
Abstract
Transgenic Populus tremula x alba (717-1B4) plants with reduced expression of a tonoplast sucrose efflux transporter, PtaSUT4, exhibit reduced shoot growth compared to wild type (WT) under sustained mild drought. The present study was undertaken to determine whether SUT4-RNAi directly or indirectly altered poplar predisposition and/or response to changes in soil water availability. While sucrose and hexose levels were constitutively elevated in shoot organs, expression responses to drought were most altered in the root tips of SUT4-RNAi plants. Prior to any drought treatment, constitutively elevated transcript levels of abscisic acid biosynthetic genes and bark/vegetative storage proteins suggested altered metabolism in root tips of RNAi plants. Stronger drought-stimulation of stress-inducible genes encoding late-embryogenesis-abundant proteins in transgenic roots was consistent with increased vulnerability to soil drying. Transcript evidence suggested an RNAi effect on intercellular water trafficking by aquaporins in stem xylem during soil drying and recovery. Co-expression network analysis predicted altered integration of abscisic acid sensing/signaling with ethylene and jasmonate sensing/signaling in RNAi compared to WT roots. The overall conclusion is that steepened shoot-root sugar gradient in RNAi plants increased sensitivity of root tips to decreasing soil water availability.
Similar content being viewed by others
Introduction
The most thoroughly documented physiological process mediated by sucrose transporters (SUTs) is the delivery of sucrose to the phloem for its long-distance transport from source leaves to non-photosynthetic sink tissues1,2,3,4,5. SUTs also mediate sucrose uptake in seeds and roots, and sucrose retrieval along the phloem transport path6,7. These intercellular trafficking functions are generally considered to be carried out by plasma-membrane-localized SUTs1, but there is growing evidence that tonoplast-localized SUTs also contribute2,4. Plasma membrane SUTs generally occur as multiple isoforms with unique or overlapping expression patterns, and their expression in source and sink organs can be linked with sucrose supply and demand via feedback mechanisms6,8. Tonoplast SUTs occur as a single isoform in most taxa, with expression patterns that vary from species to species9. Tonoplast SUT proteins mediate proton-dependent sucrose efflux into the cytosol from the vacuole, and are therefore positioned to participate in the intracellular trafficking of sucrose4,10,11.
Interestingly, promoter activity of the Arabidopsis thaliana tonoplast AtSUT4 in root tissues is sensitive to osmolyte treatments, and plant-wide changes in sugar composition during the response to salt stress are perturbed in AtSUT4 mutants12,13. Shoot expression of AtSUT4 is comparatively weak, and shoot growth is not altered in an AtSUT4 null mutant12. The Populus tremula x alba tonoplast PtaSUT4, unlike that of Arabidopsis, is well expressed throughout the shoot, and its down-regulation by RNAi leads to constitutively elevated shoot sucrose levels, and to small changes in shoot biomass allocations and wood water content4,14. In RNAi leaves, the sucrose increase was accompanied by reduced accrual of the abundant glucoside, salicortin, consistent with a downstream metabolic effect of altered sucrose compartmentalization4. When grown under conditions of chronic mild drought, height growth and leaf area were reduced in SUT4-RNAi compared to WT poplars14. This was not attributable to a difference in carbon fixation because SUT4-RNAi photosynthesis rates increased to equal those of WT plants during water-limited growth14. Increases of the drought-metabolite raffinose that normally occur in response to reduced soil moisture content were not observed in SUT4-RNAi poplars and this was attributed to altered sucrose compartmentalization14.
In PtaSUT4-RNAi poplar, sucrose concentrations were significantly elevated in bark and xylem as well as the source leaves4. That finding together with those of Frost et al.14 and of Schneider et al.12 and Gong et al.13 from AtSUT4 mutants raises the possibility that altered tonoplast sucrose trafficking can impact osmotic and metabolic status along the leaf-to-root continuum, with possible consequences to biomass accrual, depending on species growth habit and soil water availability. A plant-wide mRNA-Seq analysis coupled with sucrose and hexose profiling was therefore carried out to obtain a transcriptomic basis for how reduced tonoplast trafficking of sucrose may condition poplar sensitivity to reduced soil water content.
Results
Transgenic line and tissue selection for transcriptomic analysis
Multiple PtaSUT4-RNAi transgenic lines of Populus tremula x alba (clone 717-1B4) that exhibit altered shoot growth and drought response relative to WT have already been described4,14. To facilitate genotype selection for a comprehensive RNA-Seq characterization, three of those RNAi lines were re-sequenced for T-DNA insertion site mapping (Supplementary Table S1). For line G, T-DNA insertion site was mapped to the intergenic region downstream of Potri.004G190400 based on P. trichocarpa genome v3, suggesting that no functional genes were interrupted. In line F, T-DNA was mapped to the second intron of a nuclear cap-binding protein Potri.009G049200, whereas in line H, insufficient reads were obtained to determine the insertion site (Supplementary Table S1). Therefore, transgenic line G was chosen, along with wild type (WT), for the comprehensive transcriptomic characterization. Four organs (source leaf, developing xylem, bark and root tip) of WT and PtaSUT4-RNAi plants from three phases of a soil drying-rewetting treatment, well-watered (WW), drought (DR), and recovery (RC), were subjected to RNA-Seq. Plants ~1.5 m in height were watered daily until drought treatment was initiated by withholding water. As plants began to exhibit turgor loss in expanding leaves, mature source leaves, bark with phloem, xylem shavings, and root tips were harvested (DR samples), along with WW samples from unstressed plants. Unharvested DR plants were re-watered, regained full turgor, and were maintained in a well-watered state until harvest 48 hr later (RC samples). The acute DR and RC treatments were applied in order to obtain metabolic (sugar) and transcriptional responses that could be temporally correlated for a constructive biological inference utilizing bioinformatics and network approaches.
