
Abstract
Finding an interactive ligand-receptor pair is crucial to many applications, including the development of monoclonal antibodies. Biopanning, a commonly used technique for affinity screening, involves a series of washing steps and is lengthy and tedious. Here we present an approach termed continuous microfluidic assortment of interactive ligands, or CMAIL, for the screening and sorting of antigen-binding single-chain variable antibody fragments (scFv) displayed on bacteriophages (phages). Phages carrying native negative charges on their coat proteins were electrophoresed through a hydrogel matrix functionalized with target antigens under two alternating orthogonal electric fields. During the weak horizontal electric field phase, phages were differentially swept laterally depending on their affinity for the antigen and all phages were electrophoresed down to be collected during the strong vertical electric field phase. Phages of different affinity were spatially separated, allowing the continuous operation. More than 105 CFU (colony forming unit) antigen-interacting phages were isolated with ~100% specificity from a phage library containing 3 × 109 individual members within 40 minutes of sorting using CMAIL. CMAIL is rapid, sensitive, specific and does not employ washing, elution or magnetic beads. In conclusion, we have developed an efficient and cost-effective method for isolating and sorting affinity reagents involving phage display.
Similar content being viewed by others

Introduction
Characterization of interactions between molecule pairs (e.g., ligands to receptors, antibodies to antigens, enzymes to substrates and aptamers or drugs to their targets, etc.) provides the key to a wide spectrum of applications, including drug development, genetic engineering, biochemistry, biotechnology, molecular biology, etc.1,2,3,4,5,6. Phage display, in which the foreign amino acid sequence is genetically fused to the viral coat protein and displayed on the outer surface of the bacteriophage, provides a physical linkage of diverse phenotypes with their individual genotypes. Large libraries of proteins or peptides can be screened against target molecules, such as antigens or nucleic acids in a process analogous to natural selection. Phage display has been instrumental for conducting in vitro evolution and the development of targeted therapies, such as engineered monoclonal antibodies7,8,9.
However, the selection of phage libraries against target antigens remains a challenge and is most commonly achieved using the biopanning technique. Biopanning involves the immobilization of target molecules or antigens onto a solid support or magnetic beads followed by the incubation with the phage library. After extensive washing to remove nonadherent or weakly binding phages, bound phages are eluted and recovered using elution buffer and may be subsequently amplified in a bacteria host for further enrichment10,11,12. Three to five repeated rounds of incubation, washing, elution and amplification steps are usually carried out in order to obtain high-affinity phages, which can be quite laborious, lengthy and experimentally challenging for a number of reasons. First, non-specific phages are recovered when phage libraries are incubated with immobilized antigens. Second, removal of background by repeated washes is both labor-intensive and inefficient. Third, antigens and potential ligands can be lost during the washing steps required.
We reason that the sorting efficiency can be improved if the stochastic encounters between phages and antigens in the discrete steps of biopanning can occur in a much more frequent and controlled manner. Microfluidic technology confers advantages, including the precisely controlled fluidic environment and amiability to be automated and has been developed and applied for molecular separations13,14,15 and detections16,17,18. For example, it has been shown that microfluidic devices can provide a much more accurate and automatic handling of fluids in the biopanning procedure19,20,21,22,23,24. However, multiple rounds of biopanning are still needed to obtain desired performance and/or throughput. We have devised a method termed continuous microfluidic assortment of interactive ligands (CMAIL), exploiting the fact that most phage display work has used filamentous phage strains such as F1, Fd and M13 bacteriophages, which are closely related, small, of unique elongated shape and inherently negatively charged since the carboxyl-terminal of the major coat protein pVIII interacts with the virion DNA and is shielded from the solvent25,26,27. CMAIL allows continuous separation of antigen-interactive phages from remaining noninteractive phages; this is accomplished by differential electrophoresis under two alternating orthogonal electric fields that drive phages across the hydrogel matrix coated with antigens. A weaker electric field pulls noninteractive phages laterally, while a stronger electric field drives phages downstream to be collected at different locations as schematically shown in Fig. 1a.

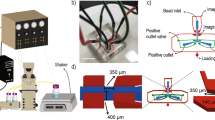
Operation and fabrication of the CMAIL device.
(a) Schematic rendition of the device; phages are loaded from the inlet and electrophoresed inside the agarose gel coated with target antigens under two alternating electric fields, EL and EV. EV is greater than EL. Negative, or antigen-noninteractive, phages migrate diagonally across the gel under both electric fields, while positive, or antigen-interactive, phages migrate only when the stronger electric field EV is applied. Phages are sorted and collected at different outlets continuously based on their affinity for the target antigen. (b) The PDMS inlet and outlets are replicated from a master mold made by using conventional lithography and are chemically bonded to a glass slide. Agarose gel is dispensed and cured in the center reservoir. (c) Image of the CMAIL device.
Alternately pulsed electric fields at an angle have been utilized to perform the size-based fractionation of DNA molecules in gels28 or an array of micropillars29. Larger DNA molecules have to reorient themselves in response to alternating electric fields and hence migrate less efficiently than smaller ones. In CMAIL, phages are of the same size, but are spatially separated based on their affinity for the target antigen. CMAIL, eliminating the need for repeated washing steps, can operate continuously. In addition, no elution of bound phages from a surface or a magnetic bead is required. Here we report in detail the performance of CMAIL using prescreened phage clones and the phage library. Our data compare favorably to results of biopanning using magnetic beads and suggest that selection antigen-interactive scFv using CMAIL may enable rapid identification of promising antibodies for targeted therapies among other applications.
