
Abstract
Combining ability is crucial for parent selection in crop hybrid breeding. The present investigation and results had revealed the underlying genetic factors which might contribute in adequate combining ability, further assisting in enhancing heterosis and stability. Here, we conducted a large-scale analysis of genomic variation in order to define genomic regions affecting the combining ability in recurrent selection population of rapeseed. A population of 175 individuals was genotyped with the Brassica60K SNP chip. 525 hybrids were assembled with three different testers and used to evaluate the general combining ability (GCA) in three environments. By detecting the changes of the genomic variation, we identified 376 potential genome regions, spanning 3.03% of rapeseed genome which provided QTL-level resolution on potentially selected variants. More than 96% of these regions were located in the C subgenome, indicating that C subgenome had sustained stronger selection pressure in the breeding program than the A subgenome. In addition, a high level of linkage disequilibrium in rapeseed genome was detected, suggesting that marker-assisted selection for the population improvement might be easily implemented. This study outlines the evidence for high GCA on a genomic level and provided underlying molecular mechanism for recurrent selection improvement in B. napus.
Similar content being viewed by others

Introduction
Crop domestication and improvement have enhanced yield, plant habits and quality. At the genetic level, these phenotypic shifts are the result of a strong selection of targeting genomic regions or genes. Most domesticated plants have experienced a “selection bottleneck” which reduces genetic diversity as compared to their precursor1. The reduction in genetic diversity across loci provides insights into the demographic history of domestication. Modern breeding has a similar effect on reducing genetic basis. With the artificial selection of crops, genetic diversity is found to be reduced faster than it was during the original domestication2. Effects of these changes have a great potential for breeding and can facilitate the consistent crop improvement. Selection of the desirable alleles leads to a more drastic loss of genetic variation because individuals carrying favored alleles contributed to each subsequent generation and those with adverse alleles were gradually eliminated from the population3. Identifying the genomic loci is essential for revealing the underlying genetic basis of the traits and improving breeding efficiency through marker-assisted selection. With the development of sequencing and DNA microarray technology, the genome wide analysis can be used to scan genes, QTLs or genome regions to obtain desirable traits4,5,6,7. The frequency of these desirable alleles can increase in the population if they are subjected to selection. Therefore, detecting the allele frequency of the breeding population before and after selection, or comparison with their wild or contrasting population can assist in identifying genes of interest5,8. For example, alleles of genes that contribute to increased fruit size in tomato9 and increased apical dominance in maize10, have a high frequency in modern varieties, whereas low frequency in their wild relatives.
Crop breeding programs have generated excellent resources that can be used to improve agronomic traits and identify favorable loci affected by artificial selection. Analysis of genetic diversity, allele frequency and heterozygosity are used to find genomic alterations and genetic effects on the traits in different generations or sub populations11. Additionally, this has been found to be a good approach for scanning genome regions, even candidate genes that underline selection7. In chicken, 82 putatively selected regions with reduced levels of heterozygosity are identified12. In a cattle population, genetic changes are detected and 13 genomic regions were found to affect milk production13. Moreover, several functional genes were verified in some selected regions in cattle14. Similar studies have been carried out in other animals15,16. In miaze, a set of genes (2~4% of 774 genes) are found to have undergone artificial selection during domestication3. Scanning of few known functional genes involved in maize domestication has indicated selection signatures on the genomic level4,17. Furthermore, several chromosome segments and genes were revealed by comparing genetic variation between wild and cultivated populations in soybean5. As for rice, a genealogical history analysis of overlapping low diversity regions can distinguish genomic backgrounds between indica and japonica rice populations and 13 additional candidate genes were identified18. Another study found 200 genomic regions, spanning 7.8% of the rice genome that had been differentially selected between two putative heterotic groups19. These studies have successfully investigated genome-wide genetic changes during domestication and modern breeding. The results can provide useful information to reveal the agronomic potential of a breeding line and genomic loci.