Shoot-root sucrose partitioning differed between WT and RNAi plants
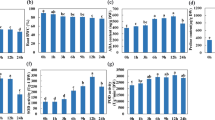
As in our previous reports, transcript levels of the single-copy tonoplast PtaSUT4 were reduced by ~75% throughout the plant while transcript levels of other SUT genes were not reduced in WW plants4 (Supplementary Fig. S1). DR treatment resulted in a trend of PtaSUT4 increases, followed by a decrease to pre-stress (WW) levels at RC in both genotypes, but with greatest significance in RNAi plants (Fig. 1). Transcript levels of the predominant plasma membrane SUT, PtaSUT3, were high in the sink organs examined (Supplementary Fig. S1), and expression changes were regarded as indicative of changes in sink demand for sucrose6,8. PtaSUT3 exhibited a strong DR-promoted increase in stem (bark and xylem) of WT, and in roots of RNAi plants (Fig. 1A). To further assess whether sucrose partitioning within or between shoot and root was sensitive to the RNAi, sucrose and its constituent hexoses were quantified (Fig. 2A). Prior to DR, sucrose and total soluble sugars (sum of sucrose and hexose) were sharply elevated in all shoot organs of RNAi plants, and those high levels were sustained throughout the treatment cycle (Fig. 2A). Sucrose and hexose levels of WT source leaves changed little at DR, but increased considerably at RC. Hexose levels trended downward in bark and xylem of both genotypes at DR, but recovered upon RC. The DR hexose decrease was significant in transgenic xylem, and resulted in a significant decrease of total sugars. While total sugar levels clearly oscillated with DR and RC in transgenic xylem, total sugar levels remained more constant in WT xylem throughout the treatment cycle (Fig. 2A). Due to the large promotive effects of SUT4-RNAi on sugar concentrations in xylem and bark, starch was also evaluated in those tissues. Xylem starch levels did not differ between genotypes or change with treatment (Fig. 2B). By contrast, starch was generally more abundant in transgenic bark relative to WT throughout the treatment cycle (Fig. 2B). Sucrose levels trended slightly lower in WT than transgenic roots before stress (P = 0.11), and exhibited a tendency to increase in WT but not RNAi plants under DR. In a follow-up experiment using a repeated measures root sampling approach, sucrose levels decreased significantly in RNAi, but not in WT root tips upon DR (Fig. 2C). Data from both experiments were consistent with reduced sucrose supply to, unloading, or retention within, transgenic compared to WT root tips during soil drying.

Heatmap illustration of DR and RC effects on transcript abundance of (A) PtaSUT4 and PtaSUT3, and (B) sucrose cleaving enzymes in various organs of wild-type and RNAi transgenic plants. Values represent log2-transformed FPKM ratios (DR/WW or RC/WW). Significant treatment effects on transcript abundance (FPKM) in (A) are denoted by bold-underline (Q ≤ 0.05) or bold-italics (P ≤ 0.05). In (B), FPKM values of individual cell wall (CIN), vacuolar (VIN), and cytosolic (NIN) invertase and sucrose synthase (SUS) gene models were summed for each biological replicate before the response ratios and statistical significance were determined. Significant treatment effects on FPKM sums are denoted by bold-underline based on the two-sample t test (n = 3 biological replicates).

(A) Sucrose (tan) and hexose (green, sum of Fru and Glc) contents in various organs. (B) Stem starch levels in xylem and bark. Darker and lighter shades are for WT and RNAi plants, respectively. Bars are means ± SD of n = 3 biological replicates. Significance testing was conducted using the two-sample t-test (*, **, *** represent P ≤ 0.05, 0.01, 0.001, respectively). Transgenic effects are indicated by asterisks: asterisks inside the color bars are for sucrose and hexoses, above the bars are for total sugars (sum of sucrose and hexoses). Treatment effects within each genotype are indicated by dashed lines with P values in the xylem panel. (C) Root sucrose changes in response to DR in an independent experiment. Data are represented by means ± SE (n = 5 WT or 6 RNAi plants). Significant treatment effects were determined using repeated measures 2-way ANOVA.
A candidate gene approach was used to mine the RNA-Seq data for transcript abundance of genes (other than SUTs) with direct relevance to sugar homeostasis. No transgenic effects on the expression of sucrose cleaving enzymes, sucrose synthases and invertases, were observed across all tissues under WW (Supplementary Fig. S2). During DR, vacuolar invertase transcript abundance increased in all organs but xylem (Fig. 1B), and the increases were generally larger in WT than RNAi plants. To summarize, the RNAi effects on sugar homeostasis and its drought response dynamics were strongest in xylem (Fig. 2A) where high transcript levels of PtaSUT3 (Supplementary Fig. S1) and sucrose metabolism genes (Supplementary Fig. S2) were commensurate with heavy sucrose trafficking.
SUT4-RNAi affected drought response trajectories of aquaporin genes
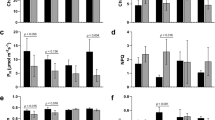
In light of differential SUT4-RNAi effects on organ water content, e.g., wood versus bark14, and potential effects of that on osmotic gradients, we examined transcript abundances in the aquaporin (AQP) gene family. AQPs, both plasma membrane (PIPs) and tonoplast intrinsic proteins (TIPs), permit the passive and selective transport of water across membranes, thereby facilitating water uptake and redistribution in response to osmotic and turgor changes15,16,17. AQPs are expressed plant-wide in Populus and their expression is sensitive to osmotic or hydrostatic pressure gradients, water availability, embolisms, and transpiration demand18,19,20,21. No transgenic effects on total AQP transcript abundance (sum of all AQP transcripts) were observed in WW plants (Fig. 3). However, trends consistent with an overall negative SUT4-RNAi effect on AQP expression in DR leaves and roots, and a promotive effect in xylem were observed (Fig. 3A). During the RC phase, AQP transcript levels trended lower in transgenics, especially in xylem and roots, organs where AQP expression is normally strongest (Fig. 3A).

(A) Illustrative depiction of total AQP expression trends. Data are means ± SD of n = 3 biological replicates, each representing the summed FPKM values of all individual PIP and TIP gene models. (B,C) Heatmap depiction of RNAi and DR effects on xylem (B) and root (C) transcript abundance. Values are log2-transformed response ratios of PIP and TIP transcript abundance. Genes are arranged by their transcript abundance (average FPKM) in each organ. Gene model names of P. trichocarpa genome v3 are abbreviated (i.e., 09G136600 for Potri.009G136600), followed by PIP (PIP1/2) or TIP (TIP1/2/4) subfamily designation. Significant treatment or transgenic effects are denoted by bold-underlined (Q ≤ 0.05) or bold-italics (P ≤ 0.05).
