
Abstract
Paralogous members of the oxidosqualene cyclase (OSC) family encode a diversity of enzymes that are important in triterpenoid biosynthesis. This report describes the isolation of the Gentiana straminea gene GsAS2 that encodes a β-amyrin synthase (βAS) enzyme. Unlike its previously isolated paralog GsAS1, GsAS2 lacks introns. Its predicted protein product was is a 759 residue polypeptide that shares high homology with other known β-amyrin synthases (βASs). Heterologously expressed GsAS2 generates more β-amyrin in yeast than does GsAS1. Constitutive over-expression of GsAS2 resulted in a 5.7 fold increase in oleanolic acid accumulation, while over-expression of GsAS1 led to a 3 fold increase. Additionally, RNAi-directed suppression of GsAS2 and GsAS1 in G. straminea decreased oleonolic acid levels by 65.9% and 21% respectively, indicating that GsAS2 plays a more important role than GsAS1 in oleanolic acid biosynthesis in G. straminea. We uses a docking model to explore the catalytic mechanism of GsAS1/2 and predicted that GsAS2, with its Y560, have higher efficiency than GsAS1 and mutated versions of GsAS2 in β-amyrin produce. When the key residue in GsAS2 was mutagenized, it produced about 41.29% and 71.15% less β-amyrin than native, while the key residue in GsAS1 was mutagenized to that in GsAS2, the mutant produced 38.02% more β-amyrin than native GsAS1.
Similar content being viewed by others
Introduction
The Gentianaceae species Gentiana straminea Maxim is endemic to western China and the Qinghai-Tibet plateau (4,000 meters above sea level), where it has been exploited as a source of medicinal compounds for at least 2,000 years1. Its major pharmaceutically active compounds are secoiridoids and triterpenoids2,3. Oleanolic acid is one of the most common such triterpenoids. It has anti-hepatitis4, antimicrobial5, anticancer6, and antiapoptotic6 activities. Given the restricted extent of growth environments of suitable for G. straminea production and limited oleanolic acid yields, the use of metabolic engineering to improve oleanolic acid production in G. straminea is an important goal. Many enzymes involved in the biosynthesis of triterpenoids have been the focus of metabolic engineering efforts. The oxidosqualene cyclases (OSCs) are the key enzymes in the cyclization of (3S) 2, 3-oxidosqualene, an important compound in the early steps of triterpenoid biosynthesis7,8. A number of genes encoding OSCs have been isolated, and their function has been determined via heterologous expression in yeast. β-amyrin synthase (βAS) genes, which encode key enzymes that catalyze the formation of the precursors of oleanolic acid, have been identified in various plant species9,10,11,12,13,14,15,16,17. The G. straminea GsAS1 gene was shown to encode a βAS18, but its function within G. straminea has not yet been established.
The majority of eukaryotic genes harbor one or more introns19,20. However, a significant proportion of plant genes are known to lack introns21. Such genes may have originated from intronless ancient genes conserved among archaebacteria, prokaryotes, and eukaryotes, or from RNA-related duplication and retropositionevents22,23,24,25. The presence of intronless genes provides an opportunity to evaluate the advantageous that the lack of introns can confer.
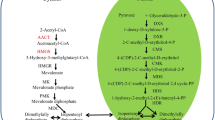
Phytosteols and triterpenes are biosynthesized initially via the condensation of acetyl-CoA by the cytosolic mevalonate (MVA) pathway in G. straminea (Fig. 1A) and the subsequent cyclization of (3S) 2, 3-oxidosqualene into, respectively, cycloartenol or β-amyrin (Fig. 1B). Triterpene cyclization in plants is more complex than sterol synthesis, as the compounds feature a pentacyclic carbon skeleton and a more extensive methyl and hydride shift26. In β-amyrin biosynthesis, the residues W259 and Y261 of the Panax ginseng enzyme PNY are important for the formation of the D/E-ring of β-amyrin27,28, and the product-determining role of the DCTAE motif has recently been demonstrated by applying a site-directed mutagenesis approach to the Euphorbia tirucalli gene that encodes β-amyrin synthase (EtAS)29. Subtle structural and hence electrostatic differences at the active site of an enzyme can radically alter its substrate and product specificity, and can also affects its catalytic efficiency30. To date, only a few studies have reported analyses of the catalytic functions of particular residues in the active site of OSCs, perhaps owing to a lack of suitable docking models of the enzyme and intermediate compounds.

(A) Early steps in biosynthesis of phytosterols and triterpenoid saponins leading to the common precursor 2, 3-oxidosqualene. HMG-CoA: 3-hydroxy-3-methylglutaryl-CoA, IPP: Isopentenyl pyrophosphate, DMPP: dimethylallyl pyrophosphate, GPP: geranyl pyrophosphate, FPP: farnesyl pyrophosphate. HMGS: 3-hydroxy-3-methylglutaryl-CoA synthase, HMGR: 3-hydroxy-3-methylglutaryl-CoA reductase, IPPI: IPP isomerase, GPPS: geranyl diphosphate synthase, FPS: farnesyl diphosphate synthase, SS: squalene synthase, SE: squalene epoxidase. (B) The biosynthetic pathway of (3S)-2, 3-oxidosqualene into phytosteol andtriterpene via cycloartenol and β-amyrin.
Most terpenoids are produced in planta in very small amounts, so their commercial exploitation is often hampered by uneconomic yields31. As our understanding of terpeniod synthesis and metabolic engineering of plants improves, new opportunities to enhance productivity and alter terpenoid product profiles are becoming available32,33. Higher product yield and recovery for compounds including carotenoids, artemisinin, patchoulol, and linalool/nerolidol have been achieved in various organisms including E. coli, Arabidopsis thaliana, Artemisia annua, tobacco, potato, and maize34,35,36,37,38,39,40,41. Since β-amyrin is a key precursor of the triterpenoid saponin pathway, over-expression of βAS may possibly result in increased saponin production. Expressing a β-amyrin synthase gene from Panax japonicushad in rice increased the accumulation ofoleanolic acid42.
No intronless OSC genes have been reported in plants43. Here, we describe the isolation of an intronless G. straminea OSC sequence (GsAS2). We compared its product profile with that of its intron-containing paralog GsAS1. Heterologously expressed GsAS2 generates relatively more β-amyrin in yeast than does GsAS1, and constitutive expression of GsAS2 in G. straminea results in an up to 5.7 fold increased in oleanolic acid yield compared to wild type plants. And a comparison between GsAS1 and GsAS2 showed that the latter’s product is the more important for the production of oleanolic acid in transgenic plants of G. straminea. We performed substrate and enzyme docking modeling and found that one key residue may ultimately explain the observed differences in oleanolic acid accumulation between GsAS2 than GsAS1. Site-directed mutagenesis and heterologous expression in yeast cells demonstrated that, relative to GsAS1 and the H560 and F560 mutant forms of GsAS2, the GsAS2 produced β-amyrin more efficiently.
