
Abstract
Lithium iron phosphate (LiFePO4) electrodes with fractal granularity are reported. They were made from a starting material prepared in water by a low cost, easy and environmentally friendly hydrothermal method, thus avoiding the use of organic solvents. Our method leads to pure olivine phase, free of the impurities commonly found after other water-based syntheses. The fractal structures consisted of nanoparticles grown into larger micro-sized formations which in turn agglomerate leading to high tap density electrodes, which is beneficial for energy density. These intricate structures could be easily and effectively coated with a thin and uniform carbon layer for increased conductivity, as it is well established for simpler microstructures. Materials and electrodes were studied by means of XRD, SEM, TEM, SAED, XPS, Raman and TGA. Last but not least, lithium transport through fractal LiFePO4 electrodes was investigated based upon fractal theory. These water-made fractal electrodes lead to high-performance lithium cells (even at high rates) tested by CV and galvanostatic charge-discharge, their performance is comparable to state of the art (but less environmentally friendly) electrodes.
Similar content being viewed by others
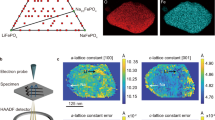


Introduction
Lithium ion batteries (LIBs) have revolutionized portable electronic devices in the past two decades, and are primed to make a great impact on transportation technology by powering electric vehicles (EVs). However, this new application demands that LIBs offer higher energy and higher power capabilities at a lower cost and with environmentally friendly materials1. Since the seminal work of Goodenough and col.2, lithium iron phosphate (LiFePO4) has been researched as cathode material for LIBs thanks to its low cost, abundant raw materials, safety, low toxicity, structural stability and excellent electrochemical properties. The active material can be reversibly charged and discharged with a stable voltage profile at 3.45 V vs. Li+/Li with a very small change in unit cell parameters during the LiFePO4/FePO4 phase transition. On the other hand, for the development of high power batteries based on this material, it is essential to understand and overcome the factors limiting lithium transport through the electrode. Indeed, despite its high theoretical specific capacity (170 mAh/g) and long cycling lifetime, the high-rate performance of the raw LiFePO4 is restricted by its poor electronic conductivity (10−9 S/cm) and slow lithium diffusion3. Many different approaches involving surface coating have been tried to improve the capacity and rate performance of LiFePO4 as cathode for LIBs. Increasing the conductivity by coating the LiFePO4 surface with carbon4 or conducting polymers5,6 have been two of the most popular.
In addition to coating, the control of surface microstructure constitutes another general approach towards faster electrode reaction for batteries. Among many possible alternatives, fractal electrode design is proposed as a very promising approach for high-performance batteries, since it greatly improves the surface to volume ratio while providing a high energy-density material with large tap density. Thus, the growth of self-assembled nanoparticles into larger microstructures can provide high surface area for high power and large bulk for high energy density. In addition, mass transfer in LIBs can be improved by spreading the reaction sites throughout the entire volume of the device. Space filling fractal networks can work to ensure that there is efficient charge transfer from a huge effective surface area to a current collector. Last but not the least; a fractal granular microstructure could minimize the internal resistance of the electrode. Recently, micro-sized yet porous LiFePO4 structures have been reported with high electronic conductivity and fast Li+ permeation. For instance, Liu et al. reported the synthesis of 3D nanoporous spherical LiFePO4/C material by spray pyrolysis techniques7. The 3D conductive carbon coating with interconnected pore networks facilitate both electron transport as well as lithium ion diffusion within the particles, leading to excellent cycling performance and rate capability7. However, most of the techniques used to grow fractal structures are based on organic solvents, surfactants and templates which make them less attractive considering production cost of the materials8. Therefore the development of effective routes for the synthesis of fractal micro-structured LiFePO4/C at an ever lower cost still represents a great challenge.
Herein, we are introducing an example of fractal granular LiFePO4 electrode with excellent electrochemical properties. Briefly, a cost-effective, water-based hydrothermal method is used to prepare micro-nano structured fractal LiFePO4 materials and electrodes. The samples were fully characterized with different physical-chemical techniques to provide proof of concept. Furthermore, the present work is aimed to investigate the boundary conditions at the electrode surface for lithium transport, and the effect of surface roughness on the diffusion-controlled lithium transport. The surface morphology of the electrodes was examined by AFM and their apparent self-similar dimensions were determined by a triangulation method.