Examining xylem and root expression more closely, a greater number of AQP genes, primarily PIPs, exhibited significant increases in SUT4-RNAi xylem, while a greater number, primarily TIPs, exhibited significant decreases in WT xylem (DR/WW in Fig. 3B). This resulted in the tendency toward higher transcript abundance of most of the well-expressed PIPs and TIPs in transgenic than WT xylem at DR (RNAi/WT in Fig. 3B). Root transcript levels of several well-expressed PIPs and TIPs were down-regulated by DR both in WT and RNAi plants (Fig. 3C). In the transgenic plants then, opposing xylem vs. root AQP transcript responses to DR were evident (Fig. 3). Following DR, there was a consistent trend toward higher AQP transcript levels in WT than RNAi roots (Fig. 3C). A correlation matrix of PIP and TIP transcript levels between organs was constructed in order to compare systemic coordination of AQP expression in WT and transgenic plants (Supplementary Fig. S3). This analysis was carried out in recognition of the potential for altered root-shoot sugar concentration gradients (Fig. 2) to osmotically affect water trafficking. Interestingly, strong inter-organ correlations of AQP transcript level were frequently observed. The analysis revealed stronger plant-wide coordination of PIP2.7, PIP1.5 and PIP1.2 than of other PIPs in both WT and transgenics. The overall AQP correlation was strongest between xylem and bark. However, correlations between leaves and other organs were frequently less robust in transgenics, especially for PIP2.5 and PIP2.6. Correlations that included roots were generally very poor in the transgenic plants (Supplementary Fig. S3).
RNA-Seq revealed latent stresses in root tips of SUT4-RNAi plants
In light of the evidence for RNAi effects on water as well as sucrose trafficking within and between organs, the potential for there to be effects on gene families known to be associated with perceived shifts in plant or organ osmotic or turgor status was considered. Approximately 100–300 genes, depending on the organ, exhibited differential expression (DE, Q ≤ 0.05 and fold-change [FC] ≥ 1.5) between pre-stress WT and RNAi plants (Fig. 4, Supplementary Dataset S1). Expression evidence for abscisic acid (ABA) involvement was anticipated, and transcript levels of NCED, which encodes nine-cis-epoxycarotenoid dioxygenase for the rate-limiting step in ABA biosynthesis22, were perturbed in RNAi plants (Fig. 4B). Under WW conditions, NCED transcript levels were more abundant in bark, xylem and roots of RNAi than WT plants. In accordance with the known drought responsiveness of NCED23, the two expressed poplar homologs were highly up-regulated by DR in all four organs of both WT and RNAi plants (Fig. 4B). DR treatment affected the transcript abundance of several thousand genes in each organ of WT and RNAi plants (Fig. 4C), with the greatest response occurring in transgenic roots (Fig. 4C). Many of the genes that encode late embryogenesis abundant (LEA) proteins are known to be induced by desiccation and ABA24,25, and a large subset were among the most strongly up-regulated in DR roots, with the response being strongest in the transgenics (Supplementary Fig. S4A). Just as striking was the magnitude of the DR induction of small heat shock proteins (HSP20s) in transgenic compared to WT roots (Supplementary Fig. S4B). Transcript levels of several nucleoside phosphorylase-like vegetative/bark storage proteins (VSPs/BSPs) were also constitutively elevated in transgenic roots, and/or exhibited stronger DR-stimulated increases in transgenic than WT roots (Supplementary Fig. S4C). Together, the results were consistent with a constitutive RNAi effect on the ABA biosynthetic pathway followed by intensified DR responses in the transgenics.

(A) Venn diagram of DE genes in response to SUT4-RNAi in various organs and treatment regimes. (B) Heatmap depiction of SUT4-RNAi and DR effects on NCED (nine-cis-epoxycarotenoid dioxygenase) transcript abundance. Values are log2-transformed ratio responses. Significant effects are denoted by bold-underlines. (C) Histogram depiction of gene numbers significantly up or down-regulated by DR treatment. Significant changes in transcript abundance were based on Q ≤ 0.05 and fold-change ≥1.5 with n = 3 biological replicates.
Self-organizing map (SOM) clustering of DE genes based on their response patterns to the three treatment phases enabled a more comprehensive assessment of RNAi-sensitive pathways than the selective approach above (Fig. 5). The DR and RC response patterns were similar in WT and RNAi plants (small genotype differentials) in six of the clusters (1, 2, 4, 8, 9 and 10). In the other six clusters (3, 5, 6, 7, 11 and 12), the genotype differentials were larger. The differential response clusters were dominated by root DE genes (~60%). About 47% of all root DE genes, compared to 23–32% of DE genes from the other organs, resided in those clusters. Gene ontology (GO) functional enrichment analysis showed that ABA, salicylic acid, jasmonic acid (JA) and ethylene biosynthesis and signaling GO terms were especially well-represented among bark, xylem and root transcripts in cluster 6 (Fig. 5B), with DR-induced increases in RNAi plants (Fig. 5A). The pattern was different for leaves, where salicylic acid and JA functions were comparatively well-represented in clusters 3 and 7, either suppressed by DR in RNAi, or stimulated by DR in WT. GO categories for cell division, monosaccharide and phenylpropanoid metabolism, response to sucrose stimulus and root development were enriched among DR-suppressed genes in transgenic roots (clusters 3 and 11). Overall, the results were consistent with comparatively strong drought stress sensing as well as reduced growth in transgenic roots, but not in leaves.

(A) SOM clusters of DE genes showing their response patterns to the three treatment phases in WT and RNAi plants. The total number of DE data points in each cluster is indicated in parenthesis on top, and tissue representation (L, leaf; B, bark; X, xylem and R, root) listed at the bottom (see Methods). Solid and dashed lines represent the group mean of WT and RNAi plants, respectively. (B) GO enrichment analysis of six SOM clusters that showed different treatment response trajectories between genotypes. Hierarchical clustering of negative log10-transformed P values for GO enrichment was visualized in heatmaps according to the color scale. DE genes within each cluster are sorted by their tissue of origin in columns and color-coded, while GO terms are shown in rows.
Analysis of major transcription factor (TF) families present in the various SOM clusters revealed their differential sensitivity to RNAi effects (Fig. 6). Overall, there was strong TF representation in cluster 10 (Fig. 6A), which contained DR-responsive genes that were relatively insensitive to RNAi effects (Fig. 5A). TFs were also well-represented in clusters 6 and 12 that exhibited RNAi-enhanced DR responses. The enrichment of ethylene response factors in these two clusters indicated a magnified sensitivity of ethylene-modulated components of the DR response in bark, xylem and especially roots of RNAi plants (Fig. 6A). Promoter cis element analysis of the SOM gene clusters was also carried out and found to corroborate the above TF observations (Fig. 6B). In general, the strongest representation of stress-related cis-elements was in cluster 10 (increased expression at DR in all organs of both genotypes). However, the classic drought-responsive element as well as W-box and ethylene-responsive element were better represented among cluster 6 than cluster 10 gene promoters, consistent with greater drought sensitivity in RNAi roots. Interestingly, several sugar-responsive elements were well-represented in cluster 10 but not in cluster 6, and one of them (SURE2) was also enriched in clusters 3 and 11 that were down-regulated by DR in transgenic roots. This suggested reduced, or otherwise altered, sucrose-involvement during the DR response of SUT4-RNAi roots.