Rapeseed (Brassica napus; AACC, 2n = 38) is one of the most important oil crops worldwide. Rapeseed originated from a doubling event between Brassica rapa (AA, 2n = 20) and Brassica oleracea (CC, 2n = 18) along the Mediterranean coastline 10,000 years ago20,21. It is considered as a young species because of a short domestication history spanning only 400–500 years22. In addition to several other factors, modern breeding has substantially increased production, especially through heterosis. In a hybrid breeding program, combining ability is a crucial factor for parental line selection and for the development of superior hybrids. Evaluation of the combining ability using traditional methods is labor intensive and time-consuming and may create a bottleneck in hybrid breeding23. Therefore, dissection and comparison of the genetic basis of combining ability can be crucial for breeding. Combining ability was defined as a complex trait in plants and was evaluated by several techniques, including molecular markers, QTL mapping and genome scan approaches24,25,26. There have been limited investigations carried out to evaluate the genetic basis of combining ability in rapeseed. During rapeseed breeding history, heterosis and double-low varieties (low erucic acid and low glucosinolate) were mainly used to produce higher yield and better quality at the cost of genetic diversity27,28. Recently, new genetic resources are used to increase the genetic basis of rapeseed, including the artificially synthesized B. napus generated from B. oleracea and B. rapa29, the subgenome materials30,31. Multigenerational improvement and a recurrent selection program are required before utilizing these new materials.
In our work, genomic SNP markers were used to analyze the breeding signatures of GCA as revealed by the genetic variation in a recurrent selection population. The objectives of our study were (1) to estimate genetic diversity of genome-wide SNPs in different groups of the rapeseed restorer population, (2) to detect the putatively selected regions and SNPs associated with breeding efforts on the genomic level and (3) to identify known important QTLs associated with rapeseed agronomic traits in selected regions. These findings might be of potential use in improving the rapeseed breeding.
Results
Phenotype variations in yield and yield-related GCA
Plant yield from the population of 175 families and 525 hybrids, were analyzed with two replicates in three different environments. GCA of each parental line was estimated statistically using the phenotype data sets. Extensive phenotype variations were observed (Table 1). The mean yield of three environments were 13.93 g, 7.41 g, 8.08 g per plant, respectively and varied from 4.36~36.87 g in Wuhan, from 1.62~15.92 g in Xiangyang and from 2.24~37.82 g in Yichang. The plant yield had high coefficients of variation in the three environments, suggesting that the yield of the rapeseed was a typical quantitative trait and was substantially affected by the environment. The mean value of GCA (Table 1) was 1.34, varied from −3.98~7.98 and the value for coefficient of variation was 172.23%.
SNP filtering and genetic analysis
After genotyping, out of 52,157 SNPs, a total of 47,986 were called successfully. SNPs with no polymorphism and missing value > 10% were removed from genotype data sets and 39,582 SNPs remained. The precise physical location of the SNPs was established by comparative analysis of the reference genome. A total of 27,049 high quality genome-wide SNPs were used for further analysis. These SNPs covered 641.91 Mb of rapeseed genome. The average physical distance between two SNPs was about 30 Kb. Out of 27,049 SNPs, 13,929 were located on the A subgenome and 13,120 SNPs were on the C subgenome. Statistical analysis of the PIC value (Table 2) revealed that the average PIC value of the A4 chromosome was the highest (0.311), whereas that of the A9 chromosome was the lowest (0.263). Approximately 76.6% of these SNPs have an over medium polymorphism level of 0.25. The polymorphic level of the A subgenome was little higher than that of the C subgenome (Table 2).
Genetic variation detecting across the regions of the specific loci
SNPs were used to detect the genetic variation of three specific loci in rapeseed genome: erucic acid related genes at the BnFAE1.1, BnFAE1.2 loci on A8 and C3 chromosomes32 and the Ogrua CMS restorer gene Rfo loci on C9 chromosome33. It consistently showed that closer the target loci, lower the genetic variation (Fig. 1). These findings indicated that our selection program have been carried out efficiently. Moreover, the evaluation method via genetic diversity could be of potential use in breeding improvement.

Genetic diversity across the regions of specific loci in the rapeseed genome.
(A) Genome regions across the erucic acid related gene loci on the chromosomes A8. (B) Genome regions across the erucic acid related gene loci on the chromosome C3. (C) Genome regions across the Ogura CMS restorer gene Rfo loci on the chromosome C9. The horizontal axis stands for the physical distance (Mb) of the chromosome, the vertical axis stands for the value of genetic diversity. The red star mark the position of gene located on chromosome; the red dotted box marks the possible inserted position of the gene Rfo.
