
Abstract
Genetic control aims to reduce the ability of insect pest populations to cause harm via the release of modified insects. One strategy is to bias the reproductive sex ratio towards males so that a population decreases in size or is eliminated altogether due to a lack of females. We have shown previously that sex ratio distortion can be generated synthetically in the main human malaria vector Anopheles gambiae, by selectively destroying the X-chromosome during spermatogenesis, through the activity of a naturally-occurring endonuclease that targets a repetitive rDNA sequence highly-conserved in a wide range of organisms. Here we describe a CRISPR-Cas9 sex distortion system that targets ribosomal sequences restricted to the member species of the Anopheles gambiae complex. Expression of Cas9 during spermatogenesis resulted in RNA-guided shredding of the X-chromosome during male meiosis and produced extreme male bias among progeny in the absence of any significant reduction in fertility. The flexibility of CRISPR-Cas9 combined with the availability of genomic data for a range of insects renders this strategy broadly applicable for the species-specific control of any pest or vector species with an XY sex-determination system by targeting sequences exclusive to the female sex chromosome.
Similar content being viewed by others


Introduction
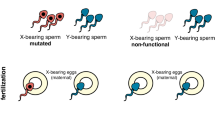
Since females largely determine the reproductive capacity of most pest species, a very attractive genetic control strategy is to distort the male-to-female reproductive sex ratio in favour of males, whereby the progressive reduction of females is anticipated to cause a dramatic contraction of the population and eventually its elimination. A synthetic sex-ratio distortion trait has been generated previously in the malaria mosquito Anopheles gambiae by expressing during the process of spermatogenesis the His Cys box endonuclease I-PpoI that cleaves a conserved sequence within the ribosomal DNA (rDNA) repeats found solely on the X-chromosome1,2,3. By restricting endonuclease activity to spermatogenesis, X-bearing sperm were selectively destroyed and eggs were predominantly fertilised by Y-bearing sperm destined to produce males3. X-shredding, unlike female-killing, operates meiotically therefore no significant reduction in male fertility is incurred. Our results thus provide the foundation for the genetic control of a host of heterogametic pest species with an XY sex-determination system. However, approaches based on the use of the endonuclease I-PpoI can be exclusively applied to a limited number of mosquito species where rDNA genes are located on the X-chromosome. In the vast majority of insects the rDNA genes are not localised on a single X-specific cluster and therefore a more flexible endonuclease platform is needed to enable the targeting of alternative X-chromosome sequences for each species.
Here we describe the first functional CRISPR-Cas9 sex-distortion system (CRISPRSD) in the malaria mosquito A. gambiae. To demonstrate the feasibility and effectiveness of this approach we utilized the CRISPR-Cas9 nuclease to target an X-linked rDNA sequence that is different from the previously utilized I-PpoI target site and conserved among the malaria vector species of the A. gambiae complex: A. arabiensis, A. gambiae, A. bwambae, A. melas and A. merus4,5, yet absent from more distantly related insects. The alignment of 28S rDNA sequences revealed a number of potential target regions meeting these criteria and of these, a single guide RNA (gRNA-T1) was chosen for its lack of predicted off-target sites in A. gambiae (Fig. 1b, Supplementary Fig. S1).

Generation of a CRISPR-Cas9 sex-ratio distortion system.
(a) CRISPRSD transformation construct. (pBac) piggyBac inverted repeats; (3xP3::DsRed) Pax promoter driving the DsRed marker to select gene integration events; (β2::Cas9) Streptococcus pyogenes Cas9 nuclease under the control of the male germline specific β2 tubulin promoter; (U6::gRNA) gRNA under the control of the ubiquitous U6 Pol III promoter. (b) Schematic representation of rDNA clusters within the Anopheles gambiae X-chromosome and the location of the multicopy gRNA target site. By shredding the X-chromosome during meiosis it is predominantly Y-bearing sperm that fertilize eggs generating a male-biased progeny.
We designed a germline transformation construct where the Cas9 endonuclease coding sequence was placed under the transcriptional control of the spermatogenesis-specific β2 tubulin promoter6. This allowed us to restrict the endonuclease activity to male meiosis despite the likely ubiquitous expression of gRNA-T1 driven by the Pol III promoter of the mosquito U6 snRNA gene7,8 (Fig. 1a). The CRISPRSD construct was contained within a piggyBac transformation vector to allow transposon-mediated insertion into the genome. A total of 4 transgenic lines each containing a unique genomic insertion of the construct were generated as previously described9. Single chromosomal integrations were selected and their respective genomic positions determined (Supplementary Table S2).
