
Abstract
MicroRNAs (miRNAs) are one class of endogenous non-coding RNAs modulating the expression of target genes involved in plant development and stress tolerance, by degrading mRNA or repressing translation. In this study, small RNA and mRNA degradome sequencing were used to identify low- and high-temperature stress-responsive miRNAs and their targets in cotton (Gossypium hirsutum). Cotton seedlings were treated under different temperature conditions (4, 12, 25, 35, and 42 °C) and then the effects were investigated. In total, 319 known miRNAs and 800 novel miRNAs were identified, and 168 miRNAs were differentially expressed between different treatments. The targets of these miRNAs were further analysed by degradome sequencing. Based on studies from Gene Ontology and Kyoto Encyclopedia of Genes and Genomes, the majority of the miRNAs are from genes that are likely involved in response to hormone stimulus, oxidation-reduction reaction, photosynthesis, plant–pathogen interaction and plant hormone signal transduction pathways. This study provides new insight into the molecular mechanisms of plant response to extreme temperature stresses, and especially the roles of miRNAs under extreme temperatures.
Similar content being viewed by others


Introduction
Temperature is an important factor limiting the geographical distribution and growing season of plants, and plants often suffer severe climatic damages such as causing tissue injury and delayed growth. Most organisms undergo optimal growth within a narrow temperature range, and can only tolerate minor fluctuations1. However, fluctuations beyond a threshold lead to low- or high-temperature stresses. Plants evolved complex mechanisms to cope with unfavourable conditions during the long-term evolutionary process2. Abiotic stresses affect plant development and production through effects on gene expression patterns under the control of different regulators such as non-coding RNAs and transcription factors3,4.
Small RNAs (sRNAs) have emerged as key post-transcriptional gene regulatory molecules in plants and animals. There are two classes of endogenous sRNAs: small interfering RNAs (siRNAs) and microRNAs (miRNAs) in plants. miRNAs are endogenous sRNAs having transcriptional and post-transcriptional regulatory effects on plant development and adaptive responses to stresses5. miRNAs predominantly play important gene-regulatory roles through base-pairing with target mRNAs for cleavage or translational repression6. They play regulatory roles in almost all aspects of plant development and response to stress7,8, and have been shown to be either up- or down-regulated in plant responses to abiotic stresses such as cold, salt, heat, drought, and oxidative stress9,10.
Low- and high-temperature stresses are two of the most important environmental factors affecting plant growth and development, but the roles of miRNAs in the plant’s response are not well understood. Although some miRNAs responding to stress are deeply conserved among various plant species, species-specific miRNAs with roles in regulatory networks associated with stress tolerance may arise from adaptation to long-term growth in stressful environments11. Recently, three conserved miRNAs and 25 predicted miRNAs have been identified by deep sequencing in response to cold stress in Brachypodium12. Several miRNAs respond to heat stress in wheat and barley, including miR160, miR166, miR16713,14.
Although temperature stress-responding miRNAs have been extensively investigated in several plant species, there has been no systematic research on miRNA profiles in cotton. Cotton is one of the most important commercial crops and is a major source of fiber for textiles in the world. Much focus has been directed to the mechanisms of cotton fiber development, quality and yield15. This includes analysis of miRNA expression during fiber development16, miRNA expression in biotic stress17 and abiotic stress responding to drought and salinity18. However, little work has been reported for cotton miRNAs in low- and high-temperature stresses, which can reduce fiber quality and yields.
The aim of this study was to profile the miRNA and their target mRNAs in cotton subjected to low- and high-temperature stresses, using high-throughput sequencing analysis and degradome sequencing. These findings reveal a putative miRNA-mediated regulatory network with a critical role in the response to low- and high-temperature stress in this important crop.
Results
Performance of seedlings under temperature stress conditions
To evaluate the biological and physiological performances during temperature treatments, we grew G. hirsutum cv.YZ1 seedlings under different temperature stress conditions (4, 12, 35, and 42 °C), each for 8 h, with the seedlings grown at 25 °C as control, all other environmental conditions being identical. Leaves of seedlings grown at 4 °C, 12 °C and 42 °C showed strong wilting, while seedlings grown at 25 °C and 35 °C showed no wilting (Fig. 1A). We measured four parameters of the leaves: H2O2 contents, proline contents, malondialdehyde (MDA) and soluble sugar contents. Overall, the contents of all physiological parameters were higher in treatments than in control especially under 4 °C conditions, except for MDA at 42 °C and proline at 12 °C, 35 °C and 42 °C (Fig. 1B–E).

Response of two-leaf-stage cotton seedlings to temperature stress.
(A) Performance of cotton seedlings under different temperature stresses for 8 h; (B) H2O2 contents of seedlings in EL, NL, CK, NH, EH; (C) Proline contents of seedlings in EL, NL, CK, NH, EH; (D) MDA contents of seedlings in EL, NL, CK, NH, EH; (E) Soluble sugar contents of seedlings in EL, NL, CK, NH, EH. Errorbars represent standard error. P-value was considered to be statistically significant with (*p < 0.05) or (**p < 0.01). EL, Extreme low temperature (4 °C); NL, Normal low temperature (12 °C); CK, Control (25 °C); NH, Normal high temperature (35 °C); EH, Extreme high temperature (42 °C).
High-throughput sequencing of small RNAs in cotton
To characterize sRNA profiles in plants grown under the different temperature regimes, five sRNA libraries were constructed from total RNA isolated from leaves of plants treated for 8 h, with two sample replicates for each treatment. These libraries were sequenced by an Illumina Solexa Sequencer and a summary of the sequencing and data analysis strategy is shown in Fig. S1. The five libraries generated 11,804,407.5 clean reads at 4 °C (Extreme low temperature; EL); 11,848,760.5 clean reads at 12 °C (Normal low temperature; NL); 11,825,387 clean reads at 25 °C (Control; CK); 11,819,102 clean reads at 35 °C (Normal high temperature; NH); and 11,802,474 clean reads at 42 °C (Extreme high temperature; EH). On average, 85.97% (unique reads, Table 1) and 92.13% (total reads, Table 1) reads were successfully matched back to the AD genome of G. hirsutum, after removing poor quality reads and adapter sequences. For annotation purposes, the sRNAs were grouped into several classes (Table 1). Overall, the clean full-length reads that generated and matched were similar among the five libraries.