Linkage disequilibrium (LD) in the R population
R2 was used to calculate the LD level. For r2 = 0.2, LD level occurred at approximately 0.8 Mb, 4.8 Mb and 2.4 Mb for A and C subgenomes and AC genome, respectively (Fig. 2). When r2 decayed to 0.1, LD values increased to 3 Mb, 8 Mb and 6.5 Mb for A and C subgenomes and AC genome, respectively. The C subgenome had a larger LD value than A subgenome. As for chromosomes of the two subgenomes, LD of chromosomes in A subgenome was highly consistent, while variation was detected in C subgenome. The LD of the C4 chromosome was higher than 6 Mb when r2 = 0.1 (Fig. S2). C1 and C2 chromosomes showed almost no LD decay. Genetic variation of the two subgenomes was also evaluated. The A subgenome had a little higher genetic diversity than the C subgenome (Table 3). By detecting the changes in genetic diversity between the selected and basic populations, we found a greater decrease in the C subgenome (2.7%) as compared to the A subgenome (1.55%).

Genome-wide LD decay for the 175 R lines.
A, C and AC represent the A and C subgenome and the whole genome of rapeseed, respectively. The unit distance is 500 Kb.
Selected regions and candidate QTLs analysis
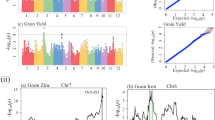
Scanning of genomic regions indicated a reduction in genetic diversity. In total, we identified 376 selected regions, covering 3.03% (21.26 Mb) of the assembled genome (Table 4; Fig. S3). More than 96% of these regions were distributed on the C subgenome (Fig. 3; Table 4). C6 chromosome had the largest size of selected regions (4.56 Mb), while A3 had the smallest (0.02 Mb). Furthermore, A1, A5, A7, A8, A9, A10 and C5 chromosomes had no distribution of selected regions. The mean size of selected regions for each chromosome on A and C subgenomes and AC genome were 0.14 Mb, 2.55 Mb and 1.52 Mb, respectively. The C subgenome had a larger distribution of selected regions than the A subgenome. Many QTLs related to yield and yield-related traits were located in these selected regions (Supplementary Table S1) which likely contributed to the increase in rapeseed yield and GCA. Among the 19 chromosomes of the rapeseed genome, we found differences in the distribution of genetic diversity in the selected regions. In particular, both the chromosomes C2 and C3 had a genome region (C2: 22.0~22.5 Mb; C3: 8.1~8.3 Mb) and distributed throughout the block which lost the genetic variation. For C2 chromosome, we detected the QTL hsy12.1 and DHy06, both previously reported to contribute in rapeseed yield34,35 (Fig. 4; Supplementary Table S1). Moreover, this region was a QTL hot spot and also contain the QTL of DH-tsm for seed weight and the QTL MPH-y06 for heterosis (Middle Parent Heterosis, MPH)35. QTL hot spots contained important QTLs for rapeseed yield and yield-related traits were also detected in the region on chromosome C3 (Supplementary Table S1). All these QTLs in the selected regions provided a potential resource for rapeseed breeding and selection for these QTLs for rapeseed genetic improvement might lead to low genetic diversity in these regions, but increase in rapeseed yield.

Statistics and distribution of the selected regions in the rapeseed genome.
The vertical axis stands for the summary size of selected regions on each chromosome.

QTLs located in the selected regions on the chromosome C2.
(A) Genetic map of QTL MPH-y06, DH-y06, DH-tsm related to rapeseed yield and seed weight (Basunanda et al. 2010). (B) Different selection on the chromosome C2 with πRatio. The horizontal axis stands for the chromosome, the vertical axis stands for the πRatio. (C) Genetic map of QTL hsy12.1 related to rapeseed yield (Quijada et al. 2006).