We then assessed whether male germline expression of the CRISPRSD unit would prevent the transmission of the X-chromosome to their progeny and thereby generate an excess of males. All the CRISPRSD lines showed a strong sex-ratio distortion, with a male bias among progeny ranging from 86.1% to 94.8% of males. The fertility of hemizygous transgenic males was also tested revealing full fertility from three of four strains, with hatching rates between 83.6% and 93.2%, whilst only one strain showed a significant reduction in hatching rate down to an average of 54.3% (Fig. 2, Supplementary Table S2). Residual somatic expression of the CRISPRSD transgene may explain reduced fertility in strain N, probably due to position effects associated with the insertion site (Supplementary Fig. S3). All CRISPRSD strains showed no difference in the average number of eggs laid per mated female when compared to the wild-type control (Fig. 2, Supplementary Table S2). Male germline expression of CRISPR-Cas9 does not appear to suffer markedly from problems related to undesired endonuclease stability and carry-over into the fertilized embryo that had complicated previous attempts to establish sex distortion based on the I-PpoI endonuclease2. This is confirmed by another set of experiments in which we find that maternally deposited Cas9 in combination with zygotic expression of the gRNA from the CRISPRSD locus is able to induce embryo lethality but we find no evidence for an effect of paternal carryover of either the gRNA or Cas9 protein on embryo viability (Supplementary Table S2, Supplementary Fig. S4). We confirmed that the observed strong sex distortion phenotype was stably inherited from males to their transgenic sons. For five consecutive generations transgenic males showed consistent levels of male-biased sex ratios in their offspring (Supplementary Table S2). In those rare females that had inherited an X-chromosome from a CRISPRSD father most of the repeats remained susceptible to cleavage, indicating that the vast majority of the rDNA copies retain the intact target sequence (Supplementary Fig. S5). In some individuals a small fraction of the repeats were not cleaved, consistent with the CRISPR-mediated generation of alleles that prevent further cleavage. A similar phenomenon was observed in previous experiments performed on the I-PpoI strains where both molecular and phenotypic analysis confirmed that enzyme-exposed alleles remained susceptible to further cleavage3.

Crosses between CRISPRSD males and wild-type females showing high sex-ratio distortion and fertility.
The adult sex ratio of the progeny of hemizygous CRISPRSD males (red squares) versus hemizygous I-PpoI males (grey dots, Galizi et al.3) and wild-type males crossed to wild-type females (black dots) with the average number of eggs per female (left panel) and the average hatching rate (right panel) shown. Error bars represent the standard error of the mean (SEM).
In conclusion three of four of the transgenic lines we tested showed high levels of sex ratio distortion as well as full male fertility demonstrating that CRISPR-Cas9 is well suited for the construction of synthetic sex ratio distortion traits. We have demonstrated proof of principle for a flexible system of CRISPR-based sex distorters whose target population can range from a species to a limited subset of genomes without affecting the other members of a sexually reproducing population. The choice of which strategy to employ will have to consider the trade-off of increased likelihood of nuclease-resistant alleles evolving at sites that are less conserved. If function is restored, gene conversion may accelerate this process when targeting multiple repeat arrays.
Two areas of further research remain: First, there is a need to develop a bioinformatic strategy to identify X-linked target sequences in the genomes of insect pest or vector species as such repeats are ipso facto absent from high-quality genome assemblies. Here again, the flexibility of CRISPR-Cas9 may be advantageous as it allows targeting multiple independent target sequences. Multiplexing of gRNAs would also hedge this approach against the rise of cleavage-resistant alleles10,11,12 (Fig. 3b). Secondly, whilst autosomal sex-distorter strains as described herein show high levels of distortion they would require repeated releases to achieve population suppression13 (Fig. 3a), e.g. in combination with an inducible expression system which would need to be established in each target species. However, genetic linkage of the sex distorter transgene with the Y-chromosome14 (Fig. 3c) would ensure that the distorter increases in frequency with each generation since it would be present in all male progeny, resulting in a self-perpetuating genetic trait that could rapidly invade and suppress, perhaps eliminate, a natural population even if initially seeded at a very low frequency15,16,17,18.

CRISPRSD system: design and applications.