Results
An intronless OSC sequence identified from G. straminea
A full length βAS cDNA sequence, named GsAS1, has already been isolated from G. straminea18. The 920 bp amplicon produced by degenerate PCR was shown by sequencing to comprise two distinct sequences, one of which was GsAS1. The other, when used to generate a full length sequence, was named GsAS2 (KJ467352); its length was 2,277 bp, encoding a predicted 759 residue polypeptide containing one DCTAE and four QW motifs (Supplementary Fig. S1A). The nucleotide sequence of the genomic locus for GsAS2 was identical to that of the full length cDNA (Supplementary Fig. S1B). Thus, GsAS2 is an intronless OSC sequence. However, 16 introns are present in the genomic sequence of GsAS1 (Fig. 2A). Phylogenetic analysis showed that GsAS2 resembled other OSCs, with the closest matches being GsAS1 and βAS enocoding genes present in Aster sp. and Artemisia sp. (Fig. 2B). The GsAS2 peptide sequence is highly homologous to that of GsAS1 (81.1% identity) and to those of the βASs present in Panax ginseng (AB014057, 76.2%), Aster sedifolius (AY836006, 73.1%), Glycyrrhiza glabra (AB037203, 73.1%), Betula platyphylla (AB055512, 74.2%), and Euphorbia tirucalli (AB206469, 74.3%).

The structure and phylogeny of GsAS1 and 2.
(A) The structure of the genomic loci for GsAS1 and 2. (B) A maximum likelihood tree based on codon alignment, annotated with relevant bootstrap values. The tree was estimated using the GTR + Γ + I substitution model implemented within PhyML software. The scale bar indicates the number of substitution sites.
The ω values associated with the GsAS1 and GsAS2 sequences were estimated for each branch using a free ratio model. Since ω represents the non-synonymous to synonymous substitution ratio, the indication was that GsAS2 has evolved under purifying selection (ω = 0.06), whereas a higher ω value (0.25) was evident for the branch leading to GsAS1 (Supplementary Table S1). The ω value per codon in the background (ω0) and foreground (ω1) lineages were, respectively, 0.11 and 75.50 (2ΔL = 434.74, P < 0.01), as estimated using branch site model A (Supplementary Table S1). GsAS1 appears to have evolved recently, at an accelerated rate,under positive selection.
Transcription of GsAS2 and GsAS1 in G. straminea
The transcription profiles of GsAS1 and GsAS2 were analyzed in different organs of G. straminea plants (Fig. 3A,B). The standard curves calculated for both GsAS1 and GsAS2 were very similar to one another (Supplementary Fig. S2). Both GsAS1 and GsAS2 were highly expressed in roots. GsAS1 transcript levels in roots were 2.3 fold greater than the levels in either the leaves or stems. GsAS2 transcript levels in roots was 27.5 fold greater than that in leaves and 18.6 fold greater than in stems. GsAS2 expression was significantly higher than GsAS1 in roots (13.3 fold increase) and in stems (1.6 fold increase). No difference in the transcript levels of GsAS1 and GsAS2 was observed in leaves (Fig. 3C).

Transcription profiles of GsAS1 and GsAS2 and oleanolic acid content in G. straminea.
(A,B) G. straminea plants used in this study. (C) The transcription of GsAS1 and GsAS2 in the root, stem, and leaf as assessed via qRT-PCR. The β-actin gene from G. straminea was used as the reference gene for quantification. The bars indicate the standard error (n = 3), *Statistically significant differences as analyzed by Student’s t-tests and ANOVA test (P ≤ 0.05). (D,E,G,H) The transcription of GsAS1 and GsAS2 in G. straminea plants treated with MeJA (D,E) and SA (G,H). The bars indicate the standard error (n = 3). *Statistically significant differences as analyzed by Student’s t-tests and ANOVA test (P ≤ 0.05). (F,I) Oleanolic acid content in G. straminea plants treated with MeJA (F) and SA (I). The bars indicate the standard error (n = 10). *Statistically significant differences as analyzed by Student’s t-tests and ANOVA test (P ≤ 0.05).
GsAS1 and GsAS2 transcription was strongly induced by methyl jasmonate (MeJA) treatment, producing, after 24 h, respectively, 9.8 and 12.2 fold increases over the level of mock-treated plants (Fig. 3D,E). The oleanolic acid content of the plants treatment with MeJA was also increased significantly (Fig. 3F). Salicylic acid (SA) did not affect the transcription level of GsAS1 or GsAS2 and did not affect oleanolic acid content (Fig. 3G–I).
GsAS2 Encodes a functional βAS with more contribution to oleanolic acid accumulation than GsAS1
Heterologous expression of GsAS1 in yeast has shown that it encodes a functional βAS18. When the GsAS2 open reading frame was placed under the control of the methanol-inducible AOX1 promoter within the pPICZA expression vector and introduced into Picha pastoris, the GsAS2 protein was detected (Fig. 4A) and it generated a product with the same retention time (27.50 min) as a β-amyrin reference standard. Control cells (carrying only an empty vector) did not express this product (Fig. 4B). The mass spectrum for this enzymatic product was consistent with that of β-amyrin (Fig. 4C). These results suggest that the enzyme encoded by GsAS2 cyclizes oxidosqualene to form β-amyrin and thus suggest that GsAS2, like GsAS1, is a functional βAS gene. Comparison of the product accumulation in the assays with the heterologously expressed forms of GsAS1 and GsAS2 showed that the yield of β-amyrin by GsAS2 was about 12.9 times greater than that of GsAS1 (Table 1) suggesting that GsAS2 has a higher potential for oleanolic acid accumulation than GsAS1 when heterologously expressed in yeast.

GsAS2 encodes a functional βAS.
(A) The expression of the wild type and mutant forms of GsAS1 and GsAS2, as assayed by western blotting. Control: protein extracted from yeast cells harboring an empty vector. Target protein: western blot profiles. Total protein: SDS-PAGE profile of total protein stained with Coomassie blue R250. (B) GC chromatographs. The trace displays the output from yeast cells carrying either an empty vector, GsAS1, GsAS1-H560Y, GsAS2, GsAS2-Y560H,or GsAS2-Y560F. (C) Mass spectra of the standard (β-amyrin) and the output of yeast cells harboring GsAS2.