(A) Heatmap distribution of TF families in the SOM clusters by organ. The number of TFs from each family is indicated for each SOM cluster-organ combination (only those with at least 10 members in any combination are shown). The number of TFs annotated for the Populus genome v3 is indicated in parentheses for each family. (B) Cis-element enrichment in SOM clusters by organ. Non-redundant cis elements were mapped onto promoter sequences and significant enrichment in the various SOM-organ gene clusters tested by chi-square statistics with false discovery correction (see Methods). The negative log10-transformed P values for cis-element enrichment were subjected to hierarchical clustering and visualized in heatmaps according to the color scale. Only SOM-organ clusters with significant cis-element enrichments were included in the heatmap.
Gene network connectivity was altered by SUT4 suppression
Based on the high number of DE genes and on SOM clustering and TF analysis, the RNAi effect was most severe in the root tips. Weighted gene correlation networks were therefore constructed for the two genotypes to assess root transcriptome reprogramming. Changes in gene connectivity (RNAi-WT) between the two networks were plotted against changes in expression (RNAi/WT) at DR, and genes were color-coded by co-expression module as determined independently for the two networks (Fig. 7, Supplementary Dataset S2). In the transgenic network, the turquoise module was the largest, with many of its members exhibiting higher connectivity as well as higher expression relative to WT at DR (Fig. 7A, top right quadrant). The most strongly enriched GO categories in the turquoise module pertained to responses to water deprivation, ABA, ethylene and JA stimuli, and oxygen-containing compound (Fig. 7C). In the WT network, the response to ABA stimulus category was enriched in the comparatively small brown module, along with fatty acid metabolic process and response to water deprivation (Fig. 7C). Fatty acid metabolic process was also co-enriched with phenylpropanoid biosynthetic processes in the WT orange module. The data are consistent with the interpretation that the ABA component of the DR response occurred against a backdrop of greater stress and stronger participation of JA and ethylene components in RNAi roots.

(A,B) Scatterplots of differential connectivity (y-axis) between WT and SUT4-RNAi roots, based on weighted gene coexpression network analysis, versus differential expression (x-axis) between the two genotypes at DR. Genes are colored by their co-expression module assignment in the SUT4-RNAi (A) or WT (B) networks. (C) Significant GO enrichments in the five largest modules of the RNAi and WT networks. Negative log10-transformed P values for GO enrichment were plotted in histograms and color-coded by module.
Besides the above-mentioned differences in NCED expression (Fig. 4B), the most striking ABA-related finding was the attenuated DR response of an ABA-stress-ripening homolog in RNAi than WT roots (Supplementary Fig. S5A). This TF is thought to partition between the nucleus and the cytosol for the orchestration of ABA-mediated stress responses in a sugar-dependent manner26,27. While DR response of other known effectors of ABA signaling appeared unchanged in RNAi roots (Supplementary Fig. S5A), expression of ethylene and JA response-related TFs was much stronger in the RNAi roots (Supplementary Fig. S5B,C).
Discussion
One underlying driver of the comprehensive RNA-Seq experiment was prior observation that SUT4-RNAi poplars exhibited greater reductions of leaf area and height growth during chronic mild drought than WT poplars14. Roots were not analyzed in the previous study, so it was unclear whether SUT4-RNAi effects on drought growth originated in the shoots, the roots, or both. The present work aimed to dissect the SUT4-RNAi effects transcriptionally, with supporting data on sugar changes. In addition, the present work utilized an acute drought treatment intended to obtain a change in plant water status without irreversibly damaging the plant, and in a time frame that precluded morphological adaptations that would complicate analysis of the response to changes in water status.
Reduced shoot-root trafficking of sucrose was sensed in SUT4-RNAi plants during DR
Transcript levels of plasma membrane SUTs can increase both in source and sink organs when demand for sucrose is high, or decrease when it is low6,8,28. We therefore examined plasma membrane PtaSUT3 expression in order to determine whether SUT4-RNAi effects on sucrose concentrations perturbed sink-source physiology in a way that affected other SUT gene expression. However, despite large organ-specific increases in sucrose levels in WW RNAi plants, plasma membrane SUT expression was not affected (Supplementary Fig. S1).). One plausible explanation for this observation is that plasma membrane SUTs are not regarded to mediate sucrose export from leaves of Populus29,30. The absence of a negative effect in xylem, a strong sucrose sink, was more surprising. However, because xylem is not a sucrose storage organ, plasma membrane SUT expression in xylem may not be under the same feedback constraints as in sucrose storing tissues such as seed embryos or certain root crops6,8,28.
As plants experienced DR, plasma membrane PtaSUT3 expression changed (Fig. 1). The DR-promoted increases were smaller in RNAi than WT stem (bark and xylem), and larger in RNAi than WT roots (Fig. 1). To adopt the feedback regulation model, the ~2-fold SUT3 expression increase in DR-stressed WT stem and root were consistent with increased sucrose demand in both organs during the onset of a water deficit. The comparatively strong DR induction in RNAi roots (~4-fold) may suggest a combination of greater root demand for sucrose and/or impaired delivery of sucrose in RNAi plants. Xylem vacuolar invertase transcripts decreased sharply in both genotypes consistent with DR-promoted changes in sucrose fluxes during transit through the stem toward roots (Fig. 2). Although decreased vacuolar invertase expression would be expected to increase xylem sucrose/hexose ratio, and perhaps vacuolar sucrose storage, sucrose only increased in WT xylem at DR (Fig. 2). A vacuolar sucrose glut apparently preceded DR in RNAi xylem despite reduced photosynthesis in WW RNAi plants14. Thus, constitutively high vacuolar sequestration of sucrose in the xylem may have been one contributing factor to reduced root provisioning and increased root demand and SUT3 expression in RNAi plants with increasingly restricted water uptake during soil drying.
