
Abstract
Bacterial lipoproteins are extracellular proteins tethered to cell membranes by covalently attached lipids. Deleting the lipoprotein signal peptidase (lsp) gene in Streptomyces coelicolor results in growth and developmental defects that cannot be restored by reintroducing lsp. This led us to hypothesise that lsp is essential and that the lsp mutant we isolated previously had acquired compensatory secondary mutations. Here we report resequencing of the genomes of wild-type M145 and the cis-complemented ∆lsp mutant (BJT1004) to map and identify these secondary mutations but we show that they do not increase the efficiency of disrupting lsp and are not lsp suppressors. We provide evidence that they are induced by introducing the cosmid St4A10∆lsp, as part of ReDirect PCR mutagenesis protocol, which transiently duplicates a number of important cell division genes. Disruption of lsp using a suicide vector (which does not result in gene duplication) still results in growth and developmental delays and we conclude that loss of Lsp function results in developmental defects due to the loss of all lipoproteins from the cell membrane. Significantly, our results also indicate the use of cosmid libraries for the genetic manipulation of bacteria can lead to phenotypes not necessarily linked to the gene(s) of interest.
Similar content being viewed by others

Introduction
Bacterial lipoproteins are essential for building and maintaining the cell envelope and they also provide a key interface with the external environment1,2,3. Most lipoprotein precursors are exported as unfolded polypeptides via the Sec (general secretory) pathway but others can be exported via the twin arginine transport (Tat) pathway, which is typically utilised for the transport of fully folded proteins4,5,6. The signal peptides of lipoproteins closely resemble other types of bacterial Sec and Tat signal peptide but they contain a characteristic lipobox motif, typically L−3-A/S−2-G/A−1-C+1, relative to the signal cleavage site, in which the cysteine residue is essential and invariant. The lipobox motif allows putative lipoproteins to be easily identified in bacterial genome sequences3,7. Following translocation, lipoprotein precursors are firstly modified by covalent attachment of a diacylglycerol molecule, derived from a membrane phospholipid, to the thiol of the conserved lipobox cysteine residue via a thioether linkage. This reaction is catalysed by an enzyme named Lgt (Lipoprotein diacylglycerol transferase) and results in a diacylated lipoprotein. Lsp (Lipoprotein signal peptidase) then cleaves the signal sequence immediately upstream of the lipidated cysteine to leave it at the +1 position. These early steps in lipoprotein biogenesis are highly conserved and unique to bacteria making them potential targets for antibacterial drug development2,8. In Gram-negative bacteria and Gram-positive Actinobacteria, lipoproteins can be further modified by addition of an amide-linked fatty acid to the amino group of the diacylated cysteine residue at the mature N-terminus. This final step is catalysed by the enzyme Lnt (Lipoprotein n-acyltransferase) and results in triacylated lipoproteins. In Gram-negative proteobacteria, Lnt modification is a pre-requisite for the recognition of lipoproteins by the Lol machinery, which transports lipoproteins to the outer membrane2,9 but its function in monoderm Gram-positive bacteria is not known. Members of the Gram-positive phyla Firmicutes and Mollicutes also N-acylate lipoproteins despite lacking Lnt homologues and S. aureus can diacylate or triacylate individual lipoproteins in an environmentally dependent manner10,11,12,13,14. These studies suggest that triacylation of lipoproteins in Gram-positive bacteria has an important role in their natural environment but is dispensable in vitro. Loss of Lnt activity in Streptomyces bacteria has no obvious effect on fitness or lipoprotein localisation in vitro but it does have a moderate effect on virulence in the plant pathogen Streptomyces scabies, supporting the idea that it has environmental importance15.