The size distribution of all sequences with lengths between 18 and 28 nt was determined. A similar size distribution was observed for each of the five libraries, and a large proportion of the sRNAs were 20~24 nt long, in which the 24 nt read was the most abundant (41.6% to 46.6% of the five libraries), followed by 21 nt (Fig. 2). The sRNA abundance and size in cotton is largely consistent with the other plant species, such as Arabidopsis thaliana19, Oryza sativa20, Glycine max21, indicating that the majority of sRNAs in plants are 24 nt.

Length distribution of small RNA reads.
EL, Extreme low temperature (4 °C); NL, Normal low temperature (12 °C); CK, Control (25 °C); NH, Normal high temperature (35 °C); EH, Extreme high temperature (42 °C).
Identification of known miRNAs
We identified known miRNAs by using homologous analysis to find sRNA sequences with criteria for selection of a length of at least 18 nt, and a maximum of two mismatches compared to all known plant miRNA sequences deposited in miRBase21.0 in the five small RNA libraries. After removing repeat sequences, 319 annotated known miRNAs belonging to 144 families were identified (Table S1); out of these, 313 were from the 4 °C treated plants, 313 from the 12 °C, 311 from the 25 °C, 311 from the 35 °C, and 312 from the 42 °C treated plants. The miRNAs had a very broad range of expression levels, from millions of sequence reads to fewer than 10. 44 miRNA families are represented by multiple members, from 2 to 20 (Fig. S2), such as the miR156 family (20 members) and the miR166 family (19 members); but 100 other miRNA families were represented by only one member. The most abundant miRNA families were found to be miR157, miR166, miR156 and miR167 (Table S2), whose expression levels were >10000 transcripts per million (TPM) clean tags, and these are highly conserved in mosses, eudicots and monocots22,23.
More than 300 miRNAs were common across the five libraries, and among these known miRNAs less than 20 miRNAs were specific to temperature treatments. For example, gh-miR397-5p in EH (42 °C) and gh-miR8767c in NH (35 °C) were expressed only in high temperature-treatment samples, while gh-miR8779, gh-miR169h-3p and gh-miR7484c were expressed only in low temperature-treatment samples (4 °C, 12 °C). In addition, high temperature-treatment libraries shared 2 miRNA families that did not occur in the 25 °C library, gh-miR7484c and gh-miR171a-2. There was only one common miRNA, gh-miR399a-2, in the two low temperature treated libraries (Fig. 3A).

Distribution of miRNAs among treatments.
(A) Known miRNAs in low-temperature treatments and control (left), known miRNAs in high-temperature treatments and control (right); (B) Novel miRNAs in low-temperature treatments and control (left); novel miRNAs in high-temperature treatments and control (right). EL, Extreme low temperature (4 °C); NL, Normal low temperature (12 °C); CK, Control (25 °C); NH, Normal high temperature (35 °C); EH, Extreme high temperature (4 °C).
Of the 144 miRNA families, 21 nt long miRNAs represented the most abundant in size, which included 52.35% known miRNAs, followed by 24 nt sequences (22.26%) and 22 nt sequences (10.97%) in the known miRNA families (Table S3).
Identification of potentially novel miRNAs
Among different RNA classifications, unannotated sRNAs accounted for the greatest proportion of the sRNAs. We selected the unknown sRNAs which could be mapped to the reference sequence to identify novel miRNAs by using Mireap software. The stem-loop structures matched to miRNA precursors were used to predict fold-back RNA secondary structure. The sequences localized inside the stem-loop structures in the 3′ or 5′ arms were regarded as mature miRNAs. We found 800 sequences supposed to be novel miRNAs with high confidence - on average 633 in the 4 °C samples, 617 in the 12 °C, 605 in the 25 °C, 630 in the 35 °C, and 613 in the 42 °C samples. Among these 800 novel miRNAs, about 350 were common across all five libraries, and about 60 miRNAs were in 4 °C, 12 °C, 35 °C and 42 °C samples (Fig. 3B). Analysis of nucleotide bias at each position in miRNAs showed that the first nucleotide of miRNAs tended to be uracil (U) (Fig. S3), which was conserved across many species24. Most of the novel miRNA in all five samples showed diversified expression levels. However, the abundance of these novel miRNAs was low, each generating fewer than 100 reads, which represents a lower expression level than that of known miRNAs. The length of these novel miRNAs ranged from 20 nt to 23 nt, in which 21 nt represented the most plentiful class, at more than 50%.
miRNA expression in response to different temperatures
Among the identified miRNAs, 306 known miRNAs and 249 novel miRNAs, whose expression levels were more than 5 TPM, were used to analyse differential expression between temperature regimes. The results presented in Table S4 show that 63 out of 306 (20.6%) known miRNAs, and 105 out of 249 (42.2%) novel miRNAs, were expressed differentially in the five libraries (log2 Ratio ≥ 1 or ≤ −1). Among the 63 differently expressed known miRNAs, only 23.8% were in both low-temperature stress (4 °C and 12 °C) and high-temperature stress (35 °C and 42 °C). There was only one miRNA common across these four different temperature stresses, gh-miR8695, which was up-regulated in all conditions. Most miRNAs were expressed differentially in specific low- or high-temperature stress, with 57.1% especially highly expressed in high-temperature stress (Fig. 4A). Interestingly, more differentially expressed miRNAs were present in high-temperature stress, with relatively few differentially expressed at 4 °C. More miRNAs were significantly down-regulated in plants under high-temperature stress than in the 25 °C reference treatment, while more miRNAs were up-regulated in low-temperature stress than at 25 °C (Fig. 4B). The most significant expression difference was seen for gh-miR828-3p, which was 3.9-fold down-regulated at 42 °C.

Comparison of known miRNAs identified between temperature stress and control libraries.
(A) Venn diagram of temperature stress responsive miRNAs at EH, EL, NH and EH; (B) The overview of miRNAs highly responsive to temperature stress; (C) Heatmaps of differently expressed known miRNAs in EL, NL, NH and EH treatments, four miRNA clusters are shown on the right. Red, up-regulated; blue, down-regulated. EL, Extreme low temperature (4 °C); NL, Normal low temperature (12 °C); NH, Normal high temperature (35 °C); EH, Extreme high temperature (42 °C).