The relative enzymatic activity of GsAS1 and GsAS2 was also assessed in terms of oleanolic acid accumulation in the GsAS1 and GsAS2 over-expression transgenic G. straminea lines and in GsAS1 and GsAS2 RNAi suppression G. straminea lines (Fig. 5A–C, Supplementary Fig. S3). Quantitative RT-PCR (qRT-PCR) analysis showed that the transcript abudance for both GsAS1 and GsAS2 was higher in the over-expression lines than in the wild type and that the transcript abudance for both GsAS1 and GsAS2 was lower in the RNAi lines than in the wild type (Supplementary Fig. S4A,B). Based on the transcript abundance results, particular over-expression transgenic lines of G. straminea were chosen for southern blot analysis. The expression levels rose as the number of transgene copies increased in transgenic G. straminea. The GsAS2 transgenic G. straminea line with the highest GsAS2 expression level had 4 copies of the transgene in its genome (Supplementary Fig. S4C). HPLC analysis showed that the oleanolic acid content of the GsAS2 over-expression lines on average 3.7 (between 1.6 and 5.7) fold higher than that of the wild type, while the GsAS1 over-expression lines, oleanolic acid accumulation was 2.3 fold higher that of the wild type (the range was 1.8–3.0 fold) (Fig. 5C). Oleanolic acid content was reduced by 65.9% in the GsAS2 RNAi plants but was reduced by just 21.0% in the GsAS1 RNAi plants (Fig. 5C). These results indicated that GsAS2 was also more efficient than GsAS1 in directing oleanolic acid accumulation in planta.

GsAS2 apparently produces oleanolic acid more efficiently than GsAS1.
(A,B) Oleanolic acid content following the up- or down-regulation of GsAS1(A) and GsAS2(B). WT: wild type, E: over-expression lines, R: RNAi lines. The bars represent the standard error (n = 10). *Statistically significant differences which were analyzed by Student’s t-tests and ANOVA (P ≤ 0.05). (C) The average increase or decrease (fold) in oleanolic acid content in transgenic plants with GsAS1 or 2 over-expression and RNAi. The bars represent the standard error (n = 3). a: Statistical significance between means was inferred by a t-test compared with wild type (P ≤ 0.05), b: Statistical significance between GsAS1 and GsAS2 transgenic plants samples means evaluated with Student’s t-test compared. (WT: wild type, E: over-expression lines, R: RNAi lines). (D–L) The effect of GsAS2 over-expresion on transcription of triterpene biosynthesis pathway genes in GsAS2 transgenic plants (WT: wild type, E: over-expression lines, R: RNAi lines).
When the transcription levels of other genes involved in the triterpene pathway were assessed in GsAS2 over-expression (E1 and E7) and RNAi (R2 and R3) lines, the abundance of SS and SE transcripts was found to be similar to that of GsAS2. For SS, the fold difference compared to the WT was 2.6 in E1 and 2.5 fold in E7, while the difference was 0.5 fold in R2 and 0.3 fold in R3 (Fig. 5D). Similarly, as compared to the WT, the abundance of SE transcripts in the E1, E7 and the R2 and R3 lines was, respectively, 2.3 and 3.5 fold higher and 0.4 and 0.3 fold lower (Fig. 5E). No differences was observed in the expression of other genes of the triterpene pathway, including HMGS, HMGR, IPPI, GPPS, FPS, CYS, or LUS (Fig. 5F–L).
The possible mechanism for the apparent increased catalytic efficiency of GsAS2 over GsAS1 for β-amyrin synthesis
To explore possible mechanisms for the apparent functional improvement of GsAS2 over GsAS1 in the production of β-amyrin, we superimposed I-TASSER derived models of GsAS1 and GsAS2 with a backbone root mean square deviation of 2.09 Å (Fig. 6A), indicating their very high overall structural likeness. This was as expected, given their similar enzymatic activity.

Modelling of GsAS1 and GsAS2 in complex with β-amyrin.
(A) Ribbon diagram representation of GsAS1 (yellow) and GsAS2 (purple). The β-amyrin molecule (black) positioned in the active site. (B) Substrate combination model of GsAS2. (C) Substrate combination model of GsAS1. Residues interacting with β-amyrin are indicated by orange colored bonds. Different residues between GsAS1 and GsAS2 interacting with β-amyrin are indicated by green colored bonds. Residues interacting indirectly with β-amyrin are indicated by pink colored bonds. The hydrogen bonds are shown as dashed lines.
Both enzymes harbored two (α/α) barrel domains connected by loops, as well as three smaller β structures (Fig. 6A). After docking with an oleanylcation, both the GsAS1 and GsAS2 active site cavities became surrounded by hydrophobic residues, except for their polar caps (Fig. 6B,C). The D484 residue is free to form a hydrogen bond with the substrate’s 3-hydroxy group, while C563 can serve as a hydrogen-bonding partner (Fig. 6B,C). Their intermediate product oleanylcations were surrounded with the aromatic residues Y259, Y412, W417, F473, W533, F551, Y560, W611, and F727 (Fig. 6B,C). The active sites of GsAS1 and GsAS2 differed with respect to H/Y560,G/W257, L/I554, G/C731, G/A532 (Fig. 6B,C). In GsAS2, the W257 indole ring lay close to (about 3 Å) the Y259 benzene ring, bringing Y259 close to C12, C13, and C18 of the oleanylcation (respectively, 2.3, 3.5, and 3.2 Å). The Y560 hydroxy group was able to interact with that of Y259 (4.5 Å) and the amido group of N610 (4.1 Å). However, in GsAS1, G257 was unable to interact with Y259, while the H560 imidazole group lay further away from the Y259 hydroxy group (>7 Å) - although it was still able to interact with N610 (3.9 Å) (Fig. 6B,C).