We previously characterised all four steps of the lipoprotein biogenesis pathway in Streptomyces spp. (Fig. 1)5,15, which is one of the best studied genera in the Gram-positive phylum Actinobacteria. Our key findings are (i) that Tat exports ~20% of lipoprotein precursors in streptomycetes; (ii) they N-acylate lipoproteins using two non-essential Lnt enzymes; (iii) Streptomyces coelicolor encodes two functional copies of Lgt which cannot be removed in the same strain; (iv) lsp mutants can be isolated at low frequencies but they acquire spontaneous secondary mutations which might be lsp suppressors. It was recently reported that Lgt is essential in Mycobacterium tuberculosis, which is also a member of the phylum Actinobacteria and that lgt deletion in the fast-growing species Mycobacterium smegmatis is accompanied by spontaneous secondary mutations16. Natural product antibiotics that target the lipoprotein biogenesis pathway include globomycin, made by Streptomyces globisporus2 and antibiotic TA made by Myxococcus xanthus1,16. Both inhibit Lsp activity and are lethal to Escherichia coli but TA resistance arises through spontaneous IS3 insertion into the lpp gene, which encodes an abundant lipoprotein that attaches the E. coli outer membrane to the peptidoglycan cell wall16,17. Over-expressing lsp also confers TA resistance in both E. coli and M. xanthus and the latter encodes additional Lsp homologues within the TA biosynthetic gene cluster17.

Lipoprotein biogenesis in Streptomyces coelicolor.
Approximately 80% of precursor lipoproteins in S. coelicolor are translocated via the general secretory (Sec) pathway with around 20% being translocated by the twin arginine transport (Tat) pathway (a). Following translocation across the cytoplasmic membrane they are diacylated on the thiol of the lipobox (+1) cysteine residue by Lgt1 or Lgt (b) and then the signal sequence is cleaved by Lsp immediately upstream of that modified cysteine (c). Lnt1 then adds a third acyl chain to the amino group on the +1 cysteine to produce a triacylated lipoprotein (d). Lnt2 is not essential for triacylation in vitro but appears to increase its efficiency. The function of the N-acyl modification is not yet known.
Deletion of S. coelicolor lsp results in very small and flat colonies that are delayed in sporulation and these lsp mutants could not be fully complemented even by reintroducing the lsp gene to its native locus. Although both cis and in trans complementation restored lipoprotein biogenesis and sporulation it did not restore the wild-type growth rate5. There are two likely reasons for this: either lsp is essential and ∆lsp mutants acquire secondary suppressor mutations or the ReDirect PCR targeting method that we used to delete the lsp gene resulted in mutations independent of lsp. ReDirect is the name given to a protocol in which the Lambda Red system is used to PCR target genes of interest in a Streptomyces cosmid library in E. coli and the mutated cosmids are conjugated into Streptomyces species to select for mutants. Here we provide evidence to support the second hypothesis by demonstrating that introduction of a cosmid carrying an ~40 kb region of the S. coelicolor chromosome, including lsp, from E. coli to S. coelicolor leads to growth and developmental defects. We further show that lsp is non-essential but that deletion of the lsp gene results in small colonies that over-produce actinorhodin, as observed previously. These phenotypes must therefore be due to the loss of lipoproteins from the cytoplasmic membrane of S. coelicolor.
Results
Mapping secondary mutations in the cis complemented Δlsp strain BJT1004
We previously reported that the S. coelicolor ∆lsp mutant BJT1001 cannot be complemented even by restoring lsp to its native locus5. Since cis complementation should effectively restore the genome to wild-type this suggests that other spontaneous mutations have occurred during the deletion of lsp. To test this we resequenced the genomes of the isogenic parent strain S. coelicolor M145 and the cis-complemented ∆lsp strain BJT1004 using two independent companies and compared them with each other and with the published M145 sequence to identify mutations (Supplementary Table S1). Across all four sequences we identified a total of 51 single nucleotide polymorphisms (SNPs) as well as a chromosomal rearrangement in BJT1004 that is not present in the parent strain M145 (Fig. 2a,b and Supplementary Text S1 and S2). Of the 51 SNPs, 13 are unique to one of the BJT1004 sequences, with four residing inside coding sequences. Only one SNP occurs in both BJT1004 sequences and this is in the intergenic region between sco5331 and sco5332 and does not affect a coding sequence. In the single chromosomal rearrangement, the IS21 insertion element (sco6393-4) has inserted into the intergenic region between sco6808 and sco6809 and this was confirmed by PCR (Fig. 2a–c). Although this could affect sco6808 expression, deletion of sco6808 has no effect on growth or development under standard laboratory conditions (Fig. 3a). This and the intergenic position of IS21 in BJT1004, led us to hypothesise that IS21 might disrupt a non-coding RNA. Examination of RNA sequence data for S. coelicolor M145 confirmed the presence of a 189 nt transcript initiating 107 bp upstream of sco6808 and reading into the last 82 nucleotides of the sco6809 gene (data from GSM1121652 and GSM1121655 RNA sequencing; Supplementary Text S1 and 2; Fig. 2a,b). Following convention we named this putative small RNA scr6809 for S. coelicolor RNA 6809. Deletion of scr6809, without disrupting the coding sequences of either sco6808 or sco6809, resulted in pleiotropic effects, including colonies that look like wild-type and colonies defective in growth, aerial hyphae formation, sporulation or antibiotic overproducers. Restreaking ∆scr6809 colonies with wild-type appearance gave rise to a range of colony morphologies, including growth and developmental defects (Fig. 3b). ∆scr6809 colonies that were already defective in growth, development or antibiotic production maintained those phenotypes after restreaking. A previous report showed that a ∆sco6808 mutant exhibits accelerated production of actinorhodin and undecylprodigiosin as well as precocious spore formation on R5 medium18. There was no observable difference between the wild-type and ∆sco6808 strains under the growth conditions used here but disruption of sco6808 in strain BJT1004 improved sporulation most likely because scr6809 has been restored to its genome (Fig. 3a).