Results
Figure 1(a) shows XRD patterns of pristine LiFePO4 and LiFePO4/C samples. All diffraction peaks are indexed to orthorhombic LiFePO4 (JCPDS card number 081-1173, space group Pnma). It is very important to note that no impurities are detected. This is in contrast with some previous reports on solvothermal syntheses making use of water or organic solvents leading to detrimental impurities9. Some researchers reported impurities such as iron phosphides Fe2P10 which often form at high temperatures (>600 °C)11 or LiFe(P2O7)12, Fe (II, III) pyrophosphates or phosphates Li3Fe2(PO4)313 and Li3PO414. This lack of impurities confirms the suitability of our water-based hydrothermal method for the successful synthesis of pure stoichiometric LiFePO4 material. Moreover, all diffraction peaks are intense and narrow for our samples, indicating a high degree of crystallinity of the LiFePO4 phase prepared both before and after carbon-coating.

(a) XRD patterns of pristine LiFePO4 and LiFePO4/C samples, (b) TGA curves of LiFePO4 and LiFePO4/C, (c) Raman spectrum of the LiFePO4/C.
Figure 1(b) shows TGA curves of pristine LiFePO4 and LiFePO4/C composite under flowing air atmosphere. These experiments were carried out to study the thermal stability of the materials and to determine the exact amount of carbon coated on LiFePO4 active phase. After an initial weight loss associated to loss of water, the TGA curves show a weight gain of 5.0% for LiFePO4 and 2.4% for LiFePO4/C between 250–650 °C. The weight uptake of pristine LiFePO4 can be explained by the following oxidation reaction15:

The amount of carbon coated on LiFePO4 was calculated by measuring the difference between the total weight gain of LiFePO4 and that of LiFePO4/C and turned out to be 2.6%.
The LiFePO4/C sample was also analyzed by Raman spectroscopy in order to investigate the nature of coated carbon. As seen in Fig. 1(c), two intense broad peaks were recorded at 1330 cm−1 and 1595 cm−1 corresponding to the A1g vibration mode of the disordered carbon (D-band) and E2g vibration mode of the ordered graphitic carbon (G-band), respectively16. The ratio of intensities of D-band to G-band (ID/IG) is 0.87, indicating sp2 carbon, which would enhance the electronic conductivity of the LiFePO4 material17.
The chemical composition and valence state of LiFePO4/C material was confirmed by XPS analysis. Figure 2(a) shows the wide range-scanning spectrum, which consists of Li, Fe, P, O and C components confirming formation of LiFePO4/C material. The Fe2p spectrum (Fig. 2(b)) exhibits two major peaks (Fe 2p3/2 and Fe 2p1/2) at binding energies of 710.9 eV and 724.2 eV indicating Fe(II) valence state which is characteristic of the olivine-type LiFePO4 products18. Figure 2(c) shows the deconvolution of the C 1 s spectrum in LiFePO4/C, clearly displaying the lower binding energy featured at 284.6 eV corresponding to C-C carbon and the higher binding energy featured at 286.1 eV followed by a shoulder at 288.9 eV, which was typically assigned to C-O-C, O-C = O arising from epoxide, carboxyl functionalities19. Figure 2(d) shows the fine structure of C KLL transition, which is strongly affected by the sp2/sp3 configuration. Parameter D can be considered as a fingerprint of the type of carbon hybridization, showing values of 13.7 eV for sp2 and 21.2 eV for sp3 hybridization states20. For extended carbon phases the D value can therefore represent a diagnose of the conducting character of a given material since sp2 is associated to graphitic carbons, better conducting than sp3 structures. The D parameter value for carbon in our LiFePO4/C sample turned out to be 16.2 eV which indicates an intermediate composition of sp2 and sp3, corresponding to a mixture of ca. 2/3 sp3 and 1/3 sp2 C in the sample. This means that although the carbon coating of LiFePO4 is not purely sp2 it does contain enough sp2 carbon to provide electrical conductivity of the material.

XPS spectra of the LiFePO4/C powder calcined at 700 °C (a) Full spectrum of LiFePO4/C (b) narrow spectrum of Fe2p (c) core-level XPS spectrum of C1s and (d) Derivate of the carbon auger peak.