Results
Immobilization of antigen molecules to the agarose gel is stable under the electric field
Agarose gels were functionalized with target antigen molecules for the sorting of interactive phages. The immobilization of antigens was achieved using hetero-crosslinking molecules whose ends were photo-reactive and amine-reactive, respectively. The conjugation of antigen molecules to the agarose gel was assessed using fluorescence-based readouts. We applied fluorescently labelled antibodies against the antigen to the functionalized agarose gel and operated the CMAIL device at an electric field of 7.4 V/cm, which was the highest intensity of electric field used in this study. As shown in Supplementary Figure 1, the fluorescence intensity remained stable for at least 120 minutes evaluated, suggesting stable immobilization of antigen molecules even under the electric filed. The initial drop in fluorescence intensity may be due to the removal of unbound antibodies by the electric field since there was no washing step conducted.
A stronger electric field is needed to electrophorese phages of higher affinity for the antigen
Filamentous phages have been shown to migrate through 2% agarose gels at pH 9.45 and the electrophoretic mobility is dependent mainly on the net charge of the pVIII coat protein for phages of the same size27,30. We examined the electrophoretic migration of M13 phages expressing fusion proteins through the 1% agarose gel at physiological pH, pH 7.4. Fluorescently labelled phage clone M+ (labelled green, antigen-interactive) and M− phages (labelled red, antigen-noninteractive) were loaded into the 1% agarose gel coated with target antigens. Fluorescent micrographs were utilized to visualize phages and suggested that both M+ and M− phages migrated toward the anode. An electric field of 3.6 V/cm or less was enough to drive M− phages, while 6.0 V/cm could drive M+ phages as shown in Supplementary Figure 2.
Phage numbers may be evaluated using nanoparticle tracking analysis (NTA)
Enumeration of phages has been demonstrated using the established technology of nanoparticle tracking analysis (NTA)31, which is more rapid than conventional phage titer analyses. NTA is an image-based method for sizing and concentration measurement of nanoscale particles. NTA provides the total count of particles under the scattering mode and has the capability to analyze fluorescently labeled particles under the fluorescence mode. We first compared phage counts obtained by using NTA and conventional phage titer analyses. Results, summarized in Supplementary Figure 3a, were in good agreement with each other for phage samples of concentrations ranging from 107 to 1011 CFU/mL. However, when the concentration of phages was below 107 CFU/mL, the number of particles per field of view was too low to be determined reliably using NTA. Next, we evaluated the concentration of fluorescently labeled phages. Phage counts obtained under the scattering mode and fluorescence mode of NTA were statistically insignificant (Supplementary Figure 3b), which validated the settings of NTA for counting fluorescently labeled phages. We then mixed together fluorescently-labeled M+ phage and unlabeled M− phage samples of known concentrations. The concentration of total phages was determined under the scattering mode, while fluorescently labelled phages were selectively identified under the fluorescence mode. The unlabeled phage count was subsequently calculated using the difference of the total phage count and the fluorescently labeled phage count and was comparable to the concentration of unlabeled M− phages measured before mixing (Supplementary Figure 3c). These results provide validation of NTA as a technique for counting both unlabeled and fluorescently-labeled phage subpopulations and was employed for assessing the performance of CMAIL.
Phage clones are sorted using CMAIL
The schematics and fabrication of the CMAIL device were shown in Fig. 1a,b, respectively. An image of the completed device was shown in Fig. 1c. We set out to study antigen binding by using CMAIL to test an established antigen-antibody fragment pair. Experiments were designed using cloned M+ and M− phages (displaying the antibody fragment selected to have, respectively, a high and low affinity for the antigen (vascular endothelial growth factor receptor 2 (VEGF-R2)). We reasoned that antigen-interactive M+ phage could interact with antigens coated in the gel strongly and migrate little under the weak horizontal electric filed, EL. Consequently, M+ phages would be recovered at outlet 1, which was directly downstream from the inlet. In contrast, antigen-noninteractive M− phages would move diagonally across the hydrogel under both electric field phases. Since phage virions collected at outlets were to be enumerated using NTA, M+ phages were fluorescently labeled and then mixed with M− phages at 1:10 ratio, or 109:1010 CFU/CFU. The sample mixture was introduced into a CMAIL device, in which two orthogonal electric fields, EL and EV, alternating every minute for 40 minutes were applied across the gel. EV was greater than EL and along the direction from inlet to outlet channels. As shown in Fig. 2a, outlet 1 collected the most phages and nearly all of them were M+ phages. Only a few M− phages were counted at outlets 4~9. A total of 8.5 × 108 CFU M+ phages were harvested from the outlets, which was equivalent to a recovery rate of 85%. In contrast, only 0.2%, or 2 × 107 CFU M− phages were collected at all outlets (Fig. 2b). To further confirm phage counts obtained by using NTA, conventional phage titer analyses and DNA sequencing were conducted. Recovered phages were isolated as described in Methods. The DNA sequences of randomly selected bacteria colonies were determined for 40 isolates; 10 each were infected from phages recovered in four outlets 1, 2, 3 and 6, respectively (Supplementary Figure 4). DNA sequence data, summarized in Table 1 and Supplementary Table 1, showed that all of the sequenced colonies infected with phages recovered at outlet 1 contained the sequence of M+ phages, while only 8 out 9 successfully sequenced colonies were positive for M+ phages from outlets 2, 3 and 6.

Antigen interactive (M+) and noninteractive (M−) phages were sorted into different outlets using CMAIL.
The input sample contained M+ and M− phages at concentrations of 109 and 1010 CFU/mL, respectively. (a) Most of the input M+ phages were recovered at outlet 1 and very few M− phages were collected at outlets 4~9. Phage counts of samples at outlets 2 and 3 were too low to be determined reliably using NTA. (b) Phage counts by using NTA indicated that more than 85% of input M+ phages were recovered, while less than 0.24% of input M− phages were collected at all the outlets combined.