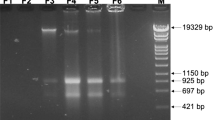
IS21 insertion into scr6809.
The sco6811-08 region of the S. coelicolor M145 genome contains 4 genes (sco6811 (Purple), sco6810 (green), sco6809 (yellow) and sco6809 (blue) and a putative sRNA scr6809 (large red) along with 3 putative promoters (broken arrows). Representations of the WT loci (a) and that sequenced from BJT1004 (b) indicate where an IS21 element (sco6393 and sco6394) has inserted within scr6809. PCR verification of the IS21 insertion with primers JM0093 and JM0094 (small red arrows) was carried out (c) using M145 (lane 1), BJ1001 (lane 2) and BJT1004 (lane 3) genomic DNA. Lanes marked L contain the size ladders (Invitrogen 1 kb plus DNA ladder), lane 1 contains the PCR product using wild-type M145 DNA (514 bp), lane 2 contains the PCR product using ∆lsp strain BJT1001 DNA and lane 3 contains the PCR product using genomic DNA from the cis complemented ∆lsp strain BJT1004 (both 2884 bp.

Analysis of the IS21 disrupted genomic region in S. coelicolor.
Colony morphology (a) shows that deletion of sco6808 has no obvious effect on growth or development in wild-type M145 but does partially restore sporulation in BJT1004 (recovery of scr6809). Disruption of scr6809 in M145 results in a range of pleiotropic morphological and developmental phenotypes (b).
To determine whether IS21 disruption of scr6809 is induced by deletion of lsp, we isolated ten more non-clonal lsp mutants. Of these 10 mutants, both single cross-over (n = 7) and double cross-over (n = 3) strains were isolated following the introduction of cosmid St4A10∆lsp into wild-type M145. Microscopy images of these strains showed a range of colony morphologies (Fig. 4a) and PCR results of the lsp loci confirm their genotypes, either single or double cross-over (Fig. 4b). These results suggest that secondary mutations are occurring in single cross-over strains containing intact lsp genes. The intergenic region between sco6808 and sco6809 was amplified to determine if these results were due to the disruption of scr6809. The size of the PCR products matched the predicted wild-type size for each strain and indicated that none of these lsp mutants contain an IS21 insertion suggesting that the original observation is not specific to lsp mutants (Fig. 4c). Consistent with this conclusion, the frequency with which lsp mutants could be isolated was not increased in BJT1004 relative to the wild-type strain suggesting that none of the mapped mutations in BJT1004 are lsp specific suppressors. All attempts to over-express scr6809 in S. coelicolor M145, S. scabies 87-22 and S. venezualae ATCC 10712 resulted in no observable phenotype but as the same vectors failed to complement the ∆scr6809 strain, this suggests that functional scr6809 was not expressed from vector pJM017. Cumulatively these results show that deletion of lsp is not solely responsible for the secondary mutations and this led us to hypothesise that these mutations accumulate as a result of duplicating cell division genes when we introduced cosmid St4A10∆lsp. These results also suggest a role for scr6809 in development, but this was not pursued in this work.

New lsp mutants generated using ReDirect do not contain the IS21 mutation.