The microstructures of the LiFePO4 and LiFePO4/C samples were investigated by SEM and are presented in Fig. 3(a,b). As seen in Fig. 3(a), our LiFePO4 sample is constituted of large microspheres a few microns in diameter formed in turn by a very large number of nanoparticles of ca. 200 nm in a configuration, which could be described as fractal. Indeed, nanosized primary particles conformed the surface of microsized secondary particles form a microstructure reminiscent of the black fractal sketch shown in the Fig. 4. This configuration presents the advantages of nanoparticulate matter and the easy handling and high tap density of microparticles. Inset of Fig. 3(a,b) show details of the closely packed primary nanoparticles featuring inter-particle slit pores. Comparing both of them allows us to conclude that this fractal granularity is retained after the pyrolysis treatment for carbon-coating (Fig. 3b). These results are in contrast to previously reported conventional hydrothermal synthesis of LiFePO4 which led to very large microcrystals (in air) and required N2 atmosphere for further growth of smaller particles21. In the present work, LiFePO4 fractal granular geometry is achieved with the simple addition of polyethyleneimine (PEI), which controls the growth of these optimal nano-microstructures without any special treatment.

(a,b) SEM images of LiFePO4, LiFePO4/C, inset shows high magnified images. For these two images isolated spheres were selected. Most abundant agglomerates of these secondary spherical particles are shown in Supporting Information 2. (c,d) HR-TEM images of LiFePO4, LiFePO4/C, inset shows low magnified images (e,f) SAED patterns of LiFePO4, LiFePO4/C, respectively.

Scheme of LiFePO4 fractal granularity design.
The samples were also studied with TEM. Figure 3(c,d) compares high-resolution images of pristine and carbon-coated LiFePO4 primary nanocrystals. From the HRTEM image (Fig. 3c,d), one can clearly see the lattice fringes with an interplanar spacing of 0.34 nm for both LiFePO4 and LiFePO4/C nano-particles, which is identified as the characteristic interplanar spacing of the (111) plane of olivine-type LiFePO4 material. The carbon layer covering the LiFePO4 surface has an average thickness of 3 nm and is clearly observed in Fig. 3(d). No long-range order is apparent in this carbon layer; yet it must be composed of conducting graphitic carbon (sp2 according to Raman) comparable to graphene domains (but not diffracting due to its small thickness). Insets of Fig. 3(c,d) show low-magnification TEM images of LiFePO4 and LiFePO4/C single crystals, confirming identical nanoparticle morphologies. Moreover, the selected area electron diffraction (SAED) patterns of both LiFePO4 and LiFePO4/C (Fig. 3e,f) were indexed as (111) plane confirming the LiFePO4 orthorhombic structure in agreement with XRD results.
The fractal granularity of LiFePO4 leads to a characteristic slit porosity formed by the primary nanosized crystals (better seen in the inset of Fig. 3(a,b). These inter-particle pores extend from the surface to the inner core of the spheres and can facilitate deep penetration of liquid electrolyte solution into the microspheres, thus providing an improved interface contact between the electrode and electrolyte. (supporting information S1).
To further investigate the surface properties of fractal LiFePO4 material, we performed Brunnauer-Emmett-Teller (BET) analysis of adsorption isotherms shown in Fig. 5(a). The LiFePO4/C sample shows a typical isotherm of type IV with hysteresis loop in a relative pressure (p/p0) range of 0.4–1.0, implying the formation of slit-like pores22, a type of porosity which can be easily understood as a result of the stacking of nanoparticles. The measured BET surface area was found to be 14.8 m2/g for LiFePO4/C. Figure 5(b) shows the Barett-Joyner-Halenda (BJH) pore size distribution curve with a distinct maximum centered at ~3.7 nm. This confirms the mesoporous nature of the LiFePO4/C fractal structure. The mesoporosity of LiFePO4/C samples results from a combination of internal space of the agglomerated nanoparticles and microspheres. Despite the modest value of the BET surface area, such a mesoporous structure facilitates diffusion of Li ions from the electrolyte into the electrode bulk by providing short diffusion lengths.

(a) Nitrogen adsorption/desorption isotherms for LiFePO4/C (b) Barret-Joyner-Halenda (BJH) pore size distribution curve for LiFePO4/C.
The microstructural characterization of LiFePO4 was completed with two more complementary techniques, namely AFM (20 × 20 microns) for electrode area analyses and SEM for cross-section (20 microns) analyses. Thus, cross-section SEM images of LiFePO4 and LiFePO4/C electrodes coated on Al substrates with elemental composition analyses were carried out and are shown and discussed in supporting information S2 and S3. The film thickness was between 15 μm and 20 μm for LiFePO4 and 5–10 μm for LiFePO4/C.