Cluster analysis revealed that four clusters were constructed based on the expression patterns of 63 known miRNAs in response to different temperature regimes (Fig. 4C). Cluster I contains miRNAs with a converse expression pattern in low- and high-temperature stress; Cluster II shows the same expression pattern both in low- and high-temperature stress. Some specific significant expression changes were observed under high-temperature stress in Cluster III; and Cluster IV contains miRNAs which are only differentially expressed in low-temperature stress (Fig. S4).
Among the 105 differentially expressed novel miRNAs identified in the five libraries, a quarter of them had the opposite expression trend in low- and high-temperature stress, and most of the expression levels were simultaneously up-regulated or down-regulated in low-temperature stress or high-temperature stress (Fig. S5). The novel miRNAs identified here were more obviously differentially expressed under temperature stress, although their expression levels were lower than those of known miRNAs. Most miRNAs (known and novel) were down-regulated significantly under high-temperature stress (Fig. S6).
Identification of known and novel miRNA targets through degradome sequencing
Plant miRNAs can bind almost perfectly to their target genes via typical complementarity matching, and regulate post-transcriptional process by transcript degradation or translational inhibition25. Accurate validation of miRNA targets is important to elucidate potential biological functions of miRNAs. In our study, degradome sequencing technology was used to detect the miRNA targets cleaved by the identified candidate miRNAs. We acquired approximately 1 × 108 raw reads from each library through the degradome sequencing. After alignment with the AD genome of G. hirsutum, when a mismatch threshold ≤2 was applied, 10,305,697 clean tags from the 4 °C library were obtained, 9,791,298 from the 12 °C library, 10,9789,76 from the 25 °C library, 9,854,917 from the 35 °C library, and 12,549,112 from the 42 °C library (Table 2).
The target mRNAs were further investigated to gain insight into the regulatory function of the miRNAs. A total of 950 target transcripts for 133 known miRNAs, and 590 candidate targets for 253 novel miRNAs, were identified. For the identification of cleavage sites, degradome peaks could be classified into five classes (categories 0, 1, 2, 3 and 4). Among the identified targets, the targets with category 0 or 1 were evaluated as the most significant, and category 0 was the most abundant category of known miRNAs (Table 3). miRNAs were found to be able to target various numbers of genes with a range of 1 to 24, of which gh-miR828a targeted the highest number of genes, reaching 24 different genes (Table S5). A total of 216 target transcripts for 63 differentially expressed miRNAs, including 32 known and 31 novel miRNAs were identified in our degradome libraries (Table S5 and Table S6).
The results revealed that the potential target genes participated in various biological processes. Targets included genes encoded transcription factors, such as GRAS, MYB, NAC-domain, and ARF (auxin response factor), which were involved in the regulation of gene expression and signal transduction; others were disease resistance-related proteins of the CC-NBS-LRR class known as stress-responsive proteins; and other stress-induced proteins, such as laccase and heat shock proteins. In addition, mRNAs encoding proteins associated adaptive responses in plant growth and development, such as copper binding protein, transferases, phosphatases as well as other proteins of unclear functions, were identified as targets of miRNAs. Based on the degradome sequencing data, Fig. 5 shows examples of some confirmed stress-related miRNA targets as ‘target plots’ (T-plots).

Cotton miRNA target alignment and its T-plot validated by degradome sequencing.
The T-plots show the distribution of the degradome tags along the full length of the target mRNA sequence. Red lines indicate signatures consistent with miRNA-directed cleavage. (A) gh-miR398b and Gh_A09G2473 (chloroplast Cu/ZnSOD); (B) gh-miR828a and Gh_D08G1815 (MYB-like DNA-binding domain protein); (C) gh-miR397a-2 and Gh_D11G3322 (laccase1b); (D) gh-miR160g and Gh_A05G0895 (auxin response factor 3).
Functional enrichment for the miRNA target genes
In the present study, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for better understanding these differentially expressed miRNAs and their target genes. According to GO classifications, the ‘biological process’ category contained the largest number of target genes. These target genes predominantly participated in 53 biological process categories, 21 molecular function categories and 1 cellular component categories (Table S7). Most specific GO classification showed that the target genes were involved in biological processes associated with response to hormone stimulus (GO:0009725), single-organism transport (GO:0044765), defense response (GO:0006952), photosynthesis, and light harvesting (GO:0009765), regulation of transcription, and DNA-dependent (GO:0006355). Sequence-specific DNA binding (GO:0003700), hydrolase activity (GO:0016787), glycine dehydrogenase (decarboxylating) activity (GO:0004375), ATP citrate synthase activity (GO:0003878), and protein dimerization activity (GO:0046983) were significantly enriched among the most abundant classes in molecular function category. Nucleus (GO:0005634) was enriched in cellular component categories.
KEGG-based analysis allows linkage of 168 miRNAs and 216 targets to 26 pathways, and the significantly enriched pathways include photosynthesis-antenna proteins (ath00196), plant-pathogen interaction (ath04626), protein processing in endoplasmic reticulum (ath04141), porphyrin and chlorophyll metabolism (ath00860) (Table S8).
Confirmation of predicted miRNAs by qRT–PCR
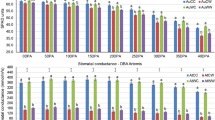
To validate the reliability of deep sequencing, we used qRT-PCR to analyse the expression of the miRNAs. Based on the high-throughput sequencing results, miRNAs were randomly selected for qRT–PCR (Fig. 6). Overall, expression patterns of these selected miRNAs obtained by qRT-PCR were consistent with the sequencing results. The miRNA expression trends are similar, indicating high reliability of the analysis. To validate whether the predicted target genes were really regulated by miRNAs under different temperature stresses, we conducted the expression analysis of the predicted target genes and their corresponding miRNAs, and the results revealed that the predicted target genes had an opposite expression profiles compared to the corresponding miRNAs (Fig. 7).

Fold-change of miRNA expression relative to CK (log2) by qRT-PCR.