Colony morphology of mutants lsp::apr 1–10 (corresponding to strains JTM008.01–JTM008.10), both single (n = 7, colonies 1–2, 4–7 and 9–10) and double cross-overs (n = 3, colonies: 3 and 7–8) show a range of phenotypes (a). PCR of the lsp loci (b) indicates colonies are either a single (wild-type and/or mutant band) or double (mutant band only) cross-overs (WT = 687 bp, mutant = 1447 bp). PCR of the scr6809 loci (c) indicates that strains 1–10 have intact scr6809 with no Insertion (WT = 514 bp, IS21 insertion = 2884 bp) using wild-type M145 and BJT1004 as controls (labeled “+” and “−” respectively).
Introduction of wild-type St4A10 results in a pleiotropic phenotype
The ReDirect PCR-targeting method19,20 used to delete the lsp gene utilized cosmid St4A10, which contains a ~40 kb region of the S. coelicolor genome spanning genes sco2069–2104 (Supplementary Table S2). Conjugation of St4A10∆lsp into S. coelicolor transiently duplicates all the genes on that cosmid (except lsp) and because this region includes cell division genes (ftsZ, ftsQ, ftsW, ftsI and ftsL) and essential cell wall synthesis genes (murG, murD, murX, murF and murE) we reasoned that transient over-expression of these genes, rather than deletion of lsp, is responsible for at least some of the spontaneous secondary mutations and the resulting pleiotropic phenotype. To test this idea we introduced an origin of transfer into the wild-type St4A10 cosmid backbone and then conjugated this cosmid into wild-type S. coelicolor M145. We used growth in the presence of kanamycin to select for single cross-over events where the whole cosmid is integrated into the chromosome, thus duplicating the S. coelicolor genes on St4A10. Analysis of these single cross-over strains revealed them to be genetically unstable, with many initially appearing similar to the observed ∆lsp phenotype, i.e. forming small colonies that are delayed in sporulation (Fig. 5). However, they do not over-produce the blue antibiotic actinorhodin which was an obvious characteristic of S. coelicolor ∆lsp. In addition, colonies arising from the M145::St4A10 strain acquired more significant developmental issues upon prolonged maintenance and restreaking onto MS agar containing kanamycin (not shown). This suggests that this strain accumulates spontaneous secondary mutations as a direct result of carrying two copies of the genes on cosmid St4A10 and also supports our hypothesis that the observed ∆lsp phenotype is at least in part due to duplication of the genes on cosmid St4A10. This is consistent with the fact that complementation of ∆lsp restored lipoprotein biogenesis and all detectable lipoproteins to the cell membrane but did not restore wild-type colony morphology5.

Introduction of wild-type St4A10 causes a pleiotropic phenotype.
Conjugation of M145 with St4A10 bla::hyg results in non-wildtype phenotypes similar to those observed in the St4A10 lsp::apr single cross-overs.
Targeted deletion of lsp results in a small colony phenotype
To test how much deletion of lsp gene alone contributed to the phenotype of BJT1001 (the ∆lsp strain generated using ReDirect) we undertook a targeted disruption of lsp in wild-type S. coelicolor M145 using a suicide vector, which does not duplicate or affect any other coding sequences. The lsp suicide vector, pJM016 (Table 1), was introduced into wild-type S. coelicolor by conjugation and ex-conjugants were selected by growing on MS agar plates containing apramycin. Following introduction of the pJM016, two colony types were observed (Fig. 6), one with wild-type appearance and the other with small colonies that over-produce actinorhodin, reminiscent of the lsp mutant BJT1001. PCR testing of the genomic DNA of both morphotypes revealed that those with the wild-type colony morphology are indeed wild-type strains with a fully functioning lsp gene whereas those with a small colony phenotype that over-produce actinorhodin have disruptions in lsp caused by pJM016. PCR amplification and sequencing of the loci in the small colony variants revealed an interesting and unexpected recombination event had occurred. The vector and almost all of the lsp gene have been removed such that all that remains is the apramycin resistance cassette (Supplementary Text S4 and S5). These data confirm that lsp is not essential in S. coelicolor and that loss of Lsp function (and the resulting loss of lipoproteins from the cell membrane) results in a growth and developmental defect and overproduction of the blue antibiotic actinorhodin, as observed previously5.

Targeted disruption of lsp using a suicide vector results in a small colony phenotype that over-produces actinorhodin.