More than 80% antigen-interactive phages are recovered when the occurrence is above 1/10,000
Antigen-interactive M+ phages were mixed with antigen-noninteractive M− phages at various ratios ranging from 1:10 to 1:108 to further assess the performance of CMAIL. Sorted phages were collected at each outlet and were counted separately using conventional phage titer techniques. Bacterial colonies infected by M+ phages were further confirmed using ELISA (see Method and Supplementary Figure 5b). As indicated in Fig. 3a and Supplementary Figure 5a, most phages were recovered at outlet 1, the outlet that was in-line with the inlet. Only a small fraction of phages were collected at other outlets. The efficiency, or the percentage of recovered M+ phages to input M+ phage remained to be greater than 80%, even when the ratio of input M+ to M− phage was 1:104. The efficiency decreased gradually with the lower mixing ratios and dropped to about 10% when the number of input M+ phages was 102 CFU. The selectivity, defined as 100% minus the percentage of harvested M− phages to input M− phages, was used to assess the capability of CMAIL to exclude antigen-noninteractive phages. As indicated in Fig. 3b, the selectivity was close to 100% for all the mixing ratios evaluated.

Sorting of antigen interactive (M+) and noninteractive (M−) phages using CMAIL is efficient and specific.
The input sample contained M+ phages ranging from 102 to 109 CFU and M− phages at a constant number of 1010 CFU. Hence, the mixing ratio of input M+ to M− phages ranged from 10−8 to 10−1 accordingly. Phages collected at outlets 1 to 9 were counted separately using conventional phage titer analyses (Supplementary Figure 5a). ELISA analysis was used to assess the percentage of bacterial colonies infected by M+ phages (Supplementary Figure 5b). (a) Most M+ phages were recovered at outlet 1 for the various mixing ratios of input M+ to M− phages evaluated. The efficiency, or the percentage of recovered M+ phages, remained greater than 80% when the mixing ratio was above 10−4 and dropped gradually to 10% when the mixing ratio was 10−8. (b) The specificity, or 100% minus the percentage of recovered M− phages, remained close to 100% for samples at all the outlets and mixing ratios evaluated.
Phages were enriched at different outlets based on their affinity
We wondered if GMAIL may sort phages into different populations based on their affinity for the antigen. To test, we chose three phage clones having a high (H), intermediate (I) and low (L) affinity for mouse serum albumin (MSA), respectively. The affinity of these phage clones, H-phage, I-phage and L-phage, was gauged using ELISA and the absorbance at 450 nm was 2.7, 1.4 and 0.45 for H-phage, I-phage and L-phage. These phage clones were mixed with M− phages at 1:10 ratio (109:1010 CFU/CFU) and loaded into and sorted in separate CMAL devices with their agarose gel coated with either the target antigen, MSA, or the control antigen, VEGF-R2. The BSA blocking step during the preparation of agarose gels was omitted in this set of experiments. As shown in Fig. 4, only scarce antigen-interactive phages were collected when the device was coated with the control antigen molecules, VEGF-R2, suggesting the CMAIL sorting was specific. Positive colony counts were calculated as the product of the colony count (Supplementary Figure 6a) and the ratio of positive ELISA result (Supplementary Figure 6b), which were proportional to the phage concentration and the percentage of antigen-interactive phages, respectively. More phages were recovered when the agarose gel of the CMAIL device was coated with target antigen molecules. Most of the H-phages were concentrated and recovered at outlet 1 with an efficiency of 93%, similar to the efficiency obtained when M+ phages and corresponding target antigens were used in CMAIL. Most of the I-phages were recovered at outlet 6 with an efficiency of 68%. In contrast, when L-phage was evaluated, the phage counts remained low at all the outlets and the total phage count was less than 0.001% to its input count. In summary, phages were sorted and recovered at different outlets of the CMAIL device based on their affinity for the antigen immobilized in the agarose gel. Phages with a strong affinity were highly concentrated at outlet 1, while phages of weaker affinity were collected at outlets further along the direction of the lateral electric field EL, or even largely swept out of the device and not retrieved.

Phage clones of different affinity for the antigen are spatially separated in the CMAIL device.
Three phage clones, H-phage, I-phage and L-phage, are of relative high, intermediate and low affinity for the mouse serum albumin (MSA), respectively. Phage clones were mixed with M− phages at 1:10 ratio, or 109:1010 CFU/CFU and introduced into CMAIL devices. Phages sorted and collected from outlets 1 to 9 were counted separately using conventional phage titer analyses and analyzed using ELISA to confirm antigen interactivity. Only scarce antigen-interactive phages were recovered in the CMAIL device coated with control antigen molecules. In contrast, H-phages were recovered with high efficiency at outlet 1, while I-phages were collected at outlet 6 when target antigens were immobilized to the agarose gel of the device. Only few L-phages were recovered after CMAIL sorting.
Five repeated CMAIL operation cycles are possible using the same device
Antigens immobilized in the agarose gel may remain present and viable under the electric field as suggested by the stable fluorescence intensity (Supplementary Figure 1). We wondered if we may reuse a CMAIL device. Phage samples were introduced into the same CMAIL device repeatedly to assess how long we can operate the device continuously without a decline in performance. Antigen-interactive M+ phages and antigen-noninteractive M− phages were mixed at 1:10 ratio (109:1010 CFU/CFU). Each cycle consisted of three steps: 1) loading of the phage sample mix into the device, 2) CMAIL sorting for 40 minutes and 3) collection of samples at each outlet. Five cycles were repeated on the same CMAIL device. Collected phages samples were subjected to conventional phage titer analyses and ELISA. Results, summarized in Fig. 5, indicated that most M+ phages were recovered at outlet 1 and the percentage of M+ phages was above 90% for all the 5 repeated cycles evaluated, suggesting the CMAIL device could be operated continuously for at least 200 minutes.