(a) The RNA-guided Cas9 is expressed from an autosomal location and gives male bias by shredding a species-specific repetitive gene sequence conserved on the X-chromosome (any endonuclease such as TALENs, ZFN or HEG could be suitable for the purpose). The X-shredding construct is inherited only by half of the progeny and it would require inundative releases to suppress or eliminate a pest population13. (b) CRISPR-Cas9 can be multiplexed to target conserved gene sequences present in one or few copies on the X-chromosome10,11,12. (c) The endonuclease is placed onto the Y-chromosome and is therefore inherited by the entire male offspring. In doing so, even a small scale release would be self-sustaining and supress the population15,16,17,18.
Methods
gRNA design
Part of the 28S rDNA consensus sequences (~1 kb) of Anopheles gambiae sensu stricto, Anopheles arabiensis, Anopheles merus, Anopheles melas and Anopheles bwambae were aligned to identify regions of complete conservation across the Anopheles gambiae species complex but absent from the rDNA of distantly related Anopheles stephensi, Anopheles funestus, Aedes Aeypti, Aedes Albopictus and Drosophila melanogaster. ZiFiT (http://zifit.partners.org/) and ChopChop (https://chopchop.rc.fas.harvard.edu) CRISPR gRNA analysis tools were used to select three different gRNAs to specifically target rDNA sequences conserved across the gambiae complex (rDNA-T1 GGGTACAAGCTTGCGTACGTCGG, rDNA-T2 GCTTGTCCGACCGTGAGCCGTGG and rDNA-T3 GGATCCGTAACTTCGGGACAAGG) (Supplementary Fig. S1) and to test for absence of off-targets within An. gambiae sensu stricto genome. gRNA-T1 GGGTACAAGCTTGCGTACGTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC targeting the rDNA-T1 locus was used for all experiments.
Generation of the CRISPRSD construct
The human codon-optimized version of the Streptococcus pyogenes Cas9 coding sequence was amplified from pX330 (AddGene, Zhang laboratory) using primers containing SalI (aacgtcgacGATCCCGGTGCCACCATGGA) and PacI (aacttaattaaTTTCGTGGCCGCCGGCCTTTT) restriction sites and subcloned into a vector containing the β2 tubulin promoter and terminator. AgeI and AscI were used to clone the β2::Cas9 expression cassette into the p165 vector containing the U6::spacer cloning site8, all flanked by piggyBac inverted repeats to obtain the p167 vector. BsaI Golden Gate cloning was used to insert the rDNA-T1 gRNA sequence GGGTACAAGCTTGCGTACGT after oligo-annealing of the primers T1F-TGCTGGGTACAAGCTTGCGTACGT and T1R-AAACACGTACGCAAGCTTGTACCC to obtain the final CRISPRSD transformation construct (Fig. 1a).
Generation and characterization of transgenic mosquito lines
Embryos from the strain G3 of An. gambiae sensu stricto (referred as wild-type) were injected with a mixture of 0.2 μg μl−1 of the CRISPRSD construct and 0.4 μg μl−1 of a vasa-driven piggyBac transposase helper plasmid19 using a Femtojet Express injector and a Narishige 202ND micromanipulator mounted on a Nikon TE-DH100W inverted microscope. The hatched larvae were screened for the transient expression of the DsRed marker on a Nikon inverted microscope (Eclipse TE200) at a wavelength of 563 nm (Filter 630/30 nm emission, 595 nm dichroic). DsRed positive mosquitoes were individually crossed to wild-type to obtain transgenic lines. Strains originated from a single integration event were selected by the inheritance pattern of the DsRed marker scored in the progeny and confirmed by inverse PCR (Supplementary Table S2) as previously described3.
Crosses and phenotype assays
Fertility and adult sex ratio were assessed by crossing male mosquitoes heterozygous for the CRISPRSD allele to an equal number of wild-type mosquitoes for 5 days. Wild-type males were crossed to equal number of wild-type females as a control. On the sixth day female mosquitoes were blood fed on an anaesthetised mouse and placed individually into 300 ml beakers 3d later. Females were given up to three days to oviposit into a 25 ml cup filled with water and lined with filter paper. For each deposition, the number of eggs laid as well as the larvae hatching were counted. The larvae were reared to adulthood and the total number of male and female mosquitoes were subsequently counted (Fig. 2, Supplementary Table S2). To determine whether the sex distortion phenotype was stably inherited from CRISPRSD fathers, transgenic sons were crossed to wild-type females for up to five consecutive generations (Supplementary Table S2). In order to assess the paternal carry-over of the CRISPR-Cas9 components into the fertilized embryos, 10 CRISPRSD males from strain I were crossed to the same number of vasa::Cas9 females and vice versa (vasa::Cas9 strain obtained from Eric Marois Lab). After individual ovipositions, number of eggs and hatched larvae were counted. Segregation of 3xP3::DsRed and 3XP3::YFP (Filter 535/30 nm emission, 515 nm dichroic) markers were used to screen respective segregation of the CRISPRSD and vasa::Cas9 alleles in first instar larval progeny (Supplementary Fig. S4).