In order to get a better understanding of Li transport and confirm the fractal nature of LiFePO4, surface roughness was analyzed in triangles of various sizes for a total area of 20 × 20 microns by AFM. Figure 6(a,b) shows AFM surfaces of LiFePO4 and LiFePO4/C film electrodes, respectively. It can be noted that the LiFePO4/C film electrode shows less roughness than LiFePO4 film electrode. In order to get quantitative insights, root mean square (rms) roughness of both electrodes was evaluated (Table 1). Since these structures appear to be fairly similar on various length scales, this surface can be regarded as a self-similar fractal. The triangulation method, reported elsewhere23, was used to determine the self-similar fractal dimensions. Figure 6(c,d) gives logarithmic scale dependence of scaled surface area (SSA) on projected triangle size (TS) obtained from the LiFePO4 and LiFePO4/C film electrodes, respectively.

(a,b) AFM images of LiFePO4 and LiFePO4/C electrodes, respectively (c,d) Dependence of surface area SA on triangle size TS obtained from AFM images of LiFePO4 and LiFePO4/C electrode, respectively. The slope s means (d log SSA/d log TS).
For both electrodes, one can find clearly a linear relationship between the log(SSA) and log(TS), indicating the self-similar scaling property of the surface. It is generally known that the self-similar fractal dimension Df, ss of a surface is given in terms of the slope of the straight line ‘s’ (=d log SSA/d log TS) as24

From Fig. 6, the outer cut-offs for their fractal structures were found to be 6.5 and 5.5 μm, respectively. Here, it should be stressed that although both LiFePO4 film electrodes show the self-similar scaling properties, the spatial outer cut-offs for their fractal structures are different from each other.
The electrochemical properties of LiFePO4 and LiFePO4/C electrodes were further investigated, results presented in Fig. 7. The ionic diffusion in the self-similar fractal electrode was studied by CV. The sharp oxidation and reduction peaks in CV curves for LiFePO4/C electrode (see Fig. 7a,b) confirm the excellent reversibility of the Li extraction-insertion reaction as compared to that for pristine LiFePO4 electrode. The well-defined oxidation and reduction peaks at ca. 3.4 V and 3.6 V are assigned to the Fe2+/Fe3+ redox couple, corresponding to the Li insertion (Fe3+ to Fe2+) and extraction (Fe2+ to Fe3+) in the LiFePO4 crystal structure, respectively. In contrast, the CV curve recorded for pristine LiFePO4 is broader and exhibits relatively low current density. This may be attributed to low conductivity and slow Li diffusion in pristine LiFePO4 electrode.

(a,b) Cyclic voltammetry (CV) curves for LiFePO4 and LiFePO4/C electrodes at different scan rates, respectively (c) Dependence of anodic peak current Ipeak on scan rate for LiFePO4 and LiFePO4/C electrodes with logarithmic scale (d) Charge/discharge curves at various C-rates for LiFePO4/C (5th cycle) (e) Discharge capacity at different C-rates of LiFePO4 and LiFePO4/C (f) Cycling performance of pristine LiFePO4 and LiFePO4/C at 1C.
Strømme et al. suggested the peak current method to determine the fractal dimension of a given electrode surface by using cyclic voltammetry: when the recorded cyclic voltammetry current is limited by diffusion of the electroactive species and away from the electrode surface, the fractal dimension df of the reaction site on the surface can be obtained by the following equation,

where

And Ipeak is the intensity of the reduction peak, and υ is the scan rate. The slope of the plot log(Ipeak) vs log (v) is αf and is called the fractal parameter, which is related to the fractal dimension of the surface as indicated in (4)
Figure 7(a–c) is also used to provide information of the ion diffusion coefficient using an improved Randles–Sevcik equation (see Supplementary Information S4) applicable to fractal electrodes25

where Ipeak is the peak current, ‘Γ’ the gamma function, ‘γ’ a geometrical factor close to π−1, λ0 the length corresponding to the outer cut-off of the fractal electrode, ‘n’ the number of transferred electrons per atom, ‘F’ Faraday’s constant, ‘v’ the scan rate, C the concentration of the electrolyte, χmax a dimensionless function of the fractal parameter given in ref. 26, R the gas constant, and T the temperature. ‘n’ is equal to 1 since the reaction is

According to equation 4, the diffusion coefficients were found to be 4.11 × 10−14 cm2/s for LiFePO4 and 1.83 × 10−13 cm2/s for LiFePO4/C, comparable to previous values found in the literature (6.56 × 10−16 to 0.52 × 10−12)27,28, indicating considerable increases in diffusivity after modifications. This study examines the effects of a carbon coating on the electrochemical performances of LiFePO4. The effects of the carbon coating as well as the mechanisms for the improved electrochemical performances after modification are discussed based on the diffusivity data and confirm better diffusion in the C-coated sample. The results show that the capacity of bare LiFePO4 decreased sharply, whereas the LiFePO4/C shows a well maintained initial capacity.