CMAIL devices can be operated for 5 repeated sorting cycles.
M+ and M− phages were mixed at a ratio of 109:1010 CFU/CFU. Five continuously repeated CMAIL operation cycles, consisting of sample loading, 40-minute sorting, sample harvesting, were conducted using the same devices. (a) Similar colony counts of phages collected from 9 individual outlets for 5 repeated CMAIL sorting cycles were obtained. (b) ELISA analysis was used to assess the percentage of bacterial colonies infected by M+ phages and similar results were obtained from samples collected in 5 repeated CMAIL sorting cycles, suggesting the CMAIL device could be operated continuously for at least 200 minutes.
CMAIL sorting of a phage library is of favorable recovery rate and purity
A phage library containing 3 × 109 individual members was screened against the same antigen (VEGF-R2) using both CMAIL and magnetic bead biopanning techniques. 1010 CFU phages were introduced into CMAIL chips and sorted for 40 minutes. Phages collected at the outlets were subjected to conventional phage titer analyses and ELISA. As shown in Fig. 6a,b, most phages were collected from outlets 1 to 4 with percent positive values above 85% as gauged using ELISA. The percent positive value was 100% for phages validated from outlet 1. The same phage library was also subjected to three rounds of biopanning using magnetic beads. Eluted phages were bacterial amplified between rounds of biopanning. As shown in Fig. 6c, the percent recovery rates of the initial capture was ~18 times higher using CMAIL than using bead biopanning. The percent recovery rate increased in successive rounds of bead biopanning, indicating the enrichment and amplification of antigen-interactive phages. The percent positive value was ~100% for phages from the outlet 1 of the CMAIL chip, which was significantly higher than that of the first round of bead biopanning (Fig. 6d). The percent positive values improved in the second and third rounds of bead biopanning. The numbers of input and output phages were summarized in Fig. 6e.

Comparison of CMAIL sorting vs. magnetic bead-based biopanning a phage library.
A phage library was screened using both CMAIL chips (n = 3) and magnetic beads against the same antigen (VEGF-R2). The interactivity of harvested phages to antigens was assessed using ELISA. (a) More than 99.99% of antigen-interactive phages harvested were collected from outlets 1 to 4 of the CMAIL chip. (b) More than 94.18% of phages collected from outlets 1 to 4 were antigen-interactive, while phages obtained from outlet 1 were 100% antigen-interactive as gauged by ELISA. (c) Percent recovery values were obtained over one round of CMAIL sorting and three rounds of biopanning using magnetic beads. Percent recoveries were determined by the number of harvested phages divided by the number of input phages. Eluted phages were bacterial amplified between rounds of biopanning and 2.0 × 1011 CFU phages were screened for each round of bead-based biopanning. (d) Percent positive values were determined by ELISA. The percent positive value was ~100% for phages from the outlet 1 of the CMAIL chip, which was significantly higher than that of the first round of bead biopanning. The percent positive values improved in the second and third rounds of bead-based biopanning. The percent positive values improved in the second and third rounds of bead-based biopanning. (e) The summary of phage counts suggested that similar number of antigen-interactive phages were selected using CMAIL chips and magnetic beads, while only ~1/18 input phages were required using CMAIL.
Discussion
Two orthogonal and alternative electric fields, EV and EL, are utilized to direct phages to navigate through the matrix of agarose fibers functionalized with target antigens in CMAIL. A stronger electric field EV was used to drive all the phages toward outlets and the electric field EL pulled phages laterally and differentially based on their affinity for the antigen. A tug-of-war between the electrophoretic force and the binding force between the antigen and the fusion protein was established on phages in CMAIL. Phages with a strong affinity moved little, while phages with a weaker affinity were pulled laterally under the electric filed EL, resulting in the spatial separation of phages based on their affinity. Both the electric fields and the gel matrix of antigens significantly increased the number of interactions between phages and antigens and hence the efficiency of CMAIL. In addition, phages are sorted into multiple populations within a single CMAIL cycle and the stringency of sorting can be precisely and conveniently adjusted by modulating the electric fields. In contrast, only two populations of phages, either bound or unbound, are obtained in biopanning. Multiple rounds of biopanning are necessary to sort phages with different stringency, if desired.
M13 phages are filamentous with 6.6 nm in diameter and 880 nm in length32. Comparing to other phages that may take shapes from spherical to sophisticated head-tail structure, M13 phages are used most commonly for phage display and are particularly well-suited for CMAIL, as their highly anisotropic shape enables them to pass through pores in the hydrogel with less entrapment. Gel electrophoresis has been employed to measure the mobility of phages and to sort different phage strains using 2% agarose gel at pH 9.530. It has been found that their mobility was largely determined by the length of the phage and the net charge on its surface. In the present study, the length of phages should be similar, since phages expressing fusion proteins contain the same size phagemid. The net charges on the surface of phages are largely determined by the major coat protein pVIII, since there are ~2700 copies per phage, outnumbering the fusion proteins. Therefore, CMAIL sorts phages mainly based on their affinity, not size or surface charge.
Continuous spatial separation. or deterministic lateral displacement, of cells33, particles34 and DNA molecules29 based on their size in an array of micropillars have been demonstrated. The interactions between the object under sorting and the micropillar can be predicted and manipulated. Hence the sorting can be accomplished much faster and with a relatively high degree of precision, compared with sorting in structure-free space or inside the gel matrix. The performance of CMAIL could be further improved by incorporating micro- or nano-pillars in the device, which is being further investigated.