AQP expression was consistent with a negative RNAi effect on water trafficking in root tips during DR and RC
The expression of various AQP genes for efficient water distribution throughout the plant is sensitive to ABA, and to osmotic or turgor changes15,21,31. AQP expression changes in roots in response to transpiration demand, in leaves for water redistribution associated with specific drought tolerance strategies, and in xylem embolism repair have all been reported in Populus18,19,21,32. One interesting observation from the present work was that xylem AQP gene expression increased in the RNAi plants at DR. The normal (WT) DR response for xylem AQP expression appears to be a transcript level decrease (Fig. 3). We might speculate that DR-increased AQP expression caused or reflected an abnormal redistribution of water in RNAi xylem due to inefficient mobilization of vacuolar sucrose. The general effect on cell turgor or intercellular space may be analogous to volume control of intercellular spaces through cell expansion mediated by concerted changes in the expression of PIPs and TIPs in legume nodules to regulate oxygen diffusion33.
AQP expression was also altered in roots of the RNAi plants, collectively trending downward in RNAi relative to WT roots at DR and remaining depressed at RC (Fig. 3A). For the plant as a whole, the AQP expression and sugars data from xylem and roots are consistent with the possibility that sugar sequestration in RNAi stems compromised root provisioning and AQP regulation of water trafficking. Compared to WT, correlations of AQP transcript levels between roots and other organs in SUT4-RNAi plants across the WW-DR-RC cycle were reduced. The reduction is consistent with compromised systemic control of water distribution due to localized sugar abundance differentials. Transpiration presumably recovered in WT and RNAi plants during RC as evidenced by restored turgidity of upper sink-source transitional leaves initially used as visual markers of DR onset, so it is not clear why root AQP expression remained low in RNAi plants once watering was resumed. Root expression of many AQPs normally correlates with transpirational demand in Populus21. Whether embolism repair, which involves both sucrose trafficking and AQP activity in Populus18,19 was compromised in the RNAi plants was not investigated here. Our results suggest that sucrose partitioning and spatial accumulation patterns affected AQP expression autonomously in multiple organs, with prolonged consequences to the roots during post-drought recovery.
Interaction of sucrose with hormone-regulated stress responses in poplar root tips
A possibility suggested by the comparatively strong HSP and LEA transcriptional responses of RNAi roots to DR (Supplementary Fig. S4) is that RNAi roots were somehow sensitized or more vulnerable to soil drying than WT roots were. Related to this, the enhanced NCED expression in RNAi roots of WW plants (Fig. 4) lent support to the idea that ABA homeostasis was altered prior to DR stress due to reduced tonoplast trafficking of sucrose and/or reduced root: shoot sucrose concentration ratios. The strong LEA response in RNAi roots is consistent with the notion of a stronger or otherwise perturbed ABA response during changes in soil moisture availability. All 27 differentially expressed LEA genes identified in our study (Supplementary Fig. S4A) contain multiple ABA response elements in their promoters. At the same time, sugar-response cis elements were under-represented compared to drought-response elements in promoters of genes that were up-regulated by DR specifically in RNAi roots (Figs 5 and 6, cluster 6). ARS1 which is thought to facilitate cross-talk between ABA and glucose26,27 exhibited a severely compromised DR response in RNAi roots. We suggest that reduced tonoplast trafficking of sucrose stimulated NCED expression regardless of stress level, while restricting long-distance transport of sucrose to roots. As soil dried, intercellular water in root tips would presumably be increasingly drawn toward tissues of greater osmolyte concentration either in shoots or in more vascularized roots, with shoot-to-root tip osmolyte gradients potentially steeper and more damaging in RNAi than WT plants.
Interestingly, transcript levels of several VSP/BSP genes were elevated in RNAi compared to WT root tips prior to, and then in an enhanced way, during DR stress (Supplementary Fig. S4). In Populus, BSP/VSP proteins accrue as storage reserves in response to stress and to seasonal changes in temperature and light34. However, BSP/VSP is also an important target of the wound signal JA in Populus and other species35. In addition, mechanical strain caused by root bending has been reported to elevate the abundance of BSPs and HSPs in poplar roots36. Other causes of mechanical strain include changes in turgor and osmotic status37. The pre-stress abundance of BSP/VSP transcripts, and their sustained high levels in RNAi roots during DR and RC may therefore be consistent with more active re-allocation of nitrogen and carbon reserves in response to multiple signaling pathways including those associated with latent mechanical stress.
During DR, TFs associated with ethylene and JA signaling increased more in root tips of RNAi than WT plants (Supplementary Fig. S5B,C). The interplay of sugars, ABA, ethylene and JA signaling during drought is extremely complex, and ethylene and JA frequently are antagonistic with ABA38,39,40. The relationship between abiotic stress, phytohormones and SUTs has also recently been explored using SUT mutants and exogenous supplements in Arabidopsis13. In citrus roots, extreme soil drying leads to a JA response which subsequently gives way to an ABA-mediated response41. In the case of the SUT4-RNAi roots, therefore, up-regulation of JAZ transcripts associated with feedback mitigation of the JA response42 may reflect the residual of a stronger DR-induced JA response than the one generated in WT roots. The apparently heightened response may also reflect altered ABA homeostasis prior to DR treatment in RNAi root tips. Together, the cis-element analysis and the root gene network data suggest that sucrose played a larger role in the WT drought response, perhaps supporting a biosynthetic response commensurate with adaptation. In contrast, there was a stronger integration of the drought response in RNAi plants with generation of oxygen-containing compounds including reactive oxygen species, and with ethylene and JA mediation
Conclusions
The data as a whole are consistent with the idea that a latent strain existed in SUT4-RNAi root tips due to altered homeostatic control of sugar osmolyte distribution. Based on altered leaf-to-root transcript level gradients of NCED (Fig. 4), ABA homeostasis may have been perturbed with possible effects on the magnitude of ethylene and JA responses during soil drying (Supplementary Fig. S5). In previous work, height growth of two independent SUT4-RNAi lines was reduced under chronic mild drought conditions more so than the WT plants14. Leaf area was also reduced in both SUT4-RNAi lines compared to WT. In light of those findings, the transcriptome results from the present study lend further support to the idea that tonoplast trafficking of sucrose is consequential for the coupling of growth with differences in soil water availability. A normal adaptation to chronic water limitation in the WT poplars appears to be a substantial reduction in leaf SUT4 expression14. In addition to the above-posited idea of latent root strain, a second point may be that SUT4 expression is necessarily dynamic in order to couple carbon use with changes in plant water status, and that constitutively reduced expression may have mitigated SUT4 function by limiting the magnitude of such expression changes.