The docking results identified two substitutions (H560Y and G257W) as potentially responsible for the apparent differences in the β-amyrin production of GsAS1 and GsAS2. The behavior of W257 has been reported by Kushiro et al.28, while the functional effect of the residue 560 has not been investigated. Previously, it has been shown that GsAS1 produces β-amyrin18, suggesting that both the H and Y560 variant may be functional with respect to β-amyrin synthesis. In this study, the two GsAS2 mutants Y560H and Y560F and GsAS1 H560Y were generated to investigate the effect of the H/Y560 residue on β-amyrin production and the catalytic properties of the enzymes. Each sample was divided into two equal portions. When one portion was processed by western blotting, the bands representing the mutant and native enzymes had very similar densities (Fig. 4A), implying a similar titer of these enzymes forms in yeast. The other sample portion was used to test triterpenoid biosynthesis in yeast extraction and analysis with GC-MS. The highest β-amyrin levels were measured in yeast cells harboring GsAS2, the relative amounts of β-amyrin in yeast cells harboring GsAS1, mGsAS1-H560Y, mGsAS2-Y560H, and -Y560F compared to cells with GsAS2, were, respectively, 7.77 ± 0.64%, 10.72 ± 0.51%, 58.71 ± 3.42%, 28.85 ± 2.30% (Table 1 and Fig. 4B). The two mutated GsAS2 sequences produced, respectively, 41.29% and 71.15% less β-amyrin than the native GsAS2, while the GsAS1 mutant produced 38.02% more β-amyrin than native GsAS1 (Table 1). These results suggest that the Y560 residue in the GsAS2 active site confers higher efficiency in β-amyrin formation than the H560 residue in the active site of GsAS1, and this difference may explain the higher efficiency of GsAS2 on oleanolic acid accumulation compared to GsAS1.
Discussion
The terpenoids form one of the largest classes of plant secondary metabolites, and the OSC genes represent an important component of their synthesis. Characterization of OSC function has been achieved indirectly via heterologous expression in yeast for genes isolated from ginseng13 and pea15, and directly in the model species A. thaliana44,45. To date, the contribution of these genes to terpenoid (especially oleanolic acid) synthesis has not been well-elucidated in a medicinal plant species. We show here that the over-expression of GsAS2 enhanced the accumulation of oleanolic acid, a finding consistent with our previous results showing that oleanolic acid accumulation is positively related to the expression level of GsASs46. The relevant transgenic G. straminea plants could therefore make a significant contribution towards the engineering of this species to produce economically-viable amounts of this valuable metabolite.
Most of the OSC gene sequences described to date feature one or more introns, reaching as many as 17 in AsbAS1 (from oat) and 16 in AtCAMS1 (from A. thaliana)13,47. Unlike GsAS2 which is lacks introns (Supplementary Fig. S1), the previously characterized G. straminea gene GsAS1 features 16 introns (Fig. 2A) and shows similar exon patterns to other OSC genes (data not shown). GsAS1 might represent the gene directly derived from the ancestor gene with a conserved structure, while the intronless GsAS2 represents more recent newer evolutionary origin. It has been suggested that a likely origin of intronless genes is retroposition48. The expression of a retroposed gene copies is expected not to correlate with its source gene, because it obtains new regulatory elements from its site of insertion48. Therefore, new function often occurs with the daughter gene (retrogene), rather than with a parental gene. However, a daughter gene may inherit promoters and enhancer elements from their parental genes, resulting in a daughter gene with regulatory properties similar to the parental gene43. Thus, both daughter gene and parental genes have the possibility to undergo neofunctionalization. There is a report of this type of case; the daughter gene actually maintained the ancestral function, while the partental gene underwent neofunctionalization49. Here, GsAS2 appeared to be more strongly transcribed than GsAS1 (Fig. 3C), with the result that alterations (either up or down) in the former’s transcription level had a greater effect on the plant’s oleanolic acid content than did the latter’s (Fig. 5A–C and Supplementary Fig. S3). An evolutionary analysis of the GsAS2 sequence suggested that its function is likely to have been conserved as a result of purifying selection (Supplementary Table S1). However, GsAS1 appears to have evolved at an accelerated rate through positive selection; it may either still be in the process of neo-functionalization or may represent a multi-functional triterpene cyclase that generates an undetected alternative product. The observation that GsAS1 makes a lesser contribution to the synthesis of oleanolic acid than GsAS2 supports the notion that OSC genes multiply via gene duplication, with positive selection driving one copy to neo-functionalize via a process of non-synonymous mutations, while the other retains the original function17.
Subtle morphological and electrostatic differences at the active site of an enzyme can radically alter their enzymatic capacities30. Here, the active sites of GsAS1 and GsAS2 differed with respect to H/Y560, G/W257, L/I554, G/C731, and G/A532. Among these, the G552, L554, and G731 residues in GsAS1 have similar chemical and physical characteristics with A552, I554 and C731 in GsAS2 (Fig. 6B,C), and may therefore contribute little to the β-amyrin synthesis activity. Substitutions (H560Y and G257W) between GsAS1 and 2 affected β-amyrin production. Here, in GsAS2, the Y560 hydroxyl group lay close to Y259, together with N610 forming a hydrophilic environment surrounding C13 of the oleanylcation, which ostensibly improves a deprotonation from C13 for the formation of β-amyrin (Fig. 6B). The GsAS1 H560 residue has a smaller side chain than that of Y560, and so may interact less strongly with Y259 in the context of deprotonation (Fig. 6C). The mutagenesis of GsAS1 and GsAS2 showed that the presence of H560 was associated with reduced β-amyrin production relative to the protein form with Y560 (Table 1). Additionally, in previous research, residue W259 in PNY (corresponding to the W257 in GsAS2) was demonstrated to play a key role in β-amyrin synthesis25, suggesting that W257 may also play a role in β-amyrin synthesis. Therefore GsAS2 was more effective than GsAS1 in β-amyrin production in yeasts, which was consistent with the results of greater accumulation of oleanolic acid upon constitutive expression of GsAS1/2.
Up-regulating a rate-limiting enzyme gene within a given metabolic pathway represents an effective means of raising productivity31. A typical example relates to the accumulation in Eleutherococcus senticosus of phytosterols and saponins via the heterologous expression of a SS-encoding gene isolated from ginseng, in which the levels of product were increased by between 2.0 and 2.5 fold50. Similarly, the artemisinin content in Artemisia annua plants over-expressing FPS exhibited a 2.5 fold increase compared to the non-transgenic control51. In the present experiment, however, it proved possible, via the over-expression of GsAS2, to enhance oleanolic acid content by nearly 6 fold (Fig. 5B).
When secondary metabolic flux is disturbed or changed by over-expression or reduced expression of genes, the expression level of other genes in the pathway can be up- or down- regulated by feedback or feed-forward effects with the products of the target genes. Over-expression of Panax ginseng squalene synthase (PgSS1) in adventitious roots of transgenic P. ginseng was followed by the up-regulation of genes of triterpene synthesis such as βAS, and this resulted in a remarkable increase in ginsenoside content52. However, there is little direct evidence for the regulatory function of βAS genes in the biosynthesis of triterpene saponins. Here, we found that the expression of SS and SE were up-regulated in plants when GsAS2 was over-expressed (Fig. 5D,E), showing that the level of GsAS2 expression affects the transcription of certain genes upstream in the triterpene pathway. The increased oleanolic acid content resulting from the over-expression of GsAS2 shows that this gene is an important component of the triterpenoid synthesis pathway in G. straminea.
