-compartment model of VEGF distribution in the mouse. PloS one 6, e27514 (2011).
Wu, F. T. et al. VEGF and soluble VEGF receptor-1 (sFlt-1) distributions in peripheral arterial disease: an in silico model. Am J Physiol Heart Circ Physiol 298, H2174–2191 (2010).
Li, C. & Wang, J. Quantifying the underlying landscape and paths of cancer. J R Soc Interface 11, 20140774 (2014).
Weddell, J. C. & Imoukhuede, P. I. Quantitative characterization of cellular membrane-receptor heterogeneity through statistical and computational modeling. Plos One 9, e97271 (2014).
Finley, S. D. & Popel, A. S. Predicting the effects of anti-angiogenic agents targeting specific VEGF isoforms. The AAPS journal 14, 500–509 (2012).
Woolard, J. et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res 64, 7822–7835 (2004).
Cebe Suarez, S. et al. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci 63, 2067–2077 (2006).
McGuigan, M. R. et al. Resistance training in patients with peripheral arterial disease: effects on myosin isoforms, fiber type distribution, and capillary supply to skeletal muscle. The journals of gerontology. Series A, Biological sciences and medical sciences 56, B302–310 (2001).
Askew, C. D. et al. Skeletal muscle phenotype is associated with exercise tolerance in patients with peripheral arterial disease. Journal of vascular surgery 41, 802–807 (2005).
McGuigan, M. R. et al. Muscle fiber characteristics in patients with peripheral arterial disease. Medicine and science in sports and exercise 33, 2016–2021 (2001).
Regensteiner, J. G. et al. Chronic changes in skeletal muscle histology and function in peripheral arterial disease. Circulation 87, 413–421 (1993).
Finley, S. D., Engel-Stefanini, M. O., Imoukhuede, P. I. & Popel, A. S. Pharmacokinetics and pharmacodynamics of VEGF-neutralizing antibodies. BMC systems biology 5, 193 (2011).
Imoukhuede, P. I. & Popel, A. S. Quantitative fluorescent profiling of VEGFRs reveals tumor cell and endothelial cell heterogeneity in breast cancer xenografts. Cancer medicine 3, 225–244 (2014).
Mac Gabhann, F. & Popel, A. S. Targeting neuropilin-1 to inhibit VEGF signaling in cancer: Comparison of therapeutic approaches. Plos Comput Biol 2, e180 (2006).
Stefanini, M. O., Wu, F. T., Mac Gabhann, F. & Popel, A. S. A compartment model of VEGF distribution in blood, healthy and diseased tissues. BMC Syst Biol 2, 77 (2008).
Bhattacharjee, G., Asplin, I. R., Wu, S. M., Gawdi, G. & Pizzo, S. V. The conformation-dependent interaction of alpha 2-macroglobulin with vascular endothelial growth factor. A novel mechanism of alpha 2-macroglobulin/growth factor binding. J Biol Chem 275, 26806–26811 (2000).
Fuh, G., Garcia, K. C. & de Vos, A. M. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem 275, 26690–26695 (2000).
Wu, F. T., Stefanini, M. O., Mac Gabhann, F. & Popel, A. S. A compartment model of VEGF distribution in humans in the presence of soluble VEGF receptor-1 acting as a ligand trap. Plos One 4, e5108 (2009).
Hudson, N. W., Kehoe, J. M. & Koo, P. H. Mouse alpha-macroglobulin. Structure, function and a molecular model. Biochem J 248, 837–845 (1987).
Imber, M. J. & Pizzo, S. V. Clearance and binding of two electrophoretic “fast” forms of human alpha 2-macroglobulin. J Biol Chem 256, 8134–8139 (1981).
Acknowledgements
The research was supported by NIH grant R01 HL101200 and R21 HL122721 (ASP, BHA). GVC acknowledges scientist development grant 16SDG30340002 from American Heart Association.
Author information
Authors and Affiliations
Contributions
L.H.C., V.C.G., B.H.A. and A.S.P. proposed the idea; L.H.C. and G.C. implemented the simulations; V.C.G. and M.H.C. conducted the experiments; L.H.C., V.C.G., S.D.F., B.H.A. and A.S.P. edited the paper. The authors thank Drs. Hojjat Bazzazi and Feilim Mac Gabhann for critical comments and suggestions.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chu, LH., Ganta, V., Choi, M. et al. A multiscale computational model predicts distribution of anti-angiogenic isoform VEGF165b in peripheral arterial disease in human and mouse. Sci Rep 6, 37030 (2016). https://doi.org/10.1038/srep37030
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37030
This article is cited by
-
VEGF-A splice variants bind VEGFRs with differential affinities
Scientific Reports (2020)
-
The Convergence of Cell-Based Surface Plasmon Resonance and Biomaterials: The Future of Quantifying Bio-molecular Interactions—A Review
Annals of Biomedical Engineering (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