Both NTA and conventional phage titer analyses have been used for assessing the concentration of phages. NTA counts phages directly based on their scattering light or fluorescence images. In contrast, phage titer analysis counts colonies of bacteria infected by phages, which is indirect, lengthy and more susceptible to errors. However, the sensitivity of both methods is limited by the number of counts per field of view or per culture plate, which is 107 and 103 counts/mL, respectively. Therefore, when phages collected at the outlet of the CMAIL device are below the detection limit, the estimated phage counts may be less reliable. Resistive-pulse sensing, detecting particles upon their passing through a pore, has been investigated to enumeration phages35. The method is real-time and label free. However, challenges associated with pore clogging and the agglomeration and coincident passing of phages have to be overcome.
ELISA has been typically used for assessing the outcome of biopanning procedure. However, it only confirms positive binding interactions, since its signal depends not only on the binding affinity, but also the heterogeneity of phage preparation as well as the level and avidity of scFv display. Further evaluation of selected clones involves DNA sequencing, scFv synthesis, as well as competitive ELISA, surface plasmon resonance or other comparable techniques for measuring the binding affinity and kinetics36. This approach is costly, time-consuming and restricts the number of clones that can be evaluated. Several work on the developments of high-throughput assessment of binding affinities has been reported and remained being assessed37,38.
Biopanning has been implemented using both plate and magnetic bead formats with different performance benchmarks, cost and time required39,40,41,42. Typically magnet beads may provide increased surface area for binding and thoroughness of washing and hence better efficiency than plate biopanning36,39,43. Compared with the initial capture efficiency of bead biopanning, the recovery rate of CMAIL is more than an order of magnitude higher. In addition, CMAIL provides a high percentage of antigen-interactive phages immediately after the initial sorting, which can effectively reduce the number of rounds and the chance of repetitive isolation of identical phages augmented from bacterial amplification. Repetitive sorting of identical phages not only waste time and resources, but also may obscure the isolation of low-abundant but valuable phage clones. In conclusion, CMAIL is an automated high-throughput platform capable of sorting phages based on their affinity for the antigen. Compared with the biopanning technique, CMAIL is rapid, cost-effective and of low reagent consumption and appears to be an approach of choice for affinity sorting of phage-display libraries.
Methods
Microfluidic device design and fabrication
The central part of the microfluidic device used in this study was an open 22 mm × 22 mm agarose gel-filled reservoir connected to a 100-μm-wide inlet on one side and nine 1.6-mm-wide outlet channels on the opposite side. The inlet and outlets were all 100 μm deep and made in polydimethylsiloxane (PDMS; Dow Corning, Midland, MI, USA) replicated from a mold fabricated in the photoresist SU-8 (MicroChem, Newton, MA, USA) using a conventional lithography process. The PDMS replica and the glass slide were bonded together after treated with oxygen plasma (18 W, 1% oxygen, 30 s) in a plasma chamber (Harrick Plasma, Ithaca, NY, USA).
Preparation and modification of agarose hydrogels
Target antigen molecules were immobilized to agarose hydrogels for the selection of interactive phages. In this work, mouse serum albumin (MSA) molecules or vascular endothelial growth factor receptor 2 (VEGF-R2) fragments were linked to agarose via a heterobifunctional photo-crosslinker, sulfosuccinimidyl-6-[4′-azido-2′-nitrophenylamino] hexanoate (sulfo-SANPAH; Merck, Darmstadt, Germany), based on the method described44. Briefly, 35 μL sulfo-SANPAH solution at 50 μg/mL in 4-(2-hydroxyethyl)-1-piperazine-ethane sulfonic acid buffer (HEPES; 50 mM, pH 8.5) was mixed with 100 μL 1% (w/v) agarose solution (Sigma Aldrich, St. Louis, MO, USA). The mixture was dispensed onto the open reservoir of the device and exposed to 365 nm UV light for 3 minutes to conjugate sulfo-SANPAH to the agarose. 100 μL solution containing antigen molecules at 8 μg/mL in phosphate buffered saline (PBS; Mediatech, Herndon, VA, USA) was subsequently added to the agarose gel and allowed to react with the NHS-ester group of the sulfo-SANPAH at 4 °C for 24 h. The ligand-modified agarose gel was incubated in 100 μL 3% (w/v) bovine serum albumin (BSA; Sigma Aldrich, St. Louis, MO, USA) solution in PBS at 37 °C for 30 minutes before use.
Microfluidic sorting of phage clones
Cloned antigen-interactive M+ phages were fluorescently labeled for optical measurements and mixed with antigen-noninteractive M− phages at various ratios before being introduced into the inlet of the CMAIL device. The CMAIL device was soaked in TBE (Tris-borate-EDTA) buffer during operation. Alternating orthogonal electric fields EL and EV were generated by using a programmable two-channel DC power supply (Keysight Technologies, Santa Rosa, CA). 15 V and 20 V, corresponding to electric fields of 3.0 V/cm and 7.4 V/cm, were applied alternatively every one minute for 40 minutes to the lateral and vertical pairs of electrodes, respectively. EV was greater and along the direction from inlet to outlets. Phages at the outlets were collected and subjected to nanoparticle tracking analysis (NTA) or bacterial amplification.
Nanoparticle tracking analysis (NTA)
Phage samples of concentration ranging from 107 to 1011 CFU/mL were introduced into the sample chamber of the NTA instrument (Model LM10 equipped with a 405 nm diode, NanoSight, Amesbury, UK). A 90-second video of scattering microscopy images was captured for each sample to determine the concentration of phages. In experiments when samples contained both M+ and M− phages, M+ phages were fluorescently labelled (Goat anti-Mouse IgG (H + L) secondary antibody; Life technologies, Carlsbad, CA) and a DAPI filter cube was used. Videos of both scattering and fluorescence microscopy images were captured for each sample to determine concentrations of the total and fluorescently-labelled phages, respectively.