In conclusion, GsAS2 exhibit more important roles on oleanolic acid accumulation than GsAS1 in G. straminea at several aspects. The transcription abundance of the GsAS2 is higher than that of GsAS1 especially in roots which have the highest oleanolic acid content. Furthermore, this function was further emphasized by lower accumulation of oleanolic acid in GsAS2 RNAi lines compared to GsAS1 RNAi lines (Fig. 5C). For apparent enzyme efficiency, GsAS2 accumulated 12.9 fold more β-amyrin that did GsAS1 when expressed heterologously in yeast (Table 1). The important residues (Y560) were identified by adocking model and mutation studies showed that the form of GsAS2 with Y560 had accumulated more oleanolic acid than both GsAS1 and various mutated forms of GsAS2. All of these results highlight the remarkable function of the intronless GsAS2 and lay a foundation for the use of this gene improving the triterpenoid production via metabolic engineering.
Materials and Methods
Plant materials and tissue culture
Embryogenic calli of G. straminea were generated from sterilized seeds cultured at 25 ± 1 °C on MS basal medium53 supplemented with 30 g/L sucrose and 2 mg/L 2, 4-dichlorophenoxyacetic acid. Calli were subcultured every two weeks. Regeneration was induced by transfer to differentiation medium (IB medium), which was MS medium supplemented with 0.5 mg/mL indole acetic acid and 0.5 mg/mL 6-benzyl adenine.
Gene isolation
Total RNA of G. straminea was extracted and reverse transcribed as described by Liu et al.18. Two degenerate primers (5′-TGGCTTTCGATA(T)CTTGGA-3′ and 5′-CCACCG(A)TTTTTG(A)CTCTGTA-3′) were designed based on highly conserved regions of reported OSCs genes from other plants including P. ginseng, Betula platyphylla, Aster sedifolius, and Euphorbia tirucalli. RT-PCR was then performed using the first strand cDNA of G. straminea as a template. The resulting 920 bp RT-PCR amplicon was cloned into the pMD18-T vector (Takara, Japan), transformed into E. coli DH5α cells, and sequenced. Overlapping 5′ and 3′ sequences were obtained using RACE PCR, based on a Full RACE Core Set (Takara, Japan) and the primer pair 5′-CTCAACCCAACAAGCAAGC-3′ and 5′-ATGGGTTGCAGAAGATGG-3′. A set of ten independent 5′ and 3′ clones was sequenced, and their sequences were aligned to obtain a consensus sequence15. For full length gene isolation, the same primer pairs of GsAS2 were amplified with the total DNA of the G. straminea as a template. The primers for GsAS1 are listed in Supplementary Table S2.
OSC sequence phylogeny
A multiple alignment of OSC sequences was performed using Muscle 3.6 software54 with some manual editing. Proteins sequence alignment was transformed into codon sequences with the help of theaa2 DNA script. Maximum likelihood phylogenies were constructed from the codon sequence alignment using PHYML software55 based on the GTR + Γ + I substitution model. The free ratio model of CODEML, implemented within the PAML4 software package56, was used to estimate the lineage-specificity of the non-synonymous to synonymous substitution ratio ω. A branch site analysis, which compared the nearly neutral model with Model A56, was performed to test the assumption that the foreground ω value of a specific branch was >1 at sites where positive selection appeared to have acted within a specific lineage. The resulting likelihood ratio tests were performed at the 5% level.
Transcription profiles of GsAS1 and GsAS2
The roots, stems, and leaves of regenerated plants in IB medium were separated, and RNA was extracted by using TRIzol reagent (Invitrogen, USA) following the manufacturer’s instructions. Then RNA was spectrophotometrically quantified and reversed transcribed into cDNA, which subsequently used as a template for qRT-PCR to analyze the expression of GsAS1 and GsAS2 in different tissues. The qRT-PCR primers targeted GsAS1 and -2; their sequences were: GsAS1-F/-R: 5′-TCCTCTGATTATATGCTTGT/5′-ACCATCCTCATTCTGAT and GsAS2-F /-R: 5′-GGAGGATTAGCAGCATCT/5′-CCATCTTGTCGTTGTGAAT. The qRT-PCR analysis used a SYBR Green I real-time PCR detection system (Applied Biosystems, USA), using the β-actin gene from G. straminea as the reference gene. Each 15 μL reaction contained 0.2 μM of each primer and 2 μL of a 1:10 dilution of the prepared first-strand cDNA. The thermalcycling program was as follows: a denaturation step of 95 °C/2min, followed by 40 cycles of 95 °C/10s, 58 °C/20s, 72 °C/20s. A melting curve analysis was performed over the range 80–95 °C at 0.5 °C intervals. An absolute quantification of GsAS1 and -2 transcript abundance was obtained through the use of a standard curvethat plotted logarithm of initial copies of template DNA against the threshold cycle number from a serial dilution of the 3.337 × 1010 copies/μL of a pPICZA-GsAS1-GsAS2 plasmid that contained atandemly linked sequences of GsAS1 and 2.
The plants treated with 50 μM MeJA or 50 μM SA for 0 h, 6 h, 12 h, and 24 h were harvested and frozen with liquid nitrogen. The treated materials were used to analyze the expression of GsAS1 and GsAS2 or the content of oleanolic acid. The treated regenerated plants used to analyze the content of oleanolic acid were air-dried, ground to a powder, suspended in ten volumes of methanol, and exposed to 60 min of ultrasonic homogenization. The material was then centrifuged at 10,000 g for 10 min, and the supernatant was filtered through a 0.45 μm membrane. 10 μL extracts of each sample was injected into an HPLC instrument with an Ultrasphere C18 column (150 mm × 4.6 mm i.d., Phenomenex). Methanol::water (9::1) was used as the mobile phase, the flow-rate was maintained at 0.8 mL·min−1, and the effluent was monitored at 209 nm. The peak area of oleanolic acid was integrated by a Classvp5.0 system, using oleanolic acid as an external standard; a standard calibration curve was generated from data measured for a range of concentrations (20–1000 mg·mL−1).