Figure 7(d) shows the charge/discharge curves at various C rates from 0.1 C to 10 C for LiFePO4/C cathodes vs. lithium anodes. The LiFePO4/C exhibits a high specific charge value of 159 mAh/g (ca. 94% of the theoretical capacity of 170 mAh/g) at 0.10 C rate (charge and discharge). This charge at a discharge voltage of 3.4 V and a tap density of 1.70 Kg/L (see below) results in energy density of 920 Wh/L. In contrast, cathodes of the pristine LiFePO4 do not show a high specific discharge capacity. Even at 0.10 C rate the pristine LiFePO4 shows a discharge capacity of 45 mAh/g wich correspond to 27% of the theoretical capacity. Both samples show good stability at diferent C-rates (Fig. 7e). The discharge capacity of LiFePO4/C at 10 C correspond to 31% of the value obtained at 0.10 C, whereas for pristine LiFePO4 the discharge capacity at 10 C represents 22% of that at 0.10 C (Fig. 7f). The pristine LiFePO4 cathode material was stable during extensive cycling at 1 C; the capacity retention over 40 cycles is 99%, which is remarkable. Figure 7d shows the very good cyclability of LiFePO4/C cathode material. This sample was stable over 100 cycles with 98% retention of capacity.
The electrochemical results achieved in the present work are comparable to other reports in the literature, while improving the synthetic procedure with a low-cost water-based simple procedure. For example, Chen et al. reported a discharge capacity of 150 mAh/g at 0.1 C rate and 100 mAh/g at 1 C22 for LiFePO4/C, Yang et al. reported discharge capacity of 140 mAh/g for porous graphene/LiFePO429 whereas Yu et al. prepared template-assisted porous LiFePO4 particles30 and reported capacity of 140 mAh/g at 0.10 C-rate.
Discussion
In summary, we have developed a low-cost and eco-friendly hydrothermal synthesis of fractal granular LiFePO4 in aqueous media leading to a pure material, free of the impurities commonly associated to other water-based syntheses. This LiFePO4 material is composed of 200 nm nanocrystals grown into hierarchically superior spherical microstructures, in turn aggregated into larger units, thus featuring fractal granularity. The size of nanoparticles in fractal micro-structure is tuned by a small amount of polyethyleneimine (PEI) (5%), which plays a triple role as i) reducing agent, preventing oxidation of Fe(II), ii) surface modifier, limiting the growth of individual nanoparticles and iii) as polymer host acting as binder. Moreover, AFM analysis confirms that both electrodes have self-similar fractal nature, although they have different spatial outer cut-offs for their fractal granularity. Carbon coating is also easily and effectively attained without altering the initial fractal granularity of LiFePO4 electrode. As expected, carbon coating improves the conductivity of LiFePO4, hence its rate capability and cycling stability as LIB electrode is greatly enhanced. Furthermore, the fractal granularity provides high tap density while maintaining a highly dispersed active material at the nanometer level for an optimized electrode-electrolyte interphase.
Methods
Synthesis of LiFePO4 fractal design
Fractal LiFePO4 materials were synthesized through an optimized hydrothermal method. The starting materials were FeSO4.7H2O (99%, Sigma-Aldrich), H3PO4 (85%, Sigma-Aldrich) and CH3COOLi.2 H2O (reagent grade, Sigma-Aldrich) and were all used as received. PEI solutions in water (50 wt%) was also purchased from Sigma-Aldrich, this solution was pre-warmed to 70 °C for faster precursor dissolution. The molar ratio of Fe:P:Li was kept at 1:1:3. In a typical reaction, 6.6618 g of lithium acetate was added to 70 ml of a preheated 5% polyethylene imine solution in water and stirred for 10 minutes. 6.6175 g of iron sulfate was dissolved in 5% PEI (70 ml) and stirred for 2 minutes. Then, 1.65 ml of 85% phosphoric acid was slowly added to the iron sulfate solution, and the resulting solution was stirred for 10 minutes. Finally, both solutions (Li and Fe/PO4 solutions) were mixed together, stirred for 10 minutes and transferred into 200 ml hydrothermal reactor (Teflon vessel sealed in a stainless-steel autoclave). The reactor was maintained at 200 °C for 24 h. The resulting LiFePO4 pale green powder was filtered; washed with deionized water/ethanol and dried at 80 °C in a vacuum oven overnight. The final weight of the powder was 3.4932 g, which represents 93% of the theoretical yield.