Methods
Plant growth and experimental treatments
Single-node cuttings were grown in 4-gal Treepots™, containing Fafard 3B potting mix supplemented with Osmocote (15-9-12 NPK 4-month release), to approximately 1.5 m in a greenhouse (August to October) under evaporative cooling and supplemental lighting (14 hr photoperiod). The acute drought treatment was conducted as described14. Briefly, water was withheld about three days until visible-yet-reversible turgor-loss of sink-source transitional leaves, leaf plastochron index43 3–5. Source leaves exhibited normal turgor at DR. WW controls and DR plants were harvested at the same time, approximately 60 hrs after the commencement of soil drying. Unharvested, DR-treated plants were then watered and harvested 48 hr later (RC). Sampling occurred under sunny conditions (10 AM to 2 PM). Source leaves (leaf plastochron index 15), phloem with bark, xylem shavings (internodes 20–30), and distal 2 cm of root tips were snap-frozen in liquid nitrogen and stored at −80 °C.
Analysis of sucrose, glucose and fructose
Freeze-dried powders (10 mg) were extracted twice with 700 μl methanol: water: chloroform (40:27:33, v/v) containing internal standards (adonitol and 2-methoxybenzoic acid). The aqueous phase was derivatized with N-methyl-N-(trimethylsilyl) trifluoroacetamide and analyzed on an Agilent 7890A GC coupled to an Agilent 5975 MS as detailed previously14. Spectra were collected in the scanning ion mode (m/z 50–500) in ChemStation (Agilent Technologies) and deconvoluted using AnalyzerPro (SpectralWorks). Retention times and mass spectra of the abundant sugars sucrose, glucose and fructose were confirmed using authentic standards. Run-to-run uniformity was monitored using a standard mixture of sucrose, glucose and fructose. Relative peak areas of each metabolite (normalized against internal standard adonitol) were used for all statistical analyses by SigmaStat 3.1.
High-throughput sequencing and data analysis
Genomic DNA was extracted from young leaves of three SUT4-RNAi transgenic lines using the E.Z.N.A. Plant DNA kit (Omega Bio-tek), quality-checked by agarose electrophoresis and quantified using a Qubit fluorometer (Life Technologies). Genomic libraries were prepared as described44 and sequenced on an Illumina NextSeq-500 at the Georgia Genomics Facility. Approximately 12–32 million (M) paired-end 75-bp reads were obtained per sample. To identify T-DNA insertion sites, individual reads were mapped to the vector as well as the Populus tremula x alba custom genome v1.145 using BLAT. Custom Perl scripts were used to filter for paired reads flanking an insertion site, e.g., one read of the pair mapped to the T-DNA and the other to the genome.
RNA was isolated using the CTAB method46 and quality-checked using a BioAnalyzer (Agilent). RNA-Seq library preparation and Illumina HiSeq-2000 sequencing were performed at BGI America. Approximately 9–17M paired-end 50-bp reads were obtained for three biological replicates (two genotypes, three treatments and four tissues). The RNA-Seq data have been submitted to NCBI Sequence Read Archive (accession number SRP041959). Filtered read pairs with adapter and rRNA sequences removed were mapped onto the Populus genome v3.0 using Tophat2 v2.0.6, and processed by Cufflinks (v2.0.2) for transcript abundance estimation and pairwise DE analysis47. The significance threshold was set at fold-change ≥1.5 and Q value ≤ 0.05. Genes with high coefficient of variation (>1.3) among replicates were excluded from further analysis. For self-organizing-map clustering, DE genes with FPKM ≥3 in at least three individual samples per tissue (two genotypes x three treatment phases x three replicates) were extracted for each tissue and then combined into one dataset for the analysis using MEV v4.948. Functional annotation of signaling and metabolic pathways was obtained from KEGG (http://www.genome.jp/kegg/pathway.html) and PoplarCyc v3.0 (http://plantcyc.org), respectively, and transcription factors from the Plant Transcription Factor Database49. Gene ontology (GO) enrichment analysis was performed using topGO50 with Fisher’s exact test, and the negative log10 transformed P values were visualized using heatmaps as described51.
Cis-element enrichment analysis
Cis-element analysis was performed as described52. Briefly, 1,032 plant cis-elements were collected from PlantCare53, Transfac54, JASPAR55 and PLACE56. Redundant cis-elements identified by MotifComparison57 with a distance score smaller than 0.3 were removed, leaving 483 cis-elements for analysis. MotifScanner57 was used to map the cis-elements onto the 2-kb upstream promoter sequences extracted from the JGI Populus genome v3. Distribution of cis-elements was summarized for all genes as a baseline for Chi-square test of cis-element enrichment in the various gene clusters. The resulting P values were corrected for false discovery rate58, and the negative log10 transformed values were visualized using heatmaps.
Co-expression gene network construction and visualization for root tips
The WGCNA (v1.29) package in R59 was used to construct a co-expression network for all root DE genes, and then for two sub-networks (WT and SUT4-RNAi) using an established pipeline51. Briefly, 8,655 DE genes across all root samples were obtained with ANOVA P ≤ 0.05 and fold-change ≥2. Pearson correlation coefficients were calculated for all gene pairs and an adjacency matrix was constructed using a power of 14. The degree distributions of the networks all followed the power law, indicating that they were scale-free. A hierarchical tree based on a topological overlap matrix was then constructed to identify discrete co-expression modules using the dynamic tree cut method59, with the minimum module size set to 140 genes. GO enrichment analysis of co-expression modules was conducted as described above.
Additional Information
How to cite this article: Xue, L.-J. et al. Drought response transcriptomes are altered in poplar with reduced tonoplast sucrose transporter expression. Sci. Rep. 6, 33655; doi: 10.1038/srep33655 (2016).
References
Ayre, B. G. Membrane-transport systems for sucrose in relation to whole-plant carbon partitioning. Molecular Plant 4, 377–394, doi: 10.1093/mp/ssr014 (2011).
Eom, J. S. et al. Impaired function of the tonoplast-localized sucrose transporter in rice, OsSUT2, limits the transport of vacuolar reserve sucrose and affects plant growth. Plant Physiol. 157, 109–119, doi: 10.1104/pp.111.176982 (2011).
Lalonde, S. et al. The dual function of sugar carriers: Transport and sugar sensing. Plant Cell 11, 707–726 (1999).
Payyavula, R. S., Tay, K. H. C., Tsai, C.-J. & Harding, S. A. The sucrose transporter family in Populus: The importance of a tonoplast PtaSUT4 to biomass and carbon partitioning. Plant J. 65, 757–770, doi: 10.1111/j.1365-313X.2010.04463.x (2011).
Riesmeier, J. W., Willmitzer, L. & Frommer, W. B. Evidence for an essential role of the sucrose transporter in phloem loading and assimilate portioning. EMBO J. 13, 1–7 (1994).