Functional analysis via heterologous expression of OSC enzymes in yeast
P. pastoris strain X33, which was cultured in YPD medium (1% yeast extract, 2% peptone and 2% glucose, supplemented with 100 μg/mL ZeocinTM (Invitrogen, USA)), was used in this study. GsAS2 with KpnI and XhoI restriction enzyme sites was amplified using the forward primer: 5′-ATCGGTACCATGTGGAGGCTAAAGATTGCA-3′ and the reverse primer: 5′-CGGCTCGAGCAGATGGCAATGGCACTCTCT-3′. GsAS1 with EcoRI and XhoI restriction enzyme sites was amplified from the pPICZA-GsAS1 plasmid using the forward primer: 5′-ACTACTAGTGAATTCATGTGGAGGCT-3′ and the reverse primer: 5′-CGGCTCGAGCGTCCGGCACTTGCTTGCGGT-3′15. The reverse primers were designed to delete the stop codons. Both fragments contained full ORFs and were subcloned into the P. pastoris expression vector pPICZA (Invitrogen, USA) under the control of the methanol-inducible promoter, 5′AOX1. The PICZA-GsAS1,2 and empty vector pPICZA were then transformed separately into P. pastoris strain X33 using electroporation according to the manufacturer’s instructions.
The extraction of membrane proteins was achieved using a Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo, USA) following the manufacturers protocol. For western blotting, 1 volume of 2 × SDS sample buffer was added to the protein extract and the mixture was incubated at 70 °C for 10 min, followed by a centrifugation at 12,000 g for 10 min. The resulting supernatant was subjected to SDS–PAGE (10% polyacrylamide) to resolve the protein species present, then transferred to a PVDF (polyvinylidene difluoride) membrane (EMD Millipore, USA) using a semi-dry electrophoretic transfer cell (Bio-Rad, USA). The subsequent immunodetection assay was based on an anti-His primary antibody (Abcam, UK) and an HRP-conjugated anti-rabbit IgG secondary antibody (CST, USA). The hybridized membrane was immersed in freshly prepared HRP reaction solution (Advansta, USA) for 1–2 min, and then exposed to X-ray film for 1 min. Yeast cells harboring GsAS2 and empty vectors were cultured at 30 °C to OD600 = 2~6 in minimal glycerol medium (MGY, 1.34% yeast nitrogen base (YNB), 1% glycerol, 4 × 10−5% biotin). The cells were collected, resuspended in minimal methanol medium (MM, 1.34%YNB, 4 × 10−5% biotin, 0.5% methanol) to OD600 = 1.0, and incubated at 30 °C for 4 d by adding 100% methanol to a final concentration of 0.5% every 24 h. The incubated yeast cells were finally collected and disrupted with 2 mL of 20%KOH/50% EtOH (1/1, v/v) for every 25 mL MM medium. The products were extracted twice with 2 mL hexane. The extracts and 1–3 μg standards β-amyrin (Sigma, USA) were analyzed by using a GCMS-QP2010 GC/MS System fitted with an Agilent DB-5MS column (29.5 m × 250 μm internal diameter, 0.25 μm film) (JW Scientific, USA). The inlet, transfer line, and ion source temperatures were set at, respectively, 270 °C, 270 °C and 200 °C, and the oven temperature was programmed to 200 °C for 2 min, raised to 270 °C at 10 °C per min, and held at 270 °C for 30 min. The flow rate of the carrier helium was 1.5 mL per min. Splitless injections (8 μL) were used, and mass spectral data in the m/z range 70–550 were acquired. A standard calibration curve for β-amyrin was generated from data measured for a range of concentrations (10–50 mg·mL−1).
Vector construction and transformation of G. straminea
The plant over-expression vector pK7WG2D and the RNAi vector pK7GWIWG2D(II) (Invitrogen, USA), both constructed using the Gateway technique, were combined with two independent GsASs cDNA sequences and transformed into seven day old G. straminea calli using particle bombardment57. After a 24 h period in darkness, the calli were transferred to selective medium containing 50 mg/L kanamycin. Surviving calli was tested for the presence of the GsASs with a PCR assay targeting the CaMV 35S promoter sequence (primer sequences: 5′-GCAGAGGCATCTTCAACG-3′ and 5′-TTCGATCATGGGCAGAAGACGAC-3′).
Characterization of regenerated transgenic plants
Surviving calli were transferred to the aforementioned regeneration medium and plants able to be successfully regenerate were evaluated for the presence of the transgenic constructs with a PCR assay targeting the CaMV 35S promoter sequence, as above. The number of copies of the transgene in plants was assessed by Southern blotting. The transcription levels of genes involved in triterpene synthesis was monitored by qRT-PCR. Oleanolic acid content was analyzed by HPLC, as mentioned detailed above.
Three dimensional modeling and mutagenesis
Three dimensional models of the GsAS1 and GsAS2 proteins were generated using I-TASSER v2.1 software58. The resulting models had C-scores 1.30 (GsAS1) and 1.37 (GsAS2). AutoDock v4.2 software59 was used to visualize the GsAS1/2/β-amyrin complex. The three dimensional structure of β-amyrin was obtained from http://zinc.docking.org (ID 3978269). In the data preprocess before docking, the structure was restrained within a grid box (40 × 86 × 60 points in each dimension) that covered the GsAS2 binding pocket. The identity of the binding pocket was inferred assuming similarity with the crystal structure of the OSC-lanosterol complex (PDB code: 1W6K). Docking searches were performed using the Lamarckian genetic algorithm, with a maximum of 25,000,000 energy evaluations and other options set as the default. Ten potential models were returned, which were then ranked on the basis of binding energy. The top ranked model was assumed to be the most likely. The models were graphically rendered using VMD software (http://www.ks.uiuc.edu/Research/vmd)60.
The H560Y substitution in GsAS1/2 was chosen for site mutagenesis experiment. The mutant was generated using a PCR-based strategy to mutate the Y in GsAS2 to H and F (Phe), as well as H to Y in GsAS1. The mutagenesis sites were designed into primers61, and the PCR strategy was followed that of Edelheit et al.62 The mutation primers were listed in Table 2.
Statistical analysis
Results arepresented as the means of three independent biological replicates in all statistical tests. Inferential statistical tests were implemented in SPSS17.0 software and included one-way ANOVA followed by Duncan’s multiple range tests and Student’s t-tests (see figure legends for specific tests for particular experiments). P values ≤0.05 were interpreted as indicating statistically significant differences.
Additional Information
How to cite this article: Liu, Y. et al. An Intronless β-amyrin Synthase Gene is More Efficient in Oleanolic Acid Accumulation than its Paralog in Gentiana straminea. Sci. Rep. 6, 33364; doi: 10.1038/srep33364 (2016).
References
Ho, T. N. & Liu, S. W. A world wide monograph of Gentiana. China: Science Press, Beijing (2001).