Expression was normalized by the level of UBQ7 in qRT-PCR. All reactions of qRT-PCR were repeated three times for each sample and the results presented as the mean ± SD. (A) Known miRNA; (B) Novel miRNA. EL, Extreme low temperature (4 °C); NL, Normal low temperature (12 °C); CK, Control (25 °C); NH, Normal high temperature (35 °C); EH, Extreme high temperature (42 °C).

Expression profiles of miRNAs and their targets under temperature stress by qRT-PCR.
All data were subjected to an analysis of variance (ANOVA) and the results presented as the mean ± SD. A P-value was considered to be statistically significant with (*p < 0.05) or (**p < 0.01). EL, Extreme low temperature (4 °C); NL, Normal low temperature (12 °C); CK, Control (25 °C); NH, Normal high temperature (35 °C); EH, Extreme high temperature (42 °C).
Discussion
miRNAs, the post-transcriptional gene regulators, are known to play an important role in response to biotic and abiotic stresses. Environmental stresses may cause certain miRNAs differentially expressed or some novel miRNAs synthesized to cope with various stresses in plants. A few of stress-responsive miRNAs have been detected in plants in various biotic and abiotic stresses, including nutrient deficiency26, drought27, heat13, cold28, and bacterial infection stress29. There are also several abiotic stress-regulated miRNAs known in cotton, but most are associated with growth30,31, and response to drought and salinity stress18.
Most plants can regulate the physiological and biochemical processes by changing H2O2, MDA, proline and soluble sugar contents to adapt to temperature stresses in which ROS generation and membrane damage were induced32,33. In our study, the physiological parameters (H2O2, MDA, proline and soluble sugar contents) in response to temperature stresses were higher than control (Fig. 1), the results herein demonstrate that seedlings might be less capable of surviving from temperature stress conditions. Therefore, we used seedlings under different temperature stress conditions (4, 12, 35, and 42 °C) to analyse the regulation by miRNAs.
We identified 319 known miRNAs and 800 novel miRNAs in total (Table S1), and their corresponding precursors and prediction targets, expressed in response to temperature stress. Among all the miRNAs, 63 known miRNAs and 105 novel miRNAs were identified to be temperature stress-responding. Degradome sequencing led to the identification of 216 target genes for 32 known and 31 novel miRNAs (Table S5 and Table S6). 156 target genes for 32 known miRNAs represent members of 27 gene families, which were enriched in transcription factors; regulatory factors; stress-responsive proteins; proteins involved in oxidation-reduction reactions and photosynthesis, as well as other proteins whose functions are currently unclear (Fig. 8).

Summary of differentially expressed miRNAs and their targets genes involved in temperature stress.
Numbers in internal circle with red color were miRNAs families. The genes in outer race represented the targets for counterparts in the line.
miRNAs might regulate cotton growth homeostasis during temperature stress
Temperature stress negatively affects plant growth and development by causing tissue injury and delaying growth, and at least several of the stress-responding miRNAs can be expected to be involved in pathways that reprogramme metabolism and physiology. For example, temperature stress is known to disrupt the plant’s nutrient, metabolite and hormone homeostasis34,35.
Copper (Cu) is an essential micronutrient required for photosynthesis, oxidative responses, and other physiological processes36. There were several Cu homeostasis related miRNAs in low- and high-temperature stress, such as miR398 and miR397. miR398 is an important regulator of Cu homeostasis, and reduces the allocation of Cu to chloroplastic copper/zinc superoxide dismutases (Cu/Zn-SOD, CSDs) in response to low copper availability and makes Cu available for some essential processes in Arabidopsis37. The different expression patterns of miR398b detected under low- and high-temperature conditions reveal that miR398b may act as a mediator in response to temperature stress. In plants, lignin is the main component of secondary cell walls and its degradation is regulated by laccases, a kind of blue copper oxidase enzyme. The relative expression levels of the two carrot laccase-encoding genes DcLac1 and DcLac2 under heat and cold treatments are similar38. In cotton, our data predict that laccase is the target of gh-miR397a-2, and gh-miR397a-2 is significantly up-regulated at 4 °C compared with the 25 °C treatment. This suggests that nutrient and metabolite homeostasis is disturbed in low- and high-temperature stress, perhaps to mobilize resources to supporting plant growth and development during stress (Fig. S7).
Cu homeostasis and hormones had a complex reciprocal relationship in plants, and hormonal regulation requires matching special copper39. It was documented that any change in temperature would modify endogenous hormone levels40. ARF transcription factors regulate the expression of auxin-inducible genes by binding auxin-responsive promoters (ARPs)41. Previous studies revealed that some ARFs are targeted by miR160 and miR16742, which play a key role in attenuating plant growth and development under stress conditions43,44. Consistent with this, the miR160 family, including miR160-1 and gh-miR160g, are significantly down-regulated in both 12 °C and 42 °C treatments, and the targeted genes were identified as encoding ARF genes. Therefore gh-miR160g might participate in cotton seedling development and seedling tolerance to temperature stress through modulation of the auxin response. Furthermore, a novel miRNA mir-119, targeting an auxin-responsive protein, was found to decrease in high-temperature stress. This suggests that auxin signalling regulation in response to low- and high-temperature stress is worthy of further study.
Therefore, such a temperature shift induced a disruption of the plant’s overall homeostatic state including nutrient, metabolite and hormone homeostasis which may impact proteins encoding regulatory factors in plant development as shown in Fig. 8 to attenuate plant growth and metabolic rate in response to temperature stresses (Fig. S7).
Chloroplast may be a sensor for temperature stress
The chloroplast is considered a sensor of environmental changes and plant stress responses45. In chloroplasts, the stabilization of the lamellar membrane systems through alterations in lipid composition is a key factor for tolerance, and damage to thylakoid membranes impairs photosynthesis and photosynthetic capacity46. Our results found significantly enriched GO terms in photosynthesis and light harvesting (GO:0009765) (Table S7), while KEGG-based pathway identifications were significantly enriched in photosynthesis-antenna proteins (ath00196) and porphyrin and chlorophyll metabolism (ath00860) (Table S8). gh-miR397a-2, which targets a chlorophyll a-b binding protein P4, was up-regulated in low-temperature stress, and gh-miR169r-3p, which targets Photosystem I reaction center subunit III (PasF), was down-regulated in high-temperature stress.