Pedigree breeding history reproduction
The genomic changes that occurred between the genealogy lines were detected. We reconstructed the recombination events that gave rise to specific inbred lines zhongshuang5 and zhongshuang4, which were both produced from zhongyou821. We traced the chromosome segments through pedigree breeding of the two lines. In total, zhongshuang5 inherited 15.41% of its genome from the ancestral line zhongyou821 while zhongshuang4 inherited 34.06% (Table 4; Fig. S4). Zhongshuang5 inherited 24.17% of the A subgenome and 10.26% of the C subgenome from zhongyou821, while zhongshuang4 inherited only 14.37% of the A subgenome but 45.63% of the C subgenome from zhongyou821. Out of the 19 chromosomes, six chromosomes (A1, A5, C1, C6, C7 and C8), showed that more than half of their chromosome fragments were inherited from zhongyou821 into zhongshuang4, particularly in C6 and C7, where almost the whole chromosomes were found to be inherited. However, 8 chromosomes (A2, A3, A4, A6, A7, A8, A9 and C2) were not inherited into zhongshuang4. In 45.63% inherited component, we observed that 84.39% was from the C subgenome. These findings were consistent with the analysis of the selected regions.
Fixed SNP provided a reference index for population improvement
By detecting the allele frequencies of genome-wide SNPs, we identified a total of 403 Fixed SNPs from the genotype data sets. There were 214 of these Fixed SNPs from the A subgenome and 189 from the C subgenome (Fig. 5). The allele frequencies of these SNPs were fixed to 0 or 1 in the selected group and subsequent generations. These loci have lost other alleles and showed monomorphism in the subsequent population.

Distribution of the Fixed SNP in the rapeseed genome.
Discussion
A yield-improving plateau occurs for a limited genetic diversity2. Demonstration and breeding program reduce the crop genetic diversity significantly1,2. To enhance the diversity, we used the contents of the subgenomes from the relation species in Brassica. By the breeding method of recurrent selection for GCA improvement, some desirable loci were maintained in the population and others undesirable loci were deleted from the population. Our analysis provided useful information to exhibit genetic base of GCA on rapeseed.
The LD value of this population was larger than the natural population reported previously. Breeding selection for the favorable alleles would increase the LD level between loci in genome36. In this study, we observed strong LD between SNPs separated up to 2.5 Mb (r2 = 0.2). This value was higher than the LD value obtained in previous studies37,38,39, which have indicated LD levels at about 500 Kb, 700 Kb and 2 cM, respectively. In these studies, researchers have used the resource populations collected from all over the world which contained higher rapeseed genetic variation. Contrastingly, in our study, the population was derived from several artificially synthesized B. napus and the subgenome materials. Afterwards, it was improved for subsequent generations which might have contributed to the higher LD. These findings might be useful for marker-assisted recurrent selection. Our results also demonstrated higher LD in the C subgenome, especially for C1, C2 and C4 chromosomes, which was consistent with the previous results40,41. Possibly, this could be explained by several reasons: the C subgenome had a lower level of genetic variability than the A subgenome and the C subgenome might be under a more intense selection pressure in our breeding program. Polygenetic analysis also showed a decreasing trend in the diversity of the C subgenome than that of the A subgenome (Table 3). It had also provided a favorable evidence for the higher LD of the C subgenome.
The C subgenome is a repository for a wider range of selected regions with favorable loci contributing to rapeseed agronomic traits. By detecting the changes in genomic diversity, we identified 376 genomic regions and covered 3.03% (21.26 Mb) of the rapeseed genome. Many important QTLs related to yield and yield-related traits were located in some of these selected regions (Supplementary Table S1). In particular, some of these genomic regions harboured QTL hotspots (for one trait or multiple traits) or significant QTL reported in other studies (Fig. 4). We noticed that more than 96.05% of these selected regions distributed on the C subgenome (Fig. 3; Table 4) and only about 3.95% was distributed on the A subgenome. This indicated that the C subgenome had sustained more pressure in the selection program or the C subgenome contributed more to the yield-related GCA than the A subgenome. The differences in the genome background between the genealogical lines further support this conclusion (Table 4; Fig. S4). In China, for improvement of the adaptive traits of the European and Japanese varieties, breeders have lead to the introgression of the A genome components of B. rapa into the B. napus genome31,42. This process has enhanced the genetic diversity of A subgenome in Chinese rapeseed. However, the breeding potential of C subgenome has not been developed and utilized much. The genetic background of our population contained European winter-type rapeseed, which has higher genetic variation of C subgenome than the A subgenome37. Furthermore, the subgenome materials (ArArCcCc) have been introgressed with the Cc genome from B. carinata and artificial synthetic materials have been introgressed with the Co genome from B. oleracea, which might also contributed to the increased genetic variation of C subgenome in B. napus. These new genetic components of the C subgenome might potentially improve the rapeseed yield. Results of the present investigation, along with a deeper understanding of heterosis and changes in breeding programs have indicated that the C subgenome needs to be fully developed in rapeseed hybrid breeding.