Zhou, Y. C. et al. Intracellular sucrose communicates metabolic demand to sucrose transporters in developing pea cotyledons. J. Exp. Bot. 60, 71–85, doi: 10.1093/jxb/ern254 (2009).
Kühn, C. & Grof, C. P. L. Sucrose transporters of higher plants. Curr. Opin. Plant Biol. 13, 287–297 (2010).
Chiou, T. J. & Bush, D. R. Sucrose is a signal molecule in assimilate partitioning. Proc. Natl. Acad. Sci. USA 95, 13997–13997 (1998).
Peng, D., Xue, L.-J., Gu, X., Leebens-Mack, J. H. & Tsai, C.-J. Bayesian phylogeny of sucrose transporters: Ancient origins, differential expansion and convergent evolution in monocots and dicots. Frontiers in Plant Science 5, 615, doi: 10.3389/fpls.2014.00615 (2014).
Chincinska, I. A. et al. Sucrose transporter StSUT4 from potato affects flowering, tuberization, and shade avoidance response. Plant Physiol. 146, 515–528, doi: 10.1104/pp.107.112334 (2008).
Reinders, A., Sivitz, A. B., Starker, C. G., Gantt, J. S. & Ward, J. M. Functional analysis of LjSUT4, a vacuolar sucrose transporter from Lotus japonicus . Plant Mol. Biol. 68, 289–299, doi: 10.1007/s11103-008-9370-0 (2008).
Schneider, S. et al. Vacuoles release sucrose via tonoplast-localised SUC4-type transporters. Plant Biol. 14, 325–336, doi: 10.1111/j.1438-8677.2011.00506.x (2012).
Gong, X. et al. Arabidopsis AtSUC2 and AtSUC4, encoding sucrose transporters, are required for abiotic stress tolerance in an ABA-dependent pathway. Physiol. Plant. 153, 119–136, doi: 10.1111/ppl.12225 (2015).
Frost, C. J., Nyamdari, B., Tsai, C.-J. & Harding, S. A. The tonoplast-localized sucrose transporter in Populus (PtaSUT4) regulates whole-plant water relations, responses to water stress, and photosynthesis. PLoS ONE 7, e44467 (2012).
Aroca, R., Porcel, R. & Ruiz-Lozano, J. M. Regulation of root water uptake under abiotic stress conditions. J. Exp. Bot. 63, 43–57, doi: 10.1093/jxb/err266 (2012).
Postaire, O. et al. A PIP1 Aquaporin Contributes to Hydrostatic Pressure-Induced Water Transport in Both the Root and Rosette of Arabidopsis. Plant Physiol. 152, 1418–1430, doi: 10.1104/pp.109.145326 (2010).
Wudick, M. M., Luu, D. T. & Maurel, C. A look inside: localization patterns and functions of intracellular plant aquaporins. New Phytol. 184, 289–302, doi: 10.1111/j.1469-8137.2009.02985.x (2009).
Secchi, F. & Zwieniecki, M. A. Down-Regulation of Plasma Intrinsic Protein1 Aquaporin in Poplar Trees Is Detrimental to Recovery from Embolism. Plant Physiol. 164, 1789–1799, doi: 10.1104/pp.114.237511 (2014).
Secchi, F. & Zwieniecki, M. A. Sensing embolism in xylem vessels: the role of sucrose as a trigger for refilling. Plant Cell Environ. 34, 514–524, doi: 10.1111/j.1365-3040.2010.02259.x (2011).
Almeida-Rodriguez, A. M., Hacke, U. G. & Laur, J. Influence of evaporative demand on aquaporin expression and root hydraulics of hybrid poplar. Plant Cell Environ. 34, 1318–1331, doi: 10.1111/j.1365-3040.2011.02331.x (2011).
Laur, J. & Hacke, U. G. Transpirational demand affects aquaporin expression in poplar roots. J. Exp. Bot. 64, 2283–2293, doi: 10.1093/jxb/ert096 (2013).
Schwartz, S. H., Tan, B. C., Gage, D. A., Zeevaart, J. A. D. & McCarty, D. R. Specific oxidative cleavage of carotenoids by VP14 of maize. Science 276, 1872–1874, doi: 10.1126/science.276.5320.1872 (1997).
Iuchi, S. et al. Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis. Plant J. 27, 325–333, doi: 10.1046/j.1365-313x.2001.01096.x (2001).
Kamisugi, Y. & Cuming, A. C. The evolution of the abscisic acid-response in land plants: comparative analysis of group 1 LEA gene expression in moss and cereals. Plant Mol.Biol. 59, 723–737, doi: 10.1007/s11103-005-0909-z (2005).
Vaseva, I. I., Grigorova, B. S., Simova-Stoilova, L. P., Demirevska, K. N. & Feller, U. Abscisic acid and late embryogenesis abundant protein profile changes in winter wheat under progressive drought stress. Plant Biol. 12, 698–707, doi: 10.1111/j.1438-8677.2009.00269.x (2010).
Dominguez, P. G. et al. ASR1 Mediates Glucose-Hormone Cross Talk by Affecting Sugar Trafficking in Tobacco Plants. Plant Physiol. 161, 1486–1500, doi: 10.1104/pp.112.208199 (2013).
Saumonneau, A. et al. Interaction of grape ASR proteins with a DREB transcription factor in the nucleus. FEBS Lett. 582, 3281–3287, doi: 10.1016/j.febslet.2008.09.015 (2008).
Vaughn, M. W., Harrington, G. N. & Bush, D. R. Sucrose-mediated transcriptional regulation of sucrose symporter activity in the phloem. Proc. Natl. Acad. Sci. USA 99, 10876–10880, doi: 10.1073/pnas.172198599 (2002).
Zhang, C. et al. Symplastic phloem loading in poplar. Plant Physiol. 166, 306–313, doi: 10.1104/pp.114.245845 (2014).
Mahboubi, A. et al. Aspen Sucrose Transporter3 Allocates Carbon into Wood Fibers. Plant Physiol. 163, 1729–1740, doi: 10.1104/pp.113.227603 (2013).
Javot, H. & Maurel, C. The role of aquaporins in root water uptake. Ann. Bot. 90, 301–313, doi: 10.1093/aob/mcf199 (2002).
Almeida-Rodriguez, A. M., Cooke, J. E. K., Yeh, F. & Zwiazek, J. J. Functional characterization of drought-responsive aquaporins in Populus balsamifera and Populus simonii x balsamifera clones with different drought resistance strategies. Physiol. Plant. 140, 321–333, doi: 10.1111/j.1399-3054.2010.01405.x (2010).