Superoxide radicals as the primary products in photo-reduction of dioxygen in chloroplasts are major consumers of Cu, which may cause lipid peroxidation and oxidative stress under stresses. Chloroplast is one of the major sites for the generation of ROS under stress, and causes damage to the photosynthetic apparatus and may function as a second messenger47. Some components in temperature stress have been found to function in protection from oxidative damage, and there were several genes related to ROS detected to be miRNA targets in cotton. Superoxide dismutases convert superoxide into molecular oxygen and hydrogen peroxidein plants. miR398 targets the mRNA of chloroplastic copper/zinc superoxide dismutase (CSD), a scavenger enzyme of ROS detoxifying superoxide radicals for degradation48,49. CSD2 is attached to the thylakoid of chloroplasts, where superoxide generated, so that it can localize and immediately sweep superoxide radicals. The decrease in expression level of miR398 led to increase in CSD expression which was important for plant tolerance to oxidative stress50. In Arabidopsis, transgenic plants expressing a form of the CSD2 showed higher ability to detoxify ROS superoxide radicals and regulation of the plant tolerance to oxidative stress conditions51. miR398 and the corresponding targets CSD genes are conserved across many plant species, and might have a ubiquitous role in oxidative stress management under various abiotic stresses. Our results show that gh-miR398b expression is up-regulated at 4 °C and down-regulated at 35 °C. The results suggested that miR398-regulated GhCSD genes play roles in temperature stress through protecting plants from oxidative damage. Interestingly, the mRNAs of a CSD gene targeted by a novel miRNA, mir-496, showed an up-regulated expression pattern at high-temperature stress. All of these results nevertheless suggest that temperature stress affects the chloroplasts and ROS, and the stress-responsive miRNAs are involved in associated pathways (Fig. S7), which is consistent with the results that H2O2, MDA, proline, and soluble sugar contents increased in response to temperature stresses.
Transcription factors play a key role in the response to temperature stress
Transcription factors regulated by miRNAs may be crucial for plant tolerance to temperature stresses. Almost one third of the temperature-responsive miRNA-targeted transcription factors were shown to be related to regulation of plant growth and development. These include auxin-responsive genes, members of the YABBY2-like transcription factor family, the HD-Zip transcription factor family, the GRAS transcription factor family, the MYB-like family, the CIPK family, the eukaryotic translation initiation factor 2c family, and the zinc finger-like family (Fig. 8).
The MYB gene family is one of the largest families in plants, and some of its members are regulated by miRNAs. For instance, an R2R3-type MYB transcription factor, MYB96, is induced by cold stress in an ABA-independent manner and subsequently activates freezing tolerance52. Over-expression of OsMYB55 in maize was found to improve plant growth and performance under high temperature and drought conditions53. In cotton, miR828 and miR858 regulate the homologous MYB2 gene during both Arabidopsis trichome and cotton fibre development54. In the present study, we show that GhMYB is targeted by gh-miR828a and gh-miR858, and is up-regulated in high-temperature stress, suggesting a role for GhMYB targeting by miR828 and miR858 in response to high-temperature stress. In addition, miR166g and its target genes class III AtHD-ZIP regulates shoot apical meristem and lateral organ formation55. In our study, we found that the HD-Zip transcription factor family was also predicted to be a target mRNA of cotton miR166, and the expression level of gh-miR166k increased at 4 °C. Therefore, there may exist a regulatory interaction between miR166 targeted HD-ZIP and temperature stress.
Abiotic and biotic stresses may somehow share common response pathway
Low- and high-temperature stresses were found to decrease the tolerance of plants to biotic stresses, so the genes encoding putative disease resistance proteins are regulated under stress conditions56,57. In the present study, miR482 family (gh-miR482-2, gh-miR482a, gh-miR482b-3p, gh-miR482c) was found to be down-regulated in high-temperature stress. Among the miR482 members, two of them targeted disease resistance proteins. gh-miR398b, which targeted a disease resistance protein RGA2, was differentially expressed both in low- and high-temperature stresses. These results indicated that stress inducible proteins were involved in temperature stresses (Fig. 8 and Fig. S7), so we propose that abiotic and biotic stresses may share some common regulatory mechanism, temperature stress may make the plant vulnerable to pathogens.
Conclusions
In summary, we identified 63 known miRNAs and 105 novel miRNAs differentially expressed in response to low- and high-temperature stress. GO terms and KEGG-based enrichment analysis showed that most of the miRNAs involved in the response to temperature stress act by regulating genes associated with growth homeostasis, ROS, chloroplast function, plant–pathogen interaction and plant hormone signal transduction pathways. This provides a valuable platform for future functional analysis.
Methods
Plant culture and treatments
Upland cotton seeds (Gossypium hirsutum cv. YZ1) were grown in a growth chamber at 25 °C under a 16 hour/8 hour (light/dark) photoperiod. When the plants reached the two-leaf stage, they were treated under different temperature conditions (4, 12, 25, 35, and 42 °C) for 8 h. All leaves were harvested and immediately frozen in liquid nitrogen, and stored at −80 °C prior to RNA isolation. Three biological replicates were carried out for each treatment. We considered the samples in 25 °C without any treatment as the reference point for comparison.
Total RNA extraction
Total RNA was extracted from leaf tissue using a modified guideline thiocyanate method58. Samples were ground in a mortar with liquid nitrogen, and RNA was extracted by ice-cold extraction buffer containing 1% β-mercaptoethanol. The supernatant was purified by phenol and chloroform and then precipitated by isopropanol and sodium acetate (3 M), washed with 75% ethanol. The RNA was air dried and dissolved in DEPC-treated water. All the RNA samples were quantified and equalised to ensure equal amounts of RNA from each treatment were available for library construction. The quality of RNA samples was evaluated using an Agilent 2100 Bioanalyzer (RIN ≥ 7.5; 28S:18S ≥ 1.3) (Agilent, Waldbronn, Germany).
Construction of small RNA and degradome libraries
The sRNA libraries were constructed following a standard protocol (Illumina, USA). Small RNAs were purified from 10 μg of total RNA by polyacrylamide gel electrophoresis, and ligated first to a 5′ RNA adaptor and then to a 3′ RNA adaptor as previously described28. Purified RNAs were reverse-transcribed to cDNA, followed by PCR amplification to generate the DNA pool16. Five DNA pooled libraries were sequenced on an Illumina Solexa Sequencer at the Beijing Genomics Institute (BGI, http://www.genomics.cn/en/index) in Wuhan. Each sample was duplicated.