Bacterial amplification
The bacterial strain E. coli TG1 was used as the host. Bacteria were grown in 2-YT medium overnight at 30 °C with shaking at 250 r.p.m. until the measured optical density at 600 nm (OD600) had reached the value of 0.5. 1 mL of bacterial culture was then incubated with sorted phage (carrying ampicillin resistance) for 30 minutes at 37 °C without shaking. Bacteria were then serial-diluted and inoculated onto 2-YT-AG (2-YT ampicillin-glucose) agar plates. Bacteria were allowed to grow on the plate for 16 h at 37 °C. Bacterial colonies, containing the ampicillin resistance gene from sorted phages, were picked randomly for DNA sequencing or ELISA (enzyme-linked immunosorbent assay).
DNA sequencing
Bacterial colonies after the bacterial amplification procedure were randomly picked for sequencing. PCR-amplified library inserts from the pIII regions of the phage genome were used for sequencing with the forward primer, 5′-CCA TGA TTA CGC CAA GCT TTG GAG CC-3′; and reverse primer, 5′-CGA TCT AAA GTT TTG TCG TCT TTC C-3′. DNA sequences of the variable regions of the complementary determining regions (CDRs), including CDRH1~3 and CDRL1~3, were extracted, aligned and translated into amino acid sequences for analysis.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was carried out to analyze the binding affinity of clonal amplified phages. E. coli TG1 that produces pIII phage proteins under the control of the wild-type lac promoter was maintained at 37 °C, 250 r.p.m unless otherwise noted during ELISA. Bacterial colonies after the bacterial amplification procedure were randomly picked and grown in 2 YT-AG medium overnight. Each culture was subsequently diluted in 1/16 with 2-YT-A (2-YT ampicillin) medium and re-grown for 2 h. Subsequently, each sample was infected with helper phages (carrying kanamycin resistance) at a final concentration of 4.8 × 109 CFU/mL in 2YT-A medium for 2 h before kanamycin was added to a final concentration of 25 μg/mL. The bacteria were allowed to grow overnight at 30 °C. 96-well immunoplates for ELISA were coated with 100 μL/well of the solution containing 4 μg/mL antigen molecules (e.g., VEGF-R2 fragments) in PBS for 16 h at 4 °C and subsequently rinsed with 300 μL/well of PBS for three times. The immunoplates were then incubated with 250 μL/well of blocking buffer containing 5% skim milk in PBS (MPBS) for 2 h at 37 °C, 250 r.p.m before rinsed with 300 μL/well of PBS for three times. The phage suspension diluted in 1% MPBS were centrifuged at 8000 × g, 4 °C for 30 min before 100 μL/well was applied into each well and incubated at 37 °C for 1 h with shaking at 250 r.p.m. The immunoplate was rinsed with 300 μL/well of PBS containing 0.05% Tween-20 (PBST) for three times to remove loosely bound phages. Phage were detected with the monoclonal HRP/anti-M13 antibody solution (100 μL/well, 1:1,000 diluted in 5% MPBS) added to wells and incubated at 37 °C for 1 h. The immunoplate was rinsed with 300 μL/well of PBST three times. TMB, a substrate of HRP, was used for detection and the enzymatic reaction was terminated with 1.0 N sulfuric acid. The ligand-binding activity of phages was quantified by measuring the absorbance at 450 nm with a reference absorbance at 655 nm. The threshold for a positive ELISA result was set to be five times the background intensity.
Biopanning of phage library
Phages were subjected to both CMAIL and a magnetic bead biopanning procedure. The phage library was prepared as described36. Briefly, E. coli TG1 cells were transformed with library DNAs by electroporation and allowed to grow to log phase. TG1 bacteria were infected with Hyperphage and incubated at 37 °C for 30 minutes. Bacteria were then spun down, resuspended in 2-YT medium containing 100 μg/mL ampicillin and 25 μg/mL kanamycin and grown overnight at 30 °C with shaking at 250 r.p.m. Phages in the supernatant were precipitated with a PEG/NaCl buffer (20% polyethylene glycol 8000, 2.5 M NaCl) and resuspended in PBS. The phage library (1010 CFU) were introduced into the inlet of the CMAIL device and sorted for 40 minutes. Phages at the outlets were collected and subjected to bacterial amplification and ELISA. For bead biopanning, the phage library was first pre-incubated with cobalt-coated paramagnetic beads to reduce the number of non-specific binding phages in the library and subjected to three rounds of biopanning. Next, the phage library was incubated with 50 nM histidine-tagged (His-Tag) VEGF-R2 with gentle rotation at 37 °C for 90 minutes. Phages bound with VEGF-R2 were isolated using cobalt coated-magnetic beads that were blocked with 2% MPBS. The beads were washed sequentially with PBST two times, 2% MPBS, PBST two times, 2% MPBS, PBS two times, 2% MPBS and PBS two times, The bound phages were eluted and bacterial amplified for the next round of biopanning as described above.
Materials and reagents
The mouse serum albumin (MSA) molecules, vascular endothelial growth factor receptor 2 (VEGF-R2) fragments, M13 bacteriophages and fluorophore-conjugated anti-M13 antibodies were provided by the Development Center for Biotechnology (New Taipei City, Taiwan)36. 2-YT medium containing yeast extract, tryptone and sodium chloride for the cultivation of M13 bacteriophage and Escherichia coli (E. coli) were obtained from Amresco (Solon, OH, USA). For conducting ELISA, 96-well immunoplates (Nunc, Rochester, NY, USA) and horseradish peroxidase labeled anti-M13 phage monoclonal antibody (HRP/anti-M13 antibody, GE27-9421-01) were from Sigma Aldrich (St Louis, MO, USA). The substrate for HRP was 3,3′.5,5′ tetramethylbenzidine (TMB) mixed with a stabilized hydrogen peroxide (1-Step Turbo TMB-ELISA, Pierce Biotechnology, Rockford, IL, USA). Cobalt-coated paramagnetic beads were obtained from Invitrogen (Dynabeads His-Tag isolation and pulldown beads, Carlsbad, CA, USA). Hyperphage was obtained from Progen (M13 K07∆pIII, Heidelberg, Germany).