Christian, Z. Altitudinal variation of secondary metabolites in flowering heads of the Asteraceae: trends and causes. Phytochem Rev 9, 197–203 (2009).
Ji, L. J. et al. Determination and evaluation of two iridoids in Gentiana straminea herbs by HPLC. Acta Botanica Boreali-Occidentalia Sinica 24, 292–295 (2004).
Kim, S., Lee, H., Lee, S., Yoon, Y. & Choi, K. H. Antimicrobial Action of Oleanolic Acid on Listeria monocytogenes, Enterococcus faecium, and Enterococcus faecalis. PLoS One 10, e0118800 (2015).
Chai, J. et al. Oral administration ofoleanolic acid, isolated from Swertia mussotii Franch, attenuates liver injury, inflammation, and cholestasis in bile duct-ligated rats. Int J Clin Exp Med 8, 1691–1702 (2015).
Zhu, Y. Y., Huang, H. Y. & Wu, Y. L. Anticancer and apoptotic activities ofoleanolic acidare mediated through cell cycle arrest and disruption of mitochondrial membrane potential in HepG2 human hepatocellular carcinoma cells. Mol Med Rep 12, 5012–5018 (2015).
Abe, I., Rohmer, M. & Prestwich, G. D. Enzymatic cyclization of squalene and oxidosqualene to sterols and triterpenes. Chem Rev 93, 2189–2208 (1993).
Haralampidis, K., Trojanowska, M. & Osbourn, A. E. Biosynthesis of triterpenoid saponins in plants. Adv. Biochem Eng Biot 75, 31–49 (2002).
Dhar, N. et al. Cloning and Functional Characterization of Three Branch Point Oxidosqualene Cyclases from Withania somnifera (L.) Dunal. J Biol Chem 289, 17249–17267 (2014).
Hayashi, H. et al. Cloning and characterization of a cDNA encoding beta amyrin synthase involved in glycyrrhizin and soyasaponin biosyntheses in licorice. Biol Pharm Bull 24, 912–916 (2001).
Iturbe-Ormaetxe, I., Haralampidis, K., Papadopoulou, K. & Osbourn, A. E. Molecular cloning and characterization of triterpene synthases from Medicago truncatula and Lotus japonicus. Plant Mol Biol 51, 731–743 (2003).
Kajikawa, M. et al. Cloning and characterization of a cDNA encoding β-amyrin synthase from petroleum plant Euphorbia tirucalli L. Phytochemistry 66, 1759–1766 (2005).
Kushiro, T., Shibuya, M. & Ebizuka, Y. Beta-amyrin synthase-cloning of oxidosqualene cyclase that catalyzes the formation of the most popular triterpene among higher plants. Eur J Biochem 256, 238–244 (1998).
Meesapyodsuk, D., Balsevich, J., Reed, D. W. & Covello, P. S. Saponin biosynthesis in Saponaria vaccaria cDNAs encoding beta-amyrin synthase and a triterpene carboxylic acid glucosyl transferase. Plant Physiol 143, 959–969 (2007).
Morita, M., Shibuja, M., Kushiro, T., Masuda, K. & Ebizuka, Y. Molecular cloning and functional expression of triterpene synthases from pea (Pisum sativum). EurJ Biochem 267, 3453–3460 (2000).
Shibuya, M. et al. Identification of a product specific β-amyrin synthase from Arabidopsis thaliana. Plant Physiol Bioch 47, 26–30 (2009).
Xue, Z. et al. Divergent evolution of oxidosqualene cyclases in plants. New Phytol 193, 1022–1038 (2012).
Liu, Y. L. et al. Cloning and Functional Analysis of a β-Amyrin Synthase Gene Associated with Oleanolic Acid Biosynthesis in Gentiana straminea MAXIM. Biol Pharm Bull 32, 818–824 (2009).
LeHir, H. H., Nott, A. & Moore, M. J. How introns influence and enhance eukaryotic gene expression. Trends Biochem Sci 28, 215–220 (2003).
Roy, S. W. & Gilbert, W. The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet 7, 211–221 (2006).
Louhichi, A., Fourati, A. & Rebai, A. IGD: a resource for intronless genes in thehuman genome. Gene 488, 35–40 (2011).
Betran, E., Emerson, J. J., Kaessmann, H. & Long, M. Sex chromosomes and male functions: Where do new genes go? Cell Cycle 3, 873–875 (2004).
Emerson, J. J., Kaessmann, H., Betran, E. & Long, M. Extensive gene traffic on the mammalian X chromosome. Science 303, 537–540 (2004).
Jain, M., Khurana, P., Tyagi, A. K. & Khurana, J. P. Genome-wide analysis of intronless genes in rice and Arabidopsis. Funct. Integr. Genomic 8, 69–78 (2008).
Long, M., Betran, E., Thornton, K. & Wang, W. The origin of new genes: Glimpses from the young and old. Nat Rev Genet 4, 865–875 (2003).
Ito, R., Hashimoto, I., Masukawa, Y. & Hoshino, T. Effect of cation-π interactions and steric bulk on the catalytic action of oxidosqualene cyclase: acase study of Phe728 of β-amyrin synthase from Euphorbia tirucalli L. Chem Eur J 19, 17150–17158 (2013a).
Kushiro, T., Shibuya, M. & Ebizuka, Y. Chimeric triterpene synthase a possible model for multifunctional triterpene synthase. J Am Chem Soc 121, 1208–1216 (1999).
Kushiro, T., Shibuya, M., Masuda, K. & Ebizuka,Y. Mutational studies on triterpene synthases: engineering lupeol synthase into β-amyrin synthase. J Am Chem Soc 122, 6816–6824 (2000).
Ito, R., Masukawa, Y. & Hoshino, T. Purification, kinetics, inhibitors and CD for recombinant β-amyrin synthase from Euphorbia tirucalli L. and functional analysis of the DCTA motif, which is highly conserved among oxidosqualene cyclases. FEBS J 280, 1267–1280 (2013b).
Wu, T. K., Chang, C. H., Liu, Y. T. & Wang, T. T. Saccharomyces cerevisiae oxidosqualene-lanosterolcyclase: a chemistry-biology interdisciplinary study of the protein structure-function-reaction mechanism relationships. ChemRec 8, 302–305 (2008).
Moses, T., Pollier, J., Thevelein, J. M. & Goossens, A. Bioengineering of plant (tri)terpenoids: frommetabolic engineering of plants to synthetic biology in vivo and in vitro. New Phytol 200, 27–43 (2013).