Recurrent selection has been established as a very useful method for plant breeding43,44. The process can break the linkage of disadvantageous alleles and pyramid favorable alleles through sustaining recombination and selection. In this study, we used the recurrent selection method to improve the GCA level of the R population. The top 20% individuals with high GCA were selected for the next generation. Genetic analyses showed that there were many genomic regions under selection. These regions might play an important role in rapeseed breeding. We suggest that most favorable alleles might be accumulated through MAS and standing selection. This might assist in the development and improvement of potential rapeseed.
In summary, we have conducted a comprehensive analysis of changes in genomic variation and identified a number of genomic regions and loci subjected to selection. Firstly, we found a slightly higher level of genetic diversity for the A subgenome as compared to the C subgenome. Both of the subgenomes had a higher LD and might be beneficial for MAS. Secondly, the program for breeding selection might decrease the genetic diversity of the population and some allelic variations would disappear or approach to fixation. Thirdly, most of the selected regions were distributed on the C subgenome, which indicated that the C subgenome might have been under stronger selection pressure, or contributed more towards GCA improvement in rapeseed hybrid breeding. Finally, we have identified several potential selection targets, genomic regions and loci, which provided further insight into rapeseed research and improvement.
Materials and Methods
Plant materials, phenotype evaluation and GCA estimation
We used two new types of rapeseed (artificial synthetic B. napus and subgenomic materials) and winter type rapeseed in the present investigation: (1) 41 artificial synthetic B. napus from the University of Goettingen, Germany, were crossed with three winter type lines (SW0736, SW0740 and SW0784) from Sweden in 1999; the F5 families were crossed with Ogura-INRA CMS lines and restorer line R2000 in 2004. (2) Seven subgenomic materials (ArArCcCc)45, 2 Pol CMS restorers (5148R and 6178R) and yellow seed coat variety No2127 were crossed with R2000. F1 obtained from (1) and (2) was used to construct the recurrent selection population in 2005 in Huazhong Agricultural University, China. A recurrent selection program was used to improve the GCA level of the population. The previous populations were randomly pollinated in an isolated environment. The sterile plants were harvested and the seeds were used to construct the next generation population. Meanwhile, seed quality (oil content, glucosinolate and erucic acid) was considered as an important breeding goal. In 2012, we randomly selected 175 plants (with the Rfo gene) and crossed them with three different testers (Yu7-120, Yu7-126 and Yu7-140) to produce hybrid seeds following NC II design46, resulting in a total of 525 hybrids. All hybrids and 178 parental lines, were sown in three semi-winter rapeseed environments (Wuhan, 29°58′N, 113° 53′E; Xiangyang, 32° 04′N, 112° 05′E and Yichang, 30′40′N, 111° 45′E) in China. Field trials were followed as completely random design with two replications at each location.
General combining ability (GCA) of each parental lines was calculated using the formula: gi = yi-ŷ, where gi stands for GCA of parental line, yi and ŷ each stand for the mean of crosses with same parent Pi and the mean of all crosses, respectively47. Based on the GCA, we set 20% as the selection intensity, which could not rapidly decrease the genetic diversity of the population. Afterwards, 35 lines with a high GCA were selected from the population, defined as the selected population or group.
The other three genealogical lines (zhongyou821, zhongshuang4 and zhongshuang5, Fig. S1) were used to detect the genome changes in pedigree breeding. The zhongyou821 is highly considered for rapeseed breeding in China. Many elite inbred lines including both open pollination cultivars and hybrid parents, were developed from this line. For example, both zhongshuang4 and zhongshuang5 are derived from zhongyou821 and bred as open pollination cultivars. Recently, F1 hybid of zhongshuang4 and Pol CMS lines is found to exhibit excellent heterosis performance. Therefore, zhongshuang4 is considered as a good restorer line and used to develop several other hybrid cultivars and restorer lines.