RT-PCR analysis of Cas9 expression in CRISPRSD strain N and I
The terminal segment containing the gonads was dissected from the rest of the body from 10 transgenic adults for each sample. The remaining carcasses were used as a control sample. Total RNA was extracted using TRI reagent (Ambion) and reverse-transcribed using Superscript II (Invitrogen) after TURBO DNA-free (Ambion) treatment following the manufacturer’s instructions. Multiplex RT–PCR assay was performed on cDNA using fast-cycling PCR master mix (Qiagen) following the manufacturer’s instructions. In the same reaction mix we added primers specific for the Cas9 gene hCas9qF3-AAAGACCGAGGTGCAGACAG and hCas9qR3-CGATCCGTGTCTCGTACAGG as well as primers specific for the ubiquitous ribosomal gene RpS7 S7fwd2-GGCGATCATCATCTACGTG and S7rev2-GTAGCTGCTGCAAACTTCG (Supplementary Fig. S3).
rDNA analysis
Genomic DNA was isolated from individual female mosquitoes originated from crosses between CRISPRSD males and wild-type females using the Wizard Genomic DNA Purification Kit (Promega). Genomic loci containing the CRISPRSD target sites were amplified with Phusion HF polymerase (Thermo Scientific) using RG236-GCAGCATAGGTGGGAGGGCTTCCTC and RG238-GTTTCTGTCCACACTGAGCTGACCT primers. Four-fifth of each PCR product were purified (Qiagen PCR purification kit) and digested with 1 μl of FastDigest Pfl23II (Thermo Fisher Scientific) for 1 hour at 37 °C. After heat inactivation at 65 °C for 15 min, the digested products were analysed on a 2% agarose gel as well as the remainder of each undigested PCR product (Supplementary Fig. S5).
Additional Information
How to cite this article: Galizi, R. et al. A CRISPR-Cas9 sex-ratio distortion system for genetic control. Sci. Rep. 6, 31139; doi: 10.1038/srep31139 (2016).
References
Windbichler, N. et al. Homing endonuclease mediated gene targeting in Anopheles gambiae cells and embryos. Nucleic Acids Res 35, 5922–5933, doi: 10.1093/nar/gkm632 (2007).
Windbichler, N., Papathanos, P. A. & Crisanti, A. Targeting the X Chromosome during Spermatogenesis Induces Y Chromosome Transmission Ratio Distortion and Early Dominant Embryo Lethality in Anopheles gambiae. Plos Genet 4, doi: 10.1371/journal.pgen.1000291 (2008).
Galizi, R. et al. A synthetic sex ratio distortion system for the control of the human malaria mosquito. Nat Commun 5, doi: 10.1038/Ncomms4977 (2014).
Besansky, N. J. et al. Molecular Phylogeny of the Anopheles-Gambiae Complex Suggests Genetic Introgression between Principal Malaria Vectors. P Natl Acad Sci USA 91, 6885–6888, doi: 10.1073/pnas.91.15.6885 (1994).
Besansky, N. J. et al. Semipermeable species boundaries between Anopheles gambiae and Anopheles arabiensis: evidence from multilocus DNA sequence variation. Proc Natl Acad Sci USA 100, 10818–10823, doi: 10.1073/pnas.1434337100 (2003).
Catteruccia, F., Benton, J. P. & Crisanti, A. An Anopheles transgenic sexing strain for vector control. Nature biotechnology 23, 1414–1417, doi: 10.1038/nbt1152 (2005).
Konet, D. S. et al. Short-hairpin RNA expressed from polymerase III promoters mediates RNA interference in mosquito cells. Insect molecular biology 16, 199–206, doi: 10.1111/j.1365-2583.2006.00714.x (2007).
Hammond, A. et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nature biotechnology 34, 78–83, doi: 10.1038/nbt.3439 (2016).