Five degradome libraries were constructed from cotton leaves to predict the potential target mRNAs following the methods described previously59. Briefly, a 5′ RNA adapter with a MmeI recognition site at the 3′ end was ligated to the resulting 42 bp fragments consisting of a free phosphate at the 5′ end, followed by reverse-transcribed to cDNA. After PCR amplification, they were digested by the enzyme MmeI, and ligated to Illumina 3′ TruSeq adaptors, followed by PCR amplification with library-specific index primers and a common 5′ primer for multiplex sequencing. Five DNA pools were sequenced on an Illumina Solexa Sequencer at BGI.
Bioinformatics analysis of sequencing data
Raw sequences obtained from the five small RNA libraries and degradome libraries were first cleaned by filtering out low-quality tags, poly (A) tags, and tags with 3′ adaptor nulls, insert nulls, 5′ adaptor contaminants, or were smaller than 18 nt. Clean reads matching other small RNAs, including rRNA, snRNA, repeat RNA, tRNA, and snoRNA, were compared with G. hirsutum non-coding RNA sequences in the NCBI GenBank and Rfam databases. The remaining sequences from 18~24 nt long were used to BLAST against the miRBase 21.0 (http://www.mirbase.org/) to identify conserved miRNAs and novel 5p- and 3p-derived miRNAs. Only the sequences that were ≤2 mismatches with known miRNAs in miRBase were considered as conserved miRNAs, otherwise reads are defined as non-conserved reads. Unannotated reads were used for prediction of novel miRNAs according to the characteristic hairpin structure of microRNA precursors by the RNA-folding software Mireap (http://sourceforge.net/projects/mireap/). We used the AD genome sequences of G. hirsutum as a miRNA positioning reference sequence to identify miRNA precursors60.
The 20~21 nt sequences of clean full-length reads collected from the degradome sequencing were used for subsequent analysis after removing low quality sequences and adapters. There were no mismatches allowed on the 10th and 11th nucleotides of mature miRNAs where the splice site on miRNA targets generally occurs in degradome analysis. Potential miRNA targets with a P-value of < 0.05 by PAREsnip software were retained, and T-plot figures were drawn. All target sequences were categorized into five classes based on the abundance of degradome tags indicating miRNA-mediated cleavage. The category 0~4 was determined as described61.
Expression pattern of miRNAs between stress and control libraries
To investigate differentially expressed miRNAs between the control and treated libraries, the fold change of each identified miRNA was calculated as the ratio of read counts in the treatment libraries to the read counts in the control library followed by transformation of log2. The value of log2 Ratio ≥1 or ≤−1, indicating the ratio of RPKM values for the treatments and control libraries, were considered as significantly differentially expressed. To show differential expression profiles, heatmaps and clusters were constructed for the miRNAs using Genesis (http://genome.tugraz.at/genesisclient/genesisclient_description.shtml).
qRT-PCR analysis
To further verify our identification results, chosen sequences were subjected to qRT-PCR. In the first step, 3 μg of total RNA was reverse-transcribed to cDNA for miRNAs using the Mir-X™ miRNA First-Strand Synthesis and SYBR Kit (Clontech, CA, USA). RNA was reverse-transcribed to cDNA for target genes using the SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). In the second step, quantitative real-time PCR (qRT-PCR) was carried out using the ABI Prism 7500 system (Applied Biosystems, Foster City, CA, USA) for 30 s at 95 °C, followed by 40 cycles of 5 s at 95 °C and 35 s at 60 °C. For qRT-PCR analysis, at least 5–10 plants of every line or treatment were sampled for each independent biological replicate62. The expression level of UBQ7 was used as the internal control to standardize the RNA samples for each reaction. Three biological replicates for each experiment were performed. Error bars represent the ± SD. The Ct (2−ΔΔCt) was used to calculate the fold changes, and relative expression levels were shown as log2 fold changes. The primers used in the study are listed in Table S9.
Measurement of physiological indexes
Endogenous H2O2 levels were detected by H2O2 Quantitative Assay Kit (Sangon Biotech, Shanghai, China). The contents of malondialdehyde (MDA) and soluble sugar and proline were measured according to the method described previously63,64.
Function enrichment analysis
To evaluate the miRNA-gene regulatory network, target sequences were annotated using Blast2GO software for assigning GO terms to investigate putative functions. GO terms among a list of genes considered all genomic genes as the background. GO-controlled vocabularies describe three categories of biological process, molecular function and cellular component. The statistical significance of GO term enrichment was measured by Fisher’s exact test with a corrected FDR of <0.05. The main metabolic pathways associated linked to target genes can be predicted by KEGG enrichment analysis using KOBAS 2.0 (http://kobas.cbi.pku.edu.cn/home.do). KEGG enrichment analysis was obtained with a P-value of < 0.05.
Additional Information
How to cite this article: Wang, Q. et al. Small RNA-mediated responses to low- and high-temperature stresses in cotton. Sci. Rep. 6, 35558; doi: 10.1038/srep35558 (2016).
References
Peleg, Z. & Blumwald, E. Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 14, 290–295, 10.1016/j.pbi.2011.02.001 (2011).
Huang, G. et al. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 39, 969–987, 10.1007/s11033-011-0823-1 (2012).
Ci, D., Song, Y., Tian, M. & Zhang, D. Methylation of miRNA genes in the response to temperature stress in Populus simonii. Front. Plant Sci. 6, 921, 10.3389/fpls.2015.00921 (2015).
Chiasson, D. M. et al. Soybean SAT1 (Symbiotic Ammonium Transporter 1) encodes a bHLH transcription factor involved in nodule growth and NH4+ transport. Proc. Natl. Acad. Sci. USA 111, 4814–4819, 10.1073/pnas.1312801111 (2014).
Chen, X. Small RNAs and their roles in plant development. Annu Rev Cell Dev. 25, 21–44, 10.1146/annurev.cellbio.042308.113417 (2009).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233, 10.1016/j.cell.2009.01.002 (2009).