In the next step, LiFePO4 particles were coated with carbon using glucose as carbon source. A solution of 10 wt% glucose and 90 wt% LiFePO4 in water was prepared. This solution was stirred for 5 hours, and then sonicated for 10 minutes. Later, the sample was filtered and dried overnight at 90 °C in a vacuum oven. The sample was initially calcined at 350 °C for 3 h and then sintered at 700 °C for 10 h, all under nitrogen atmosphere. The tap density of LiFePO4 samples was 1.40 g/cm3 (pristine) and 1.70 g/cm3 (carbon-coated)31,32, which are above the average tap-density values reported in the literature for LiFePO431,33,34.
Materials characterization
The phase purity and crystalline structure of the samples were determined by X-ray diffraction (XRD) by means of PANalytical X′Pert PRO diffractometer using a CuKα radiation source (λ = 1.5418 Å). The morphology of the samples was studied by scanning electron microscopy (SEM, FE Quanta 650 F ESEM) and transmission electron microscopy (TEM, Tecnai G2 F20 HRTEM) operated at an acceleration voltage of 200 keV. N2 adsorption/desorption was determined by Brunauer-Emmett-Teller (BET) measurements using Micromeritics instrument (Data Master V4.00Q, Serial#:2000/2400). Thermal gravimetric analysis (TGA 500Q) was carried out under air atmosphere between room temperature and 850 °C, and the flow rate of the synthetic air was 10 ml/min. Raman spectra were recorded on a HORIBA Scientific LabRAM HR Raman spectrometer system using Ar laser. The X-ray photoelectron spectra (XPS) analyses were obtained by X-ray photoelectron spectroscopy (XPS, SPECS Germany, PHOIBOS 150). Surface morphologies of the self-affine fractal electrodes were obtained with a SPA 400 equipped with a SPI3800N Probe Station (Seiko Instruments Inc.) in the atomic force microscope (AFM) mode using commercial silicon nitride cantilevers.
Electrode preparation and electrochemical characterization
The cathodes were prepared by pressing a mixture of the active materials with carbon Super P (Timcal) and polyvinylidene fluoride (PVDF) binder in a weight ratio 85:10:5. They were mixed in a mortar for 5 minutes and then dispersed in N-Methyl-2-pyrrolidone and coated onto Al foil. Electrochemical test cells (Swagelok-type) were assembled in an argon-filled glove box with the coated Al disk as working electrode, lithium metal foil as the counter/reference electrode, and 1 M solution of LiPF6 in a 1:1 vol/vol mixture of ethylene carbonate and diethyl carbonate as the electrolyte. Glass microfiber filter paper was used as separator. For electrochemical battery test, the cells were charged and discharged galvanostatically within a fixed voltage window between 2.5 V and 4.0 V (with identical charge and discharge rates). Cyclic Voltammetry (CV) was performed in the same potential window at a scan rate between 5 mV/s to 0.1 mV/s. All electrochemical measurements were performed with Biologic VMP3 potentiostat/galvanostat.
Additional Information
How to cite this article: Cabán-Huertas, Z. et al. Aqueous synthesis of LiFePO4 with Fractal Granularity. Sci. Rep. 6, 27024; doi: 10.1038/srep27024 (2016).
References
Armand, M. & Tarascon, J. M. Building better batteries. Nature 451, 652 (2008).
Padhi, A. K., Nanjundaswamy, K. S. & Goodenough, J. B. Phospho‐olivines as Positive‐Electrode Materials for Rechargeable Lithium Batteries. J. Electrochem. Soc. 144, 1188, 10.1149/1.1837571 (1997).
Xu, Y.-N., Chung, S.-Y., Bloking, J. T., Chiang, Y.-M. & Ching, W. Y. Electronic Structure and Electrical Conductivity of Undoped LiFePO4 . Electrochem. Solid-State Lett. 7, A131, 10.1149/1.1703470 (2004).
Ravet, N. et al. Electroactivity of natural and synthetic triphylite. J. Power Sources 97–98, 503, 10.1016/S0378-7753(01)00727-3 (2001).