To test how much of the BJT1001 phenotype is due to loss of lsp we disrupted the lsp gene using a suicide vector which does not affect or duplicate any other target genes. Plate images (a) show two distinct phenotypes following insertion of the suicide vector (pJM016) into M145, either a wildtype appearance (M145 pJM016 (1), n = 3 corresponding to JTM018.03-04 and 08) or a small colony phenotype over producing actinorhodin (M145 pJM016 (2), n = 7, corresponding to strains JTM018.01-2, 05-07 and 09-10) similar to our original observation of the lsp phenotype5, alongside lsp loci PCR results (b). All strains with intact lsp show a wild-type phenotype while those with disrupted lsp have the reported lsp phenotype.
Discussion
The results presented here show that the pleiotropic nature of S. coelicolor ∆lsp strain BJT1001 resulted from the introduction of cosmid St4A10∆lsp and this was most likely caused by over-expression of the cell division and cell wall biosynthesis genes carried on that cosmid (Supplementary Table S2). We have further shown that the resulting secondary mutations do not make it easier to delete lsp suggesting they are not lsp-specific suppressors. Genetic manipulation has always been challenging in Streptomyces bacteria and the ReDirect PCR targeting method 13 years ago was a significant development but our work is a cautionary tale to others to consider the effects of using large insert cosmid libraries in the genetic manipulation of bacteria. Fortunately, recent advances in CRISPR/Cas9 editing of Streptomyces genomes21 negate the need for a cosmid library and techniques such as this will further accelerate research into the basic biology of Streptomyces and other filamentous actinomycetes. This is vital because the secondary metabolites derived from these bacteria are an under utilised reservoir from which new anti-infectives and other drugs can and must be developed. Moreover, our identification of a new small RNA scr6809 and the demonstration that its deletion results in a range of growth and developmental defects add to the growing appreciation of the significance of small RNAs in streptomycetes22,23,24.
Materials and Methods
Bacterial strains and culture conditions
All primers, plasmids and strains used are listed in Table 1. Strains were routinely grown as previously described5 following the recipes of Kieser et al.25. E. coli was grown in LB or LB –NaCl for Hygromycin selection and S. coelicolor M145 and its derivatives were grown on Soya Flour Mannitol (SFM) medium to study growth and development or LB culture for genomic isolations.
Gene deletions and complementation
Gene deletions were carried out following the ReDirect method of PCR-targeting26 as previously described Hutchings et al.27. Disruption of lsp (sco2074::apr) on cosmid St4A10 (pJM013, St4A10∆lsp) using the pIJ773 apramycin disruption-cassette and sco6808 (sco6808::apr) and scr6809 (scr6809::apr) on cosmid St1A2 (pJM010-St1A2∆sco6808 and pJM012-St1A2∆sco6808 respectively) using primers JM0101-2, JM0083-84 and JM0091-2 respectively were confirmed by PCR using primers JM0150-1, JM0085-6 and JM0093-4 respectively. Introduction of the wild-type cosmid St4A10 was facilitated by introducing an oriT by disruption and replacement of the Supercos-1 backbone bla resistance gene (pJM014–St4A10bla::hyg) using primers JM0095-6 and the hygromycin disruption cassette from pIJ10701, confirmed using primers JM0099-100. The lsp suicide vector pJM016 was produced by introducing a 411 bp fragment of the lsp gene with an N-terminal BamHI site, amplified with primers JM0117-8 and cloned into pGEM T-Eazy. The BamHI site was then used to subclone the BamHI fragment from a pIJ773 digest, containing an apr disruption cassette. An overexpression construct, pJM017 was synthesised by Genscript to include the pMC500 MCS and terminators28 with scr6809 (sequence included in Supplementary Text S5). All constructs were subsequently conjugated into S. coelicolor following the method described by Gust et al.26.
Genomic DNA isolation
Genomic DNA was isolated from M145 and BJT1004 following the Pospiech and Neumann (1995) salting out method as described by Keiser et al. (2000). Mycelium from a 30 ml culture was resuspended in 5 ml SET buffer containing 1 mg/ml lysozyme and incubated at 37 °C 30–60 min. To this lysate, 140 μl of proteinase K solution (20 mg ml−1) was added, mixed, then 600 μl of 10% SDS added, mixed and incubated at 55 °C for 2 h, with occasional mixing throughout. After this incubation 2 ml of 5 M NaCl was added, mixed and left to cool to 37 °C before adding 5 ml chloroform, mixed at 20 °C for 30 min. Samples were centrifuged at 4500 × g for 15 min at 20 °C. The supernatant was removed to a fresh tube and DNA precipitated by adding 0.6 volumes of 100% isopropanol. Tubes were mixed by inversion and after at least 3 min DNA spooled out using a sterile Pasteur pipette. The DNA was rinced in 70% ethanol, air dried and dissolved in 1–2 ml TE buffer (10 mM Tris-HCl pH 7.8, 1 mM EDTA) at 55 °C.