SNP filtering and genotype analysis
Genomic DNA was extracted from young leaves using the cetyl triethyl ammnonium bromide (CTAB) method48. The Illumina BrassicaSNP60 Bead Chip containing 52,157 SNPs was employed to genotype this panel of rapeseed. The experiment followed the manufacturer’s protocol as described by Illumina Company. (http://www.illumina.com/technology/infinium_hd_assay.ilmn). The SNP data was clustered and called automatically using the Illumina GenomeStudio genotyping software. SNPs with no polymorphism and missing value > 10% were excluded. The source sequences of the remaining SNPs were identified through BlastN searches against the reference genome sequence of Darmor-bzh49 (http://www.genoscope.cns.fr/brassicanapus). SNPs with an ambiguous physical position or multiple blast-hits were also excluded from the genotype data sets.
Polygenetic and linkage disequilibrium analysis
Genetic diversity (π), polymorphism information content (PIC) and alleles frequencies of each SNP on 19 chromosomes were estimated by the PowerMarker software50. Linkage disequilibrium (LD) between SNPs was calculated by all markers using the TASSEL software version 5.151. LD decay was evaluated on the basis of the r2 value and corresponding distance between two SNPs.
Selected regions, Fixed SNP and candidate QTL detecting
To calculate diversity changes across the genome, a sliding window method was used to analyze each chromosome separately, with a window size of five SNPs and a sliding step of two SNPs. Ratio of the genetic diversity value of each window between selected and basic populations was used to identify genomic regions affected by selection, which was estimated by the formula: πRatio = πbasic/πselected. We selected the top 5% windows as candidate regions for further analysis. In addition, we analyzed many reported QTLs of rapeseed yield and yield- related traits. If the closely linked markers or the mapped interval were located in or overlapped with selected regions, we considered them to be candidate selected QTLs. We also calculated the allele frequencies of each SNP on the 19 chromosomes and identified the SNPs which allele frequencies were changed to a hundred percent in the selected population and defined such SNPs as Fixed SNP.
Genome changes detecting during the pedigree breeding
We used the Beagle4.1 software52 to detect the chromosome segments of identity by descent (IBD)53 between the two half-sib sister lines (zhuangshuang4 and zhongshuang5) and their common ancestor (zhongyou821) by genome-wide SNP markers. The P value of the significant level was set as 1 × 10−7. Uncertain regions (not defined IBD segments) were equally appropriate into the two adjacent blocks. We surveyed the inherited proportion of their genome from zhongyou821 and set different colors for chromosome segments according to the type of IBD.
Additional Information
How to cite this article: Zhao, X. et al. Breeding signature of combining ability improvement revealed by a genomic variation map from recurrent selection population in Brassica napus. Sci. Rep. 6, 29553; doi: 10.1038/srep29553 (2016).
References
Buckler, E. S., Thornsberry, J. M. & Kresovich, S. Molecular diversity, structure and domestication of grasses. Genet Res 77, 213–218 (2001).
Innan, H. & Kim, Y. Pattern of polymorphism after strong artificial selection in a domestication event. P Natl Acad Sci USA 101, 10667–10672 (2004).
Wright, S. I. et al. The effects of artificial selection on the maize genome. Science 308, 1310–1314 (2005).
Hufford, M. B. et al. Comparative population genomics of maize domestication and improvement. Nat Genet 44, 808–U118, doi: 10.1038/ng.2309 (2012).
Lam, H. M. et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat Genet 42, 1053–1059 (2010).
Lin, T. et al. Genomic analyses provide insights into the history of tomato breeding. Nat Genet 46, 1220–1226 (2014).
Strasburg, J. L. et al. What can patterns of differentiation across plant genomes tell us about adaptation and speciation? Philos T R Soc B 367, 364–373 (2012).
Doebley, J. F., Gaut, B. S. & Smith, B. D. The molecular genetics of crop domestication. Cell 127, 1309–1321 (2006).
Nesbitt, T. C. & Tanksley, S. D. Comparative sequencing in the genus Lycopersicon: implications for the evolution of fruit size in the domestication of cultivated tomatoes. Genetics 162, 365–379 (2002).
Clark, R. M., Linton, E., Messing, J. & Doebley, J. F. Pattern of diversity in the genomic region near the maize domestication gene tb1. P Natl Acad Sci USA 101, 700–707 (2004).