Additional Information
How to cite this article: Hsiao, Y.-H. et al. Continuous microfluidic assortment of interactive ligands (CMAIL). Sci. Rep. 6, 32454; doi: 10.1038/srep32454 (2016).
References
Xu, W., Lucke, A. J. & Fairlie, D. P. Comparing sixteen scoring functions for predicting biological activities of ligands for protein targets. J. Mol. Graph. Model. 57, 76–88, 10.1016/j.jmgm.2015.01.009 (2015).
Umlauf, B. J., McGuire, M. J. & Brown, K. C. Introduction of plasmid encoding for rare tRNAs reduces amplification bias in phage display biopanning. BioTechniques 58, 81–84, 10.2144/000114256 (2015).
Chin, S. E. et al. Isolation of high-affinity, neutralizing anti-idiotype antibodies by phage and ribosome display for application in immunogenicity and pharmacokinetic analyses. J. Immunol. Methods. 416, 49–58, 10.1016/j.jim.2014.10.013 (2015).
Yang, M. et al. Phage-display library biopanning and bioinformatic analysis yielded a high-affinity peptide to inflamed vascular endothelium both in vitro and in vivo. J. Control. Release. 174, 72–80, 10.1016/j.jconrel.2013.11.009 (2014).
Ebrahimizadeh, W. & Rajabibazl, M. Bacteriophage vehicles for phage display: biology, mechanism and application. Curr. Microbiol. 69, 109–120, 10.1007/s00284-014-0557-0 (2014).
Lee, B. H. & Kwon, T. H. Application of phage display for ligand peptidomics to identify peptide ligands binding to AQP2-expressing membrane fractions. Method. Mol. Biol. 1023, 181–189, 10.1007/978-1-4614-7209-4_12 (2013).
Becker, M. et al. Application of M13 phage display for identifying immunogenic proteins from tick (Ixodes scapularis) saliva. BMC Biotechnol. 15, 43, 10.1186/s12896-015-0167-3 (2015).
Padmanaban, G. et al. Identification of peptides that selectively bind to myoglobin by biopanning of phage displayed-peptide library. J. Biotechnol. 187, 43–50, 10.1016/j.jbiotec.2014.07.435 (2014).
t Hoen, P. A. et al. Phage display screening without repetitious selection rounds. Anal. Biochem. 421, 622–631, 10.1016/j.ab.2011.11.005 (2012).
Lang, Q. et al. Specific probe selection from landscape phage display library and its application in enzyme-linked immunosorbent assay of free prostate-specific antigen. Anal. Chem. 86, 2767–2774, 10.1021/ac404189k (2014).
Ellis, S. E., Newlands, G. F., Nisbet, A. J. & Matthews, J. B. Phage-display library biopanning as a novel approach to identifying nematode vaccine antigens. Parasite Immunol. 34, 285–295, 10.1111/j.1365-3024.2011.01317.x (2012).
Zahid, M. et al. Identification of a cardiac specific protein transduction domain by in vivo biopanning using a M13 phage peptide display library in mice. PloS One 5, e12252, 10.1371/journal.pone.0012252 (2010).
Persson, J., Augustsson, P., Laurell, T. & Ohlin, M. Acoustic microfluidic chip technology to facilitate automation of phage display selection. FEBS J. 275, 5657–5666, 10.1111/j.1742-4658.2008.06691.x (2008).
Melin, J. & Quake, S. R. Microfluidic large-scale integration: the evolution of design rules for biological automation. Annu. Rev. Bioph. Biom. 36, 213–231, 10.1146/annurev.biophys.36.040306.132646 (2007).
Bessette, P. H., Hu, X., Soh, H. T. & Daugherty, P. S. Microfluidic library screening for mapping antibody epitopes. Anal. Chem. 79, 2174–2178, 10.1021/ac0616916 (2007).
Charles, P. T. et al. Multi-channeled single chain variable fragment (scFv) based microfluidic device for explosives detection. Talanta 144, 439–444, 10.1016/j.talanta.2015.06.039 (2015).
Wang, Y. et al. Duplex microfluidic SERS detection of pathogen antigens with nanoyeast single-chain variable fragments. Anal. Chem. 86, 9930–9938, 10.1021/ac5027012 (2014).
Vistas, C. R. et al. An amorphous silicon photodiode microfluidic chip to detect nanomolar quantities of HIV-1 virion infectivity factor. Analyst 139, 3709–3713, 10.1039/c4an00695j (2014).
Cung, K. et al. Rapid, multiplexed microfluidic phage display. Lab Chip 12, 562–565, 10.1039/c2lc21129g (2012).
Liu, Y. et al. Controlling the selection stringency of phage display using a microfluidic device. Lab Chip 9, 1033–1036, 10.1039/b820985e (2009).
Wang, J. P. et al. Selection of phage-displayed peptides on live adherent cells in microfluidic channels. P. Natl. Acad. Sci. USA 108, 6909–6914, 10.1073/pnas.1014753108 (2011).
Wang, C. H., Weng, C. H., Che, Y. J., Wang, K. & Lee, G. B. Cancer cell-specific oligopeptides selected by an integrated microfluidic system from a phage display library for ovarian cancer diagnosis. Theranostics 5, 431–442, 10.7150/thno.10891 (2015).