Genome resequencing and secondary mutation identification
The isolated DNA from our wild-type S. coelicolor M145 parent strain and BJT1004 were sent to both GATC Biotech and The Genome Analysis Centre (TGAC) for 35 bp paired end HiSeq Illumina sequencing. Assembly mapping and SNP identification was carried out with MIRA (Chevreux et al., 2004) using the reference genome NC_003888 (Bentley et al.29) as a scaffold for mapping each of the resequenced genomes. Putative SNPs were detected in each sample independently reporting the SNP position, the nucleotide change, the number of reads that sequence the region, those containing wild-type or mutated nucleotides and a percentage change. Each set of results was then compared by eye to determine the likely hood that a SNP was real by number of reads containing the mutation and its presence in each sample. Larger mutations (rearrangements) were identified in the same fashion.
Microscopy
Brightfield images were acquired using a Zeiss M2 Bio Quad SV11 stereomicroscope. Samples were illuminated from above using a halogen lamp images captured with an AxioCam HRc CCD camera. The AxioVision software (Carl Zeiss, Welwyn Garden City, UK) was used for image capture and processing.
Additional Information
How to cite this article: Munnoch, J. T. et al. Cosmid based mutagenesis causes genetic instability in Streptomyces coelicolor, as shown by targeting of the lipoprotein signal peptidase gene. Sci. Rep. 6, 29495; doi: 10.1038/srep29495 (2016).
References
Hutchings, M. I., Palmer, T., Harrington, D. J. & Sutcliffe, I. C. Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold ’em, knowing when to fold 'em. Trends Microbiol. 17, 13–21 (2009).
Buddelmeijer, N. The molecular mechanism of bacterial lipoprotein modification–How, when and why? FEMS Microbiol. Rev. 39, 246–261 (2015).
Sutcliffe, I. C., Harrington, D. J. & Hutchings, M. I. A phylum level analysis reveals lipoprotein biosynthesis to be a fundamental property of bacteria. Protein Cell 3, 163–170 (2012).
Gralnick, J. A., Vali, H., Lies, D. P. & Newman, D. K. Extracellular respiration of dimethyl sulfoxide by Shewanella oneidensis strain MR-1. Proc. Natl. Acad. Sci. USA 103, 4669–4674 (2006).
Thompson, B. J. et al. Investigating lipoprotein biogenesis and function in the model Gram-positive bacterium Streptomyces coelicolor. Mol. Microbiol. 77, 943–957 (2010).
Shruthi, H., Babu, M. M. & Sankaran, K. TAT-pathway-dependent lipoproteins as a niche-based adaptation in prokaryotes. J. Mol. Evol. 70, 359–370 (2010).
Rahman, O., Cummings, S. P., Harrington, D. J. & Sutcliffe, I. C. Methods for the bioinformatic identification of bacterial lipoproteins encoded in the genomes of Gram-positive bacteria. World J. Microbiol. Biotechnol. 24, 2377–2382 (2008).
Okuda, S. & Tokuda, H. Lipoprotein Sorting in Bacteria. Annu. Rev. Microbiol. 65, 239–259 (2011).
Narita, S. I., Matsuyama, S. I. & Tokuda, H. Lipoprotein trafficking in Escherichia coli. Arch. Microbiol. 182, 1–6 (2004).
Jan, G., Fontenelle, C., Le Hénaff, M. & Wróblewski, H. Acylation and immunological properties of Mycoplasma gallisepticum membrane proteins. Res. Microbiol. 146, 739–750 (1995).
Asanuma, M. et al. Structural evidence of α-aminoacylated lipoproteins of Staphylococcus aureus. FEBS J. 278, 716–728 (2011).
Nakayama, H., Kurokawa, K. & Lee, B. L. Lipoproteins in bacteria: Structures and biosynthetic pathways. FEBS J. 279, 4247–4268 (2012).