Dudareva, N., Klempien, A., Muhlemann, J. K. & Kaplan, I. Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytol 198, 16–32 (2013).
Roberts, S. C. Production and engineering of terpenoids in plant cell culture. Nat Chem Bio 3, 387–395 (2007).
Aharoni, A. et al. Terpenoid metabolism in wild-type and transgenic Arabidopsis plants. Plant Cell 15, 2866–2884 (2003).
Harjes, E. C. et al. Natural genetic variation in lycopene epsilon cyclase tapped for maize biofortification. Science 319, 330–333 (2008).
Kim, S. W. & Keasling, J. D. Metabolic engineering of the nonme-valonateisopentenyl diphosphate synthesis pathway in Escherichia coli enhances lycopene production. Biotechnol Bioeng 72, 408–415 (2001).
Lucker, J., Verhoeven, H. A., van der Plas, L. H. & Bouwmeester, H. J. Molecularengineering of floral scent. In: Dudareva N., Pichersky E., eds. Biology of floral scent. Boca Raton, FL, USA: CRC Press, 321–337 (2006).
Martin, V. J. J., Pitera, D. J., Withers, S. T., Newman, J. D. & Keasling, J. D. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nat Biotechnol 21, 796–802 (2003).
Schmidt-Dannert, C., Umeno, D. & Arnold, F. H. Molecular breeding of carotenoid biosynthetic pathways. Nat Biotechnol 18, 750–753 (2000).
Wu, S. et al. Engineering triterpene metabolism in tobacco. Planta 236, 867–877 (2012).
Wu, S. et al. Redirection of cytosolic or plastidic isoprenoid precursors elevates terpene production in plants. Nat Biotechnol 24, 1441–1447 (2006).
Huang, Z. W. et al. Production of oleanane-type sapogenin in transgenic rice via expression of β-amyrin synthase gene from Panax japonicus C. A. Mey. BMC Biotechnol 15, 45 (2015).
Wang, W. et al. High Rate of Chimeric Gene Origination by Retroposition in Plant Genomes. Plant Cell 18, 1791–1802 (2006).
Husselstein M. T., Schaller, H. & Benveniste, P. Molecular cloning and expression in yeast of (3S) 2, 3-oxidosqualene-triterpenoid cyclases from Arabidopsis thaliana. Plant Mol Bio 45, 75–82 (2001).
Segura, M. J., Meyer, M. M. & Matsuda, S. P. Arabidopsis thaliana LUP1 converts oxidosqualene to multiple triterpene alcohols and a triterpenediol. Org Lett 2, 2257–2262 (2000).
Zhao, Z., Song, Y., Liu, Y., Qiao, M. & Xiang, F. The effect of elicitors on oleanolic acid accumulation and expression of triterpenoid synthesis genes in Gentiana straminea. Biol Plantarum 57, 139–143 (2013).
Qi, X. et al. A gene cluster for secondary metabolism in oat: implications for the evolution of metabolic diversity in plants. Proc Natl Acid Sci USA 101, 8233–8238 (2004).
Kaessmann, H., Vinckenbosch, N. & Long, M. RNA-based gene duplication: mechanistic and evolutionary insights. Nat Rev Genet 10, 19–31 (2009).
Krasnov, A. N., Kurshakova, M. M., Ramensky, V. E., Mardanov, P. V. & Nabirochkina, E. N. A retrocopy of a gene can functionally displace the source gene in evolution. Nucleic Acids Res 33, 6654–6661 (2005).
Seo, J. W., Jeong, J. H. & Shin, C. G. Ovevexpression of squalene synthase in Eleutheeococcus senticosus increases phytosterol and trierpene accumulation. Phytochemistry 66, 869–877 (2005).
Banyai, W., Kirdmanee, C., Mii, M. & Supaibulwatana, K. Overexpression of farnesyl farnesyl pyrophosphate synthase (FPS) gene affected artemisinin content and growth of Artemisia annua L. Plant Cell Tiss Org 103, 255–265 (2010).
Lee, M. H. et al. Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol 45, 976–984 (2004).
Murashige, T. & Skoog, F. A revised medium for rapid growth and bioassays for tobacco tissue cultures. Physiol Plantarum 15, 473–497 (1962).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Bio 52, 696–704 (2003).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24, 1586–1591 (2007).
Karimi, M., Inzé, D. & Depicker, A. Gateway vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7, 193–195 (2002).
Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008).
Morris, G. M. et al. Auto Dock 4 and Auto Dock Tools 4: automated docking with selective receptor flexibility. J Comput Chem 30, 2785–2791 (2009).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J Mol Graph Model 14, 27–38 (1996).
Karnik, A., Karnik, R. & Grefen, C. SDM-Assist software to design site-directed mutagenesis primers introducing “silent” restriction sites. BMC Bioinformatics 14, 105 (2013).
Edelheit, O., Hanukoglu, A. & Hanukoglu, I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol 9, 61 (2009).
Acknowledgements
This research was financially supported by the National Key Research and Development Program of China (grant No. 2016YFD0101902), the National High Technology Research and Development Program “863” (grant No. 2013AA102602-4), the National Special Science Research Program of China (grant No. 2013CB967303), the National Natural Science Foundation of China (grant Nos 31270328, 31471515, 31200226, 31500232 and 31370337), the National Transgenic Project of China (grant No. 2013ZX08010002-002), the China Postdoctoral Science Foundation funded project (grant No. 2014M551893), the Science & Technology Plan of Shandong Province (grant No. 2013GNC11010), the Natural Science Foundation of Shandong Province (grant No. ZR2015PC009), and the Research Program for International S&T Cooperation Projects of Shandong Province (grant No. 2011176). We thank Doctor John Huge Snyder for his help in editing this manuscript.
Author information
Authors and Affiliations
Contributions
Y.L., Z.Z., Z.X. and F.X. designed research; Y.L., Z.Z., Z.X., L.W., P.W. and J.L. performed research; T.W., J.G. and Z.L. contributed new reagents/analytic tools; Z.Z., Z.X., Y.C. and S.L. analyzed data; Z.Z., Z.X., Y.C. and F.X. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Liu, Y., Zhao, Z., Xue, Z. et al. An Intronless β-amyrin Synthase Gene is More Efficient in Oleanolic Acid Accumulation than its Paralog in Gentiana straminea. Sci Rep 6, 33364 (2016). https://doi.org/10.1038/srep33364
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33364
This article is cited by
-
Functional characterization of the β-amyrin synthase gene involved in platycoside biosynthesis in Platycodon grandiflorum
Horticulture, Environment, and Biotechnology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