Che, Y. J. et al. An integrated microfluidic system for screening of phage-displayed peptides specific to colon cancer cells and colon cancer stem cells. Biomicrofluidics 9, 10.1063/1.4933067 (2015).
Park, H. S., Lee, W. & Nam, Y. S. Elution dynamics of M13 bacteriophage bound to streptavidin immobilized in a microfluidic channel. Biochip J. 10, 48–55, 10.1007/s13206-016-0107-x (2016).
Smith, G. P. & Petrenko, V. A. Phage display. Chem. Rev. 97, 391–410, 10.1021/cr960065d (1997).
Marvin, D. A. & Hohn, B. Filamentousbacterial viruses. Bacteriol. Rev. 33, 172–209 (1969).
Boeke, J. D., Russel, M. & Model, P. Processing of filamentous phage pre-coat protein - effect of sequence variations near the signal peptidase cleavage site. J. Mol. Biol. 144, 103–116, 10.1016/0022-2836(80)90027-3 (1980).
Schwartz, D. C. & Cantor, C. R. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel-electrophoresis. Cell 37, 67–75, 10.1016/0092-8674(84)90301-5 (1984).
Bakajin, O. et al. Separation of 100-kilobase DNA molecules in 10 seconds. Anal. Chem. 73, 6053–6056, 10.1021/ac015527o (2001).
Moses, P. B., Boeke, J. D., Horiuchi, K. & Zinder, N. D. Restruturing the bacteriophage f1 genome - expression of gene 8 in the intergenic space. Virology 104, 267–278, 10.1016/0042-6822(80)90332-3 (1980).
Carr, B., Siupa, A., Hole, P., Malloy, A. & Hannell, C. Nanoparticle tracking analysis for the rapid and direct visualisation, sizing and counting of viruses, phage and VLPs in suspension. Hum. Gene Ther. 23, A8–A9 (2012).
Dogic, Z. & Fraden, S. Smectic phase in a colloidal suspension of semiflexible virus particles. Phys. Rev. Lett. 78, 2417–2420, 10.1103/PhysRevLett.78.2417 (1997).
Karabacak, N. M. et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 9, 694–710, 10.1038/nprot.2014.044 (2014).
Huang, L. R., Cox, E. C., Austin, R. H. & Sturm, J. C. Continuous particle separation through deterministic lateral displacement. Science 304, 987–990, 10.1126/science.1094567 (2004).
Lee, C. Y. et al. In Solid-State Electronics and Photonics in Biology and Medicine, Vol. 64 ECS Transactions (ed. Wang, Y. L. ) 51–56 (2014).
Lai, Y. D. et al. Generation of potent anti-vascular endothelial growth factor neutralizing antibodies from mouse phage display library for cancer therapy. Int. J. Mol. Sci. 17, doi: 10.3390/ijms17020214 (2016).
Gonzalez-Techera, A. et al. High-throughput method for ranking the affinity of peptide ligands selected from phage display libraries. Bioconjugate Chem. 19, 993–1000, 10.1021/bc700279y (2008).
Lad, L. et al. High-throughput kinetic screening of hybridomas to identify high-affinity antibodies using bio-layer interferometry. J. Biomol. Screen. 20, 498–507, 10.1177/1087057114560123 (2015).
McConnell, S. J., Dinh, T., Le, M. H. & Spinella, D. G. Biopanning phage display libraries using magnetic beads vs. polystyrene plates. BioTechniques 26, 208–214 (1999).
Walter, G., Konthur, Z. & Lehrach, H. High-throughput screening of surface displayed gene products. Comb. Chem. High T. Scr. 4, 193–205 (2001).
Konthur, Z., Wilde, J. & Lim, T. S. In Antibody Engineering 2nd edn, Vol 1 (eds Kontermann, R. & Dubel, S. ) Ch. 18, 267–287 (Springer-Verlag, 2010).
Krebs, B. et al. High-throughput generation and engineering of recombinant human antibodies. J. Immunol. Methods 254, 67–84, 10.1016/s0022-1759(01)00398-2 (2001).
Kala, M., Bajaj, K. & Sinha, S. Magnetic bead enzyme-linked immunosorbent assay (ELISA) detects antigen-specific binding by phage-displayed scFv antibodies that are not detected with conventional ELISA. Anal. Biochem. 254, 263–266, 10.1006/abio.1997.2378 (1997).
Dodla, M. C. & Bellamkonda, R. V. Anisotropic scaffolds facilitate enhanced neurite extension in vitro. J. Biomed. Mater. Res. A 78, 213–221 (2006).
Acknowledgements
We thank T.L. Shen, who is with the National Taiwan University for optical measurements. This work was supported in part by the Taiwan Ministry of Science and Technology grants MOST 104-2221-E-007-143 (C.C.) and 105-2221-E-007-009 (C.C.).
Author information
Authors and Affiliations
Contributions
C.C. and Y.-H.H. conceived of the study and analyzed data. C.C., Y.-H.H., C.-Y.H., C.-Y. H., C.-H.W. and C.-H.H. designed experiments. Y.-H.H. performed experiments. Y.-Y.W performed bead biopanning experiments. C.C. and Y.-H.H. wrote the manuscript with input from all authors.
Ethics declarations
Competing interests
C.C. and Y-.H.H. are inventors on a patent application related to microfluidicbased sorting of phages.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hsiao, YH., Huang, CY., Hu, CY. et al. Continuous microfluidic assortment of interactive ligands (CMAIL). Sci Rep 6, 32454 (2016). https://doi.org/10.1038/srep32454
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep32454
This article is cited by
-
A mini review on recent progress of microfluidic systems for antibody development
Journal of Diabetes & Metabolic Disorders (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