Kurokawa, K. et al. The Triacylated ATP Binding Cluster Transporter Substrate-binding Lipoprotein of Staphylococcus aureus Functions as a Native Ligand for Toll-like Receptor 2. J. Biol. Chem. 284, 8406–8411 (2009).
Serebryakova, M. V. et al. The acylation state of surface lipoproteins of mollicute Acholeplasma laidlawii. J. Biol. Chem. 286, 22769–22776 (2011).
Widdick, D. a. et al. Dissecting the complete lipoprotein biogenesis pathway in Streptomyces scabies. Mol. Microbiol. 80, 1395–1412 (2011).
Tschumi, A. et al. Functional analyses of mycobacterial lipoprotein diacylglyceryl transferase and comparative secretome analysis of a mycobacterial lgt mutant. J. Bacteriol. 194, 3938–3949 (2012).
Xiao, Y. & Wall, D. Genetic redundancy, proximity and functionality of lspA, the target of antibiotic TA, in the Myxococcus xanthus producer strain. J. Bacteriol. 196, 1174–1183 (2014).
Yang, Y. H. et al. Finding new pathway-specific regulators by clustering method using threshold standard deviation based on DNA chip data of Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 80, 709–717 (2008).
Gust, B., Challis, G. L., Fowler, K., Kieser, T. & Chater, K. F. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. USA 100, 1541–1546 (2003).
Redenbach, M. et al. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3(2) chromosome. Mol. Microbiol. 21, 77–96 (1996).
Cobb, R. E., Wang, Y. & Zhao, H. High-Efficiency Multiplex Genome Editing of Streptomyces Species Using an Engineered CRISPR/Cas System. ACS Synth. Biol. 4, 723–728 (2014).
Vockenhuber, M. P. et al. Deep sequencing-based identification of small non-coding RNAs in Streptomyces coelicolor. RNA Biol. 8, 468–477 (2011).
Moody, M. J., Young, R. A., Jones, S. E. & Elliot, M. A. Comparative analysis of non-coding RNAs in the antibiotic-producing Streptomyces bacteria. BMC Genomics 14, 558 (2013).
Romero, D. A. et al. A comparison of key aspects of gene regulation in Streptomyces coelicolor and Escherichia coli using nucleotide-resolution transcription maps produced in parallel by global and differential RNA sequencing. Mol. Microbiol. 94, 963–987 (2014).
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A. Practical Streptomyces Genetics. John Innes Cent. Ltd. 529, doi: 10.4016/28481.01 (2000).
Gust, B., Kieser, T. & Chater, K. PCR targeting system in Streptomyces coelicolor A3(2). John Innes Cent. 3, 1–39 (2002).
Hutchings, M. I., Hong, H. J., Leibovitz, E., Sutcliffe, I. C. & Buttner, M. J. The sigma(E) cell envelope stress response of Streptomyces coelicolor is influenced by a novel lipoprotein, CseA. J. Bacteriol. 188, 7222–7229 (2006).
Gao, C., Mulder, D., Yin, C. & Elliot, M. a. Crp Is a Global Regulator of Antibiotic Production in Streptomyces. MBio 3, 1–12 (2012).
Bentley, S. D. et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147 (2002).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645 (2000).
MacNeil, D. J. et al. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68 (1992).
Hopwood, D. A. et al. Genetic manipulation of Streptomyces: a laboratory manual. (1985).
Acknowledgements
We thank Marie Elliot for sharing unpublished RNA sequence data for Streptomyces coelicolor M145. This work was supported by a NERC PhD studentship to JTM and BBSRC grants BB/F009429/1 and BB/F009224/1 to MIH and TP, respectively.
Author information
Authors and Affiliations
Contributions
J.T.M. designed and carried out the experiments, J.T.M., I.C.S., T.P. and M.I.H. designed experiments, J.T.M., D.A.W., I.C.S., T.P. and M.I.H. analysed data and wrote the manuscript, D.A.W. prepared DNA for sequencing, G.C. analysed sequencing data.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Munnoch, J., Widdick, D., Chandra, G. et al. Cosmid based mutagenesis causes genetic instability in Streptomyces coelicolor, as shown by targeting of the lipoprotein signal peptidase gene. Sci Rep 6, 29495 (2016). https://doi.org/10.1038/srep29495
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep29495
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