Beissinger, T. M. et al. A genome-wide scan for evidence of selection in a maize population under long-term artificial selection for ear number. Genetics 196, 829–840 (2014).
Qanbari, S. et al. A high resolution genome-wide scan for significant selective sweeps: an application to pooled sequence data in laying chickens. PloS One 7, e49525 (2012).
Flori, L. et al. The genome response to artificial selection: a case study in dairy cattle. PloS One 4, e6595 (2009).
Pan, D. et al. Genome-wide detection of selective signature in Chinese Holstein. PloS One 8 (2013).
Pollinger, J. P. et al. Selective sweep mapping of genes with large phenotypic effects. Genome Res 15, 1809–1819 (2005).
Rubin, C. J. et al. Strong signatures of selection in the domestic pig genome. P Natl Acad Sci USA 109, 19529–19536 (2012).
Jiao, Y. et al. Genome-wide genetic changes during modern breeding of maize. Nat Genet 44, 812–815 (2012).
He, Z. et al. Two evolutionary histories in the genome of rice: the roles of domestication genes. PLoS Genet 7, e1002100 (2011).
Xie, W. et al. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. P Natl Acad Sci USA 112, E5411–E5419 (2015).
Song, K. & Osborn, T. C. Polyphyletic origins of Brassica napus: new evidence based on organelle and nuclear RFLP analyses. Genome 35, 992–1001 (1992).
Nagaharu, U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jap J Bot 7, 389–452 (1935).
Gómez-Campo, C. & Prakash, S. Origin and domestication. Biology of Brassica coenospecies, 33–58 (1999).
Smith, J. et al. Use of doubled haploids in maize breeding: implications for intellectual property protection and genetic diversity in hybrid crops. Mol Breeding 22, 51–59 (2008).
Qu, Z. et al. QTL mapping of combining ability and heterosis of agronomic traits in rice backcross recombinant inbred lines and hybrid crosses. PLoS One 7, e28463 (2012).
Qi, H. et al. Identification of combining ability loci for five yield-related traits in maize using a set of testcrosses with introgression lines. Theor Appl Genet 126, 369–377 (2013).
Huang, J. et al. General combining ability of most yield-related traits had a genetic basis different from their corresponding traits per se in a set of maize introgression lines. Genetica 141, 453–461, doi: 10.1007/s10709-013-9744-3 (2013).
Bus, A., Körber, N., Snowdon, R. J. & Stich, B. Patterns of molecular variation in a species-wide germplasm set of Brassica napus. Theor Appl Genet 123, 1413–1423 (2011).
Reif, J. C. et al. Trends in genetic diversity among European maize cultivars and their parental components during the past 50 years. Theor Appl Genet 111, 838–845 (2005).
Wu, J. et al. Assessing and broadening genetic diversity of a rapeseed germplasm collection. Breeding Sci 64, 321 (2014).
Rahman, H., Bennett, R. A. & Séguin-Swartz, G. Broadening genetic diversity in Brassica napus canola: Development of canola-quality spring B. napus from B. napus×B. oleracea var. alboglabra interspecific crosses. Can J Plant Sci 95, 29–41 (2015).
Qian, W. et al. Introgression of genomic components from Chinese Brassica rapa contributes to widening the genetic diversity in rapeseed (B. napus L.), with emphasis on the evolution of Chinese rapeseed. Theor App Gene 113, 49–54 (2006).
Fourmann, M. et al. The two genes homologous to Arabidopsis FAE1 co-segregate with the two loci governing erucic acid content in Brassica napus. Theor Appl Genet 96, 852–858 (1998).
Hu, X. et al. Mapping of the Ogura fertility restorer gene Rfo and development of Rfo allele-specific markers in canola (Brassica napus L.). Mol Breeding 22, 663–674 (2008).
Quijada, P. A., Udall, J. A., Lambert, B. & Osborn, T. C. Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 1. Identification of genomic regions from winter germplasm. Theor Appl Genet 113, 549–561 (2006).
Basunanda, P. et al. Comparative mapping of quantitative trait loci involved in heterosis for seedling and yield traits in oilseed rape (Brassica napus L.). Theor Appl Genet 120, 271–281 (2010).