Fedorková, A., Nacher-Alejos, A., Gómez-Romero, P., Oriňáková, R. & Kaniansky, D. Structural and electrochemical studies of PPy/PEG-LiFePO4 cathode material for Li-ion batteries. Electrochim. Acta 55, 943, 10.1016/j.electacta.2009.09.060 (2010).
Murugan, A. V., Muraliganth, T. & Manthiram, A. Microwave-Irradiated Solvothermal Synthesis of LiFePO4 Nanorods and their Nanocomposites for Lithium Ion Batteries. ECS Transactions 16, 49, 10.1149/1.3115307 (2009).
Liu, J., Conry, T. E., Song, X., Doeff, M. M. & Richardson, T. J. Nanoporous spherical LiFePO4 for high performance cathodes. Energy & Environmental Science 4, 885, 10.1039/C0EE00662A (2011).
Yang, J. et al. 3D porous LiFePO4/graphene hybrid cathodes with enhanced performance for Li-ion batteries. J. Power Sources 208, 340, 10.1016/j.jpowsour.2012.02.032 (2012).
Yu, D. Y. W. et al. Impurities in LiFePO4 and Their Influence on Material Characteristics. J. Electrochem. Soc. 155, A526, 10.1149/1.2919105 (2008).
Ojczyk, W. et al. Studies of selected synthesis procedures of the conducting LiFePO4-based composite cathode materials for Li-ion batteries. J. Power Sources 173, 700, 10.1016/j.jpowsour.2007.05.055 (2007).
Ait Salah, A. et al. Reduction Fe3+ of Impurities in LiFePO4 from Pyrolysis of Organic Precursor Used for Carbon Deposition. J. Electrochem. Soc. 153, A1692, 10.1149/1.2213527 (2006).
Kim, D.-K. et al. Effect of synthesis conditions on the properties of LiFePO4 for secondary lithium batteries. J. Power Sources 159, 237, 10.1016/j.jpowsour.2006.04.086 (2006).
Herle, P. S., Ellis, B., Coombs, N. & Nazar, L. F. Nano-network electronic conduction in iron and nickel olivine phosphates. Nat Mater 3, 147, 10.1038/nmat1063 (2004).
Arnold, G. et al. Fine-particle lithium iron phosphate LiFePO4 synthesized by a new low-cost aqueous precipitation technique. J. Power Sources 119–121, 247, 10.1016/S0378-7753(03)00241-6 (2003).
Belharouak, I., Johnson, C. & Amine, K. Synthesis and electrochemical analysis of vapor-deposited carbon-coated LiFePO4 . Electrochem. Commun. 7, 983, 10.1016/j.elecom.2005.06.019 (2005).
Kim, U. J., Furtado, C. A., Liu, X., Chen, G. & Eklund, P. C. Raman and IR Spectroscopy of Chemically Processed Single-Walled(sic) Carbon Nanotubes. J. Am. Chem. Soc. 127, 15437, 10.1021/ja052951o (2005).
Zaghib, K., Mauger, A., Gendron, F. & Julien, C. M. Magnetic studies of phospho-olivine electrodes in relation with their electrochemical performance in Li-ion batteries. Solid State Ionics 179, 16, 10.1016/j.ssi.2007.12.071 (2008).
Xiangcheng Sun Caiyun Chen, H. S., Bo, C. & Kai, S. Controlled Preparation and Surface Structure Characterization of Carbon-Coated Lithium Iron Phosphate and Electrochemical Studies as Cathode Materials for Lithium Ion Battery. International Journal of Materials and Chemistry 2, 218, 10.5923/j.ijmc.20120205.06 (2012).
Fedorková, A. et al. Electrochemical and XPS study of LiFePO4 cathode nanocomposite with PPy/PEG conductive network. Solid State Sciences 14, 1238, 10.1016/j.solidstatesciences.2012.06.010 (2012).
Mezzi, A. & Kaciulis, S. Surface investigation of carbon films: from diamond to graphite. Surf. Interface Anal. 42, 1082, 10.1002/sia.3348 (2010).
Kuwahara, A., Suzuki, S. & Miyayama, M. Hydrothermal synthesis of LiFePO4 with small particle size and its electrochemical properties. J. Electroceram. 24, 69, 10.1007/s10832-008-9442-1 (2010).
Chen, R., Wu, Y. & Kong, X. Y. Monodisperse porous LiFePO4/C microspheres derived by microwave-assisted hydrothermal process combined with carbothermal reduction for high power lithium-ion batteries. J. Power Sources 258, 246, 10.1016/j.jpowsour.2014.02.068 (2014).