Lobo, N. F., Clayton, J. R., Fraser, M. J., Kafatos, F. C. & Collins, F. H. High efficiency germ-line transformation of mosquitoes. Nature protocols 1, 1312–1317, doi: 10.1038/nprot.2006.221 (2006).
Nissim, L., Perli, S. D., Fridkin, A., Perez-Pinera, P. & Lu, T. K. Multiplexed and Programmable Regulation of Gene Networks with an Integrated RNA and CRISPR/Cas Toolkit in Human Cells. Mol Cell 54, 698–710, doi: 10.1016/j.molcel.2014.04.022 (2014).
Xie, K. B., Minkenberg, B. & Yang, Y. N. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. P Natl Acad Sci USA 112, 3570–3575, doi: 10.1073/pnas.1420294112 (2015).
Esvelt, K. M., Smidler, A. L., Catteruccia, F. & Church, G. M. Concerning RNA-guided gene drives for the alteration of wild populations. eLife e03401, doi: 10.7554/eLife.03401 (2014).
Gould, F. & Schliekelman, P. Population genetics of autocidal control and strain replacement. Annual review of entomology 49, 193–217, doi: 10.1146/annurev.ento.49.061802.123344 (2004).
Bernardini, F. et al. Site-specific genetic engineering of the Anopheles gambiae Y chromosome. Proc Natl Acad Sci USA 111, 7600–7605, doi: 10.1073/pnas.1404996111 (2014).
Hamilton, W. D. Extraordinary sex ratios. A sex-ratio theory for sex linkage and inbreeding has new implications in cytogenetics and entomology. Science 156, 477–488 (1967).
Deredec, A., Burt, A. & Godfray, H. C. The population genetics of using homing endonuclease genes in vector and pest management. Genetics 179, 2013–2026, doi: 10.1534/genetics.108.089037 (2008).
Beaghton, A., Beaghton, P. J. & Burt, A. Gene drive through a landscape: Reaction-diffusion models of population suppression and elimination by a sex ratio distorter. Theoretical population biology 108, 51–69, doi: 10.1016/j.tpb.2015.11.005 (2016).
Deredec, A., Godfray, H. C. & Burt, A. Requirements for effective malaria control with homing endonuclease genes. Proc Natl Acad Sci USA 108, E874–880, doi: 10.1073/pnas.1110717108 (2011).
Volohonsky, G. et al. Tools for Anopheles gambiae Transgenesis. G3-Genes Genom Genet 5, 1151–1163, doi: 10.1534/g3.115.016808 (2015).
Acknowledgements
We thank Kriezis Antonios, Matthew Gribble, Samson W Q Lee, Xenia Karlsson, Dario Meacci, Giulia Morselli, Ann Hall, Christopher O A Bamikole, Louise A Marston, Mariana Reis Wunderlich and Carla Siniscalchi for experimental assistance and Alekos Simoni, Silke Fuchs and Roya E Haghighat-Khah for useful discussion. This study was supported by a grant from the Foundation for the National Institutes of Health through the Vector-Based Control of Transmission: Discovery Research (VCTR) program of the Grand Challenges in Global Health initiative of the Bill & Melinda Gates Foundation.
Author information
Authors and Affiliations
Contributions
R.G., A.H., K.K., C.T. and F.B. performed the experiments. R.G., A.H., T.N., N.W. and A.C. designed the experiments. R.G., A.H., S.M.O., P.-A.P., T.N. and N.W. performed analyses. R.G., A.H., T.N., N.W. and A.C. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Galizi, R., Hammond, A., Kyrou, K. et al. A CRISPR-Cas9 sex-ratio distortion system for genetic control. Sci Rep 6, 31139 (2016). https://doi.org/10.1038/srep31139
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31139
This article is cited by
-
Targeting mosquito X-chromosomes reveals complex transmission dynamics of sex ratio distorting gene drives
Nature Communications (2024)
-
Gene drive and genetic sex conversion in the global agricultural pest Ceratitis capitata
Nature Communications (2024)
-
Single-cell profiling of Anopheles gambiae spermatogenesis defines the onset of meiotic silencing and premeiotic overexpression of the X chromosome
Communications Biology (2023)
-
CRISPR-mediated knockout of cardinal and cinnabar eye pigmentation genes in the western tarnished plant bug
Scientific Reports (2022)
-
Gene drives gaining speed
Nature Reviews Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