Smith, J. M. & Haigh, J. The hitch-hiking effect of a favourable gene. Genet Res 23, 23–35 (1974).
Wang, N. et al. Genome-wide investigation of genetic changes during modern breeding of Brassica napus. Theor Appl Genet 127, 1817–1829 (2014).
Xu, L. et al. Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res dsv035 (2015).
Ecke, W., Clemens, R., Honsdorf, N. & Becker, H. C. Extent and structure of linkage disequilibrium in canola quality winter rapeseed (Brassica napus L.). Theor Appl Genet 120, 921–931 (2010).
Hatzig, S. V. et al. Genome-wide association mapping unravels the genetic control of seed germination and vigor in Brassica napus. Front Plant Sci 6, 221, doi: 10.3389/fpls.2015.00221 (2015).
Schiessl, S., Iniguez-Luy, F. & Qian, W. & Snowdon, R. J. Diverse regulatory factors associate with flowering time and yield responses in winter-type Brassica napus. BMC Genomics 16, 737, doi: 10.1186/s12864-015-1950-1 (2015).
Shiga, T. Rape breeding by interspecific crossing between Brassica napus and Brassica campestris in Japan. Jpn Agric Res Quart 5, 5–10 (1970).
Hallauer, A. R. Recurrent selection in maize. Plant Breed. Rev 9, 115–179 (1992).
Labate, J. A., Lamkey, K. R., Lee, M. & Woodman, W. L. Molecular genetic diversity after reciprocal recurrent selection in BSSS and BSCB1 maize populations. Crop Sci 37, 416–423 (1997).
Li, M., Chen, X. & Meng, J. Intersubgenomic Heterosis in Rapeseed Production with a Partial New-Typed Containing Subgenome A from and C from. Crop Sci 46, 234, doi: 10.2135/cropsci2004.0759 (2006).
Zhu, J. & Weir, B. Analysis of cytoplasmic and maternal effects I. A genetic model for diploid plant seeds and animals. Theor Appl Genet 89, 153–159 (1994).
Kebede, B., Thiagarajah, M., Zimmerli, C. & Rahman, M. H. Improvement of Open-Pollinated Spring Rapeseed (L.) Through Introgression of Genetic Diversity from Winter Rapeseed. Crop Sci 50, 1236–1243 (2010).
Jarret, N. J. G. a. R. L. A Modified CTAB DNA Extraction Procedure for Musa and Ipomoea. Plant Mol Biol Rep. 9(3), 262–266 (1991).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Liu, K. & Muse, S. V. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129 (2005).
Bradbury, P. J. et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Browning, S. R. & Browning, B. L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81, 1084–1097 (2007).
Lai, J. et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat genet 42, 1027–1030 (2010).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFD0100300), National Science-technology Support Plan Projects (2014BAD01B07) and Natural Science Foundation of Hubei province Key Program (2014CFA008). We are thankful to Mr. Mayank Gautam for editing the English version of the manuscript.
Author information
Authors and Affiliations
Contributions
X.Z. and J.T. designed the study. X.Z., B.L. and K.Z. performed the field experiments and traits investigation. X.Z. and K.H. performed the SNP analysis in the R population. X.Z., B.Y., J.W., C.M., J.S., T.F. and J.T. performed the linkage disequilibrium and genetic diversity analysis of the R population. X.Z. and J.T. performed the genomic change analysis of the R population and the pedigree breeding history analysis of the genealogical lines. X.Z. analysed all the data and wrote the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, X., Li, B., Zhang, K. et al. Breeding signature of combining ability improvement revealed by a genomic variation map from recurrent selection population in Brassica napus. Sci Rep 6, 29553 (2016). https://doi.org/10.1038/srep29553
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep29553
This article is cited by
-
Prospects of Marker-Assisted Recurrent Selection: Current Insights and Future Implications
Tropical Plant Biology (2023)
-
Fine mapping of BnDM1—the gene regulating indeterminate inflorescence in Brassica napus
Theoretical and Applied Genetics (2023)
-
Comparative transcriptomic analysis reveals the molecular mechanism underlying seedling biomass heterosis in Brassica napus
BMC Plant Biology (2022)
-
Identification and fine mapping of a major locus controlling branching in Brassica napus
Theoretical and Applied Genetics (2020)
-
Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus
Nature Communications (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