Fleurat-Lessard, P., Michonneau, P., Maeshima, M., Drevon, J. J. & Serraj, R. The distribution of aquaporin subtypes (PIP1, PIP2 and gamma-TIP) is tissue dependent in soybean (Glycine max) root nodules. Ann. Bot. 96, 457–460, doi: 10.1093/aob/mci195 (2005).
Pettengill, E. A., Pettengill, J. B. & Coleman, G. D. Elucidating the evolutionary history and expression patterns of nucleoside phosphorylase paralogs (vegetative storage proteins) in Populus and the plant kingdom. BMC Plant Biol. 13, 17, doi: 10.1186/1471-2229-13-118 (2013).
Beardmore, T., Wetzel, S. & Kalous, M. Interactions of airborne methyl jasmonate with vegetative storage protein gene and protein accumulation and biomass partitioning in Populus plants. Canadian Journal of Forest Research 30, 1106–1113, doi: 10.1139/cjfr-30-7-1106 (2000).
Trupiano, D. et al. The proteome of Populus nigra woody root: response to bending. Ann. Bot. 110, 415–432, doi: 10.1093/aob/mcs040 (2012).
Pereira, C. S. & Hunenberger, P. H. Effect of trehalose on a phospholipid membrane under mechanical stress. Biophys. J. 95, 3525–3534, doi: 10.1529/biophysj.108.131656 (2008).
Lackman, P. et al. Jasmonate signaling involves the abscisic acid receptor PYL4 to regulate metabolic reprogramming in Arabidopsis and tobacco. Proc. Natl. Acad. Sci. USA 108, 5891–5896, doi: 10.1073/pnas.1103010108 (2011).
LeNoble, M. E., Spollen, W. G. & Sharp, R. E. Maintenance of shoot growth by endogenous ABA: genetic assessment of the involvement of ethylene suppression. J. Exp. Bot. 55, 237–245, doi: 10.1093/jxb/erh031 (2004).
Cheng, M. C., Liao, P. M., Kuo, W. W. & Lin, T. P. The Arabidopsis Ethylene Response Factor1 Regulates Abiotic Stress-Responsive Gene Expression by Binding to Different cis-Acting Elements in Response to Different Stress Signals. Plant Physiol. 162, 1566–1582, doi: 10.1104/pp.113.221911 (2013).
de Ollas, C., Hernando, B., Arbona, V. & Gomez-Cadenas, A. Jasmonic acid transient accumulation is needed for abscisic acid increase in citrus roots under drought stress conditions. Physiol. Plant. 147, 296–306, doi: 10.1111/j.1399-3054.2012.01659.x (2013).
Chini, A. et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 448, 666–U664, doi: 10.1038/nature06006 (2007).
Larson, P. R. & Isebrands, J. G. The plastochron index as applied to developmental studies of cottonwood. Can. J. Forest Res. 1, 1–11 (1971).
Schmitz, R. J. et al. Patterns of population epigenomic diversity. Nature 495, 193–198, doi: 10.1038/nature11968 (2013).
Xue, L.-J., Alabady, M. S., Mohebbi, M. & Tsai, C.-J. Exploiting genome variation to improve next-generation sequencing data analysis and genome editing efficiency in Populus tremula x alba 717-1B4, Tree Genetics & Genomes 11, 82, doi: 10.1007/s11295-015-0907-5 (2015).
Tsai, C. J., Cseke, L. J. & Harding, S. A. In Handbook of Molecular and Cellular Methods In Biology and Medicine (eds Cseke, L. J., Kirakosyan, A., Kaufman, P. B. & Westfall, M. V. ) 167–186 (CRC Press, 2011).
Trapnell, C. et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature biotechnology 31, 46–53, doi: 10.1038/nbt.2450 (2013).
Saeed, A. I. et al. TM4: a free, open-source system for microarray data management and analysis. BioTechniques 34, 374–378 (2003).
Zhang, H. et al. PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic acids research 39, D1114–D1117, doi: 10.1093/nar/gkq1141 (2011).
Alexa, A., Rahnenfuhrer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006).
Xue, L.-J. et al. Constitutively elevated salicylic acid levels alter photosynthesis and oxidative state, but not growth in transgenic Populus . Plant Cell 25, 2714–2730 (2013).
Street, N. R., Jansson, S. & Hvidsten, T. R. A systems biology model of the regulatory network in Populus leaves reveals interacting regulators and conserved regulation. BMC Plant Biol 11, 13, doi: 10.1186/1471-2229-11-13 (2011).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic acids research 30, 325–327 (2002).
Wingender, E., Dietze, P., Karas, H. & Knuppel, R. Transfac: a database on transcription factors and their DNA binding sites. Nucleic acids research 24, 238–241 (1996).
Portales-Casamar, E. et al. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic acids research 38, D105–D110, doi: 10.1093/nar/gkp950 (2010).
Higo, K., Ugawa, Y., Iwamoto, M. & Korenaga, T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic acids research 27, 297–300 (1999).
Aerts, S. et al. Toucan: deciphering the cis-regulatory logic of coregulated genes. Nucleic acids research 31, 1753–1764 (2003).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 57, 289–300 (1995).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics 9, 559, doi: 10.1186/1471-2105-9-559 (2008).
Acknowledgements
The authors thank Kavita Aulakh for assistance with genomic DNA extraction, and Nicholas Rohr and Bob Schmitz for Illumina DNA library preparation. This research was supported by the Office of Biological and Environmental Research within the Department of Energy (grant no. DE-SC0005140) to SAH and CJT.
Author information
Authors and Affiliations
Contributions
S.A.H. and C.-J.T. conceived the study, C.J.F. designed and executed the experiments, L.-J.X. performed bioinformatics analyses, L.-J.X., S.A.H. and C.-J.T. analyzed the data, S.A.H., L.-J.X. and C.-J.T. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xue, LJ., Frost, C., Tsai, CJ. et al. Drought response transcriptomes are altered in poplar with reduced tonoplast sucrose transporter expression. Sci Rep 6, 33655 (2016). https://doi.org/10.1038/srep33655
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33655
This article is cited by
-
Transcriptome analysis of response strategy in Hemerocallis fulva under drought stress
Genes & Genomics (2023)
-
The effect of long-term dehydration and subsequent rehydration on markers of inflammation, oxidative stress and apoptosis in the camel kidney
BMC Veterinary Research (2020)
-
Physiological and transcriptome analysis of He-Ne laser pretreated wheat seedlings in response to drought stress
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