Zhai, L. et al. Transcriptional identification and characterization of differentially expressed genes associated with embryogenesis in radish (Raphanus sativus L.). Sci. Rep. 6, 21652, 10.1038/srep21652 (2016).
Xu, S. et al. Identification of chilling-responsive microRNAs and their targets in vegetable soybean (Glycine max L.). Sci. Rep. 6, 26619, 10.1038/srep26619 (2016).
Cakir, O., Candar‐Cakir, B. & Zhang, B. Small RNA and degradome sequencing reveals important microRNA function in Astragalus chrysochlorus response to selenium stimuli. Plant Biotechnol. J. 14, 543–556, 10.1111/pbi.12397 (2016).
Wang, Y. et al. Combined small RNA and degradome sequencing to identify miRNAs and their targets in response to drought in foxtail millet. BMC Genet. 17, 57, 10.1186/s12863-016-0364-7 (2016).
Gan, Q., Chi, X. & Qin, S. Roles of microRNA in plant defense and virus offense interaction. Plant Cell Rep. 27, 1571–1579, 10.1007/s00299-008-0584-z (2008).
Zhang, J., Xu, Y., Huan, Q. & Chong, K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics 10, 449, 10.1186/1471-2164-10-449 (2009).
Xin, M. et al. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 10, 123, 10.1186/1471-2229-10-123 (2010).
Kruszka, K. et al. Transcriptionally and post-transcriptionally regulated microRNAs in heat stress response in barley. J. Exp. Bot. 65, 6123–6135, 10.1093/jxb/eru353 (2014).
Zhu, Y. et al. Transcriptome analysis reveals crosstalk of responsive genes to multiple abiotic stresses in cotton (Gossypium hirsutum L.). PloS One 8, e80218, 10.1371/journal.pone.0080218 (2013).
Liu, N. et al. Small RNA and degradome profiling reveals a role for miRNAs and their targets in the developing fibers of Gossypium barbadense. Plant J. 80, 331–344, 10.1111/tpj.12636 (2014).
He, X. et al. Identification of novel microRNAs in the Verticillium wilt-resistant upland cotton variety KV-1 by high-throughput sequencing. SpringerPlus 3, 564, 10.1186/2193-1801-3-564 (2014).
Xie, F., Wang, Q., Sun, R. & Zhang, B. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J. Exp. Bot. 66, 789–804, 10.1093/jxb/eru437 (2015).
Rajagopalan, R., Vaucheret, H., Trejo, J. & Bartel, D. P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 20, 3407–3425, 10.1101/gad.1476406 (2006).
Wei, L. Q., Yan, L. F. & Wang, T. Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol. 12, R53, 10.1186/gb-2011-12-6-r53 (2011).
Song, Q. et al. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 11, 5, 10.1186/1471-2229-11-5 (2011).
Axtella, M. J. & Bartela, D. P. Antiquity of MicroRNAs and their targets in land plants. Plant Cell 17, 1658–1673, 10.1105/tpc.105.032185 (2005).
Cui, L., Shan, J., Shi, M., Gao, J. & Lin, H. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 80, 1108–1117, 10.1111/tpj.12712 (2014).
Mi, S. et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133, 116–127, 10.1016/j.cell.2008.02.034 (2008).
Djuranovic, S., Nahvi, A. & Green, R. miRNA-Mediated Gene Silencing by Translational Repression Followed by mRNA Deadenylation and Decay. Science 336, 237–240, 10.1126/science.1215691 (2012).
Fujii, H., Chiou, T., Lin, S., Aung, K. & Zhu, J. A miRNA involved in phosphate-starvation response in Arabidopsis. Curr. Biol. 15, 2038–2043, 10.1016/j.cub.2005.10.016 (2005).
Zhou, L. et al. Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 61, 4157–4168, 10.1093/jxb/erq237 (2010).
Lv, D. et al. Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene 459, 39–47, 10.1016/j.gene.2010.03.011 (2010).
Navarro, L. et al. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 312, 436–439, 10.1126/science.1126088 (2006).
Yang, X., Wang, L., Yuan, D., Lindsey, K. & Zhang, X. Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J. Exp. Bot. 64, 1521–1536, 10.1093/jxb/ert013 (2013).
Xie, F., Don-C, J., Wang, Q., Sun, R. & Zhang, B. Small RNA sequencing identifies miRNA roles in ovule and fibre development. Plant Biotechnol. J. 13, 355–369, 10.1111/pbi.12296 (2015).
Nahar, K., Hasanuzzaman, M., Mahabub-Alam, M. & Fujita, M. Exogenous spermidine alleviates low temperature injury in Mung Bean (Vignaradiata L.) seedlings by modulating ascorbate-glutathione andglyoxalase Pathway. Int. J. Mol. Sci. 16, 30117–30132, 10.3390/ijms161226220 (2015).
Peng, T., Zhu, X., Duan, N. & Liu, J. PtrBAM1, a β-amylase-coding gene of Poncirust rifoliata, is a CBF regulon member with function in cold tolerance by modulating soluble sugar levels. Plant Cell Environ. 37, 2754–2767, 10.1111/pce.12384 (2014).
Charles, G., Fatma K., Joachim K., Joachim S. & Dirk K. H. Metabolomics of temperature stress. Physiol. Plant. 2, 220–235, 10.1111/j.1399-3054.2007.00999.x (2007).
Julia, K. & Claudia, J. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 63, 1593–1608, 10.1093/jxb/err460 (2012).
Yamasaki, H. et al. Regulation of copper homeostasis by micro-RNA in Arabidopsis. J. Biol. Chem. 282, 16369–16378, 10.1074/jbc.M700138200 (2007).
Abdel-Ghany, S. E. & Pilon, M. MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J. Biol. Chem. 283, 15932–15945, 10.1074/jbc.M801406200 (2008).
Ma, J., Xu, Z., Wang, F. & Xiong, A. Isolation, purification and characterization of two laccases from carrot (Daucus carota L.) and their response to abiotic and metal ions stresses. Protein J. 34, 444–452, 10.1007/s10930-015-9639-5 (2015).
Peñarrubia, L. et al. Temporal aspects of copper homeostasis and its crosstalk with hormones. Front. Plant Sci. 6, 255, 10.3389/fpls.2015.00255 (2015).