Annadhason, A. Methods of Fractal Dimension Computation. International Journal of Computer Science and Information Technology & Security 2, 166 (2012).
Shin, H.-C., Pyun, S.-I. & Go, J.-Y. A study on the simulated diffusion-limited current transient of a self-affine fractal electrode based upon the scaling property. J. Electroanal. Chem. 531, 101, 10.1016/S0022-0728(02)01068-9 (2002).
Strømme, M., Niklasson, G. A. & Granqvist, C. G. Voltammetry on fractals. Solid State Commun. 96, 151, 10.1016/0038-1098(95)00363-0 (1995).
Nicholson, R. S. & Shain, I. Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem. 36, 706, 10.1021/ac60210a007 (1964).
Park, C.-K., Park, S.-B., Oh, S.-H., Jang, H. & Cho, W.-I. Li Ion Diffusivity and Improved Electrochemical Performances of the Carbon Coated LiFePO4 . Bulletin of the Korean Chemical Society 32, 836, 10.5012/bkcs.2011.32.3.836 (2011).
Satyavani, T. V. S. L., Ramya Kiran, B., Rajesh Kumar, V., Srinivas Kumar, A. & Naidu, S. V. Effect of particle size on dc conductivity, activation energy and diffusion coefficient of lithium iron phosphate in Li-ion cells. Engineering Science and Technology, an International Journa. 10.1016/j.jestch.2015.05.011 (2015).
Yang, S., Zhou, X., Zhang, J. & Liu, Z. Morphology-controlled solvothermal synthesis of LiFePO4 as a cathode material for lithium-ion batteries. J. Mater. Chem. 20, 8086, 10.1039/C0JM01346C (2010).
Yu, F., Zhang, J., Yang, Y. & Song, G. Porous micro-spherical aggregates of LiFePO4/C nanocomposites: A novel and simple template-free concept and synthesis via sol–gel-spray drying method. J. Power Sources 195, 6873, 10.1016/j.jpowsour.2010.01.042 (2010).
Prosini, P. P. Iron Phosphate Materials as Cathodes for Lithium Batteries: The Use of Environmentally Friendly Iron in Lithium Batteries. 85 (Springer-Verlag London, 2011).
Oh, S. W. et al. Double Carbon Coating of LiFePO4 as High Rate Electrode for Rechargeable Lithium Batteries. Adv. Mater. 22, 4842, 10.1002/adma.200904027 (2010).
Oh, S. W. et al. Polyvinylpyrrolidone-assisted synthesis of microscale C-LiFePO4 with high tap density as positive electrode materials for lithium batteries. Electrochim. Acta 55, 1193, 10.1016/j.electacta.2009.10.007 (2010).
Chen, J. Recent Progress in Advanced Materials for Lithium Ion Batteries. Materials 6, 156 (2013).
Acknowledgements
Funding from the E C Seventh Framework program (FP7/2007-2013) under grant Agreement 266090 (SOMABAT) is gratefully acknowledged. ICN2 acknowledges support of the Spanish MINECO through the Severo Ochoa Centers of Excellence Program under Grant SEV-2013-0295. Authors appreciate the award to DPD of a Marie-Curie Fellowship through Beatriu de Pinos Program (BP-DGR-2013) for Catalan system of science and technology, Spain. We thank AGAUR (Generalitat de Catalunya) for project NESTOR Nanomaterials for Energy STORage. 2014_SGR_1505.
Author information
Authors and Affiliations
Contributions
Z.C.H., O.A. and P.G.R. designed the experiments and analyzed the data. Z.C.H., D.P.D. and P.G.R. wrote the manuscript. To preparation and reviewing manuscript all authors contributed equally.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cabán-Huertas, Z., Ayyad, O., Dubal, D. et al. Aqueous synthesis of LiFePO4 with Fractal Granularity. Sci Rep 6, 27024 (2016). https://doi.org/10.1038/srep27024
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27024
This article is cited by
-
Insights of cobalt doping on carbon-coated LiFePO4 olivine nanoparticles prepared by citric acid combustion route as cathodes for lithium batteries
Ionics (2019)
-
Optimizing the particle-size distribution and tap density of LiFePO4/C composites containing excess lithium
Ionics (2019)
-
Nano-scale hollow structure carbon-coated LiFePO4 as cathode material for lithium ion battery
Ionics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