Achard, P. et al. The cold-inducible CBF1 factor–dependent signaling pathway modulates the accumulation of the growth-repressing DELLA proteins via its effect on gibberellin metabolism. Plant Cell 20, 2117–2129, 10.1105/tpc.108.058941 (2008).
Chapman, E. J. & Estelle, M. Mechanism of auxin-regulated gene expression in plants. Annu. Rev. Genet. 43, 265–285, 10.1146/annurev-genet-102108-134148 (2009).
Gutierrez, L. et al. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 21, 3119–3132, 10.1105/tpc.108.064758 (2009).
Sunkar, R., Li, Y. & Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 17, 196–203, 10.1016/j.tplants.2012.01.010 (2012).
Sattar, S., Addo‐Quaye, C. & Thompson, G. A. miRNA‐mediated auxin signalling repression during Vat‐mediated aphid resistance in Cucumis melo. Plant Cell Environ., 39, 1216–1227, 10.1111/pce.12645 (2016).
Fernández, A. P. & Strand, Å. Retrograde signaling and plant stress: plastid signals initiate cellular stress responses. Curr. Opin. Plant Biol. 11, 509–513, 10.1016/j.pbi.2008.06.002 (2008).
Crosatti, C., Rizza, F., Badeck, F. W., Mazzucotelli, E. & Cattivelli, L. Harden the chloroplast to protect the plant. Physiol. Plant. 147, 55–63, 10.1111/j.1399-3054.2012.01689.x (2013).
Gill, S. S. & Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Bioch. 48, 909–930, 10.1016/j.plaphy.2010.08.016 (2010).
Jagadeeswaran, G., Saini, A. & Sunkar, R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 229, 1009–1014, 10.1007/s00425-009-0889-3 (2009).
Lu, Y., Feng, Z., Bian, L., Xie, H. & Liang, J. miR398 regulation in rice of the responses to abiotic and biotic stresses depends on CSD1 and CSD2 expression. Funct. Plant Biol. 38, 44–53, 10.1071/FP10178 (2011).
Ding, Y., Tao, Y. & Zhu, C. Emerging roles of microRNAs in the mediation of drought stress response in plants. J. Exp. Bot. 64, 3077–3086, 10.1093/jxb/ert164 (2013).
Sunkar, R., Kapoor, A. & Zhu, J. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 18, 2051–2065, 10.1105/tpc.106.041673 (2006).
Lee, H. G. & Seo, P. J. The MYB96–HHP module integrates cold and abscisic acid signaling to activate the CBF–COR pathway in Arabidopsis. Plant J. 82, 962–977, 10.1111/tpj.12866 (2015).
Casaretto, J. A. et al. Expression of OsMYB55 in maize activates stress-responsive genes and enhances heat and drought tolerance. BMC Genomics 17, 312, 10.1186/s12864-016-2659-5 (2016).
Guan, X. et al. miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat. Commun. 5, 3050, 10.1038/ncomms4050 (2014).
Williams, L., Grigg, S. P., Xie, M., Christensen, S. & Fletcher, J. C. Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 132, 3657–3668, 10.1242/dev.01942 (2005).
Chen, L. et al. Genome-wide identification and expression analysis of heat-responsive and novel microRNAs in Populus tomentosa. Gene 504, 160–165, 10.1016/j.gene.2012.05.034 (2012).
Fan, G., Li, X., Deng, M., Zhao, Z. & Yang, L. Comparative analysis and identification ofmiRNAs and their target genes responsiveto salt stress in diploid and tetraploidPaulownia fortuneiseedlings. Plos One 11, e0149617, 10.1371/journal.pone.0149617 (2016).
Zhu, L., Tu, L. & Zeng, F. An improved simple protocol for isolation of high quality RNA from Gossypium spp. suitable for cDNA library construction. Acta Agr. Sinica 31, 1657–1659 (2005).
German, M. A. et al. Global identification of microRNA–target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 26, 941–946, 10.1038/nbt1417 (2008).
Xie, F. & Zhang, B. microRNA evolution and expression analysis in polyploidized cotton genome. Plant Biotechnol. J. 13, 421–434, 10.1111/pbi.12295 (2015).
Liu, H. et al. Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics 15, 25, 10.1186/1471-2164-15-25 (2014).
Sun, L. et al. Cotton cytochrome P450 CYP82D regulates systemic cell death by modulating the octadecanoid pathway. Nat. Commun. 5, 5372, 10.1038/ncomms6372 (2014).
Zhou, M. et al. Improvement of drought and salt tolerance in Arabidopsis and Lotus corniculatus by overexpression of a novel DREB transcription factor from Populus euphratica. Gene 10, 10–17, 10.1016/j.gene.2012.06.089 (2012).
Deng, Y., Chen, S., Chen, F., Cheng, X. & Zhang, F. The embryo rescue derived intergeneric hybrid between chrysanthemum and Ajania przewalskii shows enhanced cold tolerance. Plant Cell Rep. 12, 2177–2186, 10.1007/s00299-011-1123-x (2011).
Acknowledgements
This work was supported by China Agricultural Research System (CARS-18-09).
Author information
Authors and Affiliations
Contributions
X.Z. designed the research. Q.W. performed the experiments and analyzed the data. Q.W. drafted the manuscript. N.L., X.Y., L.T. and X.Z. provided valuable suggestions and advice concerning the manuscript. All of the authors carefully checked and approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, Q., Liu, N., Yang, X. et al. Small RNA-mediated responses to low- and high-temperature stresses in cotton. Sci Rep 6, 35558 (2016). https://doi.org/10.1038/srep35558
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35558
This article is cited by
-
Genetic manipulation of microRNAs: approaches and limitations
Journal of Plant Biochemistry and Biotechnology (2023)
-
Morpho-physiological integrators, transcriptome and coexpression network analyses signify the novel molecular signatures associated with axillary bud in chrysanthemum
BMC Plant Biology (2020)
-
High temperature triggered plant responses from whole plant to cellular level
Plant Physiology Reports (2020)
-
Transcriptome analysis reveals new microRNAs-mediated pathway involved in anther development in male sterile wheat
BMC Genomics (2018)
-
Genome-wide analysis of GRAS transcription factor gene family in Gossypium hirsutum L.
BMC Genomics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
