
Abstract
Chicken, Gallus gallus, is a valuable species both as a food source and as a model organism for scientific research. Here, we sequenced the genome of Gyeongbuk Araucana, a rare chicken breed with unique phenotypic characteristics including flight ability, large body size, and laying blue-shelled eggs, to identify its genomic features. We generated genomes of Gyeongbuk Araucana, Leghorn, and Korean Native Chicken at a total of 33.5, 35.82, and 33.23 coverage depth, respectively. Along with the genomes of 12 Chinese breeds, we identified genomic variants of 16.3 million SNVs and 2.3 million InDels in mapped regions. Additionally, through assembly of unmapped reads and selective sweep, we identified candidate genes that fall into heart, vasculature and muscle development and body growth categories, which provided insight into Gyeongbuk Araucana’s phenotypic traits. Finally, genetic variation based on the transposable element insertion pattern was investigated to elucidate the features of transposable elements related to blue egg shell formation. This study presents results of the first genomic study on the Gyeongbuk Araucana breed; it has potential to serve as an invaluable resource for future research on the genomic characteristics of this chicken breed as well as others.
Similar content being viewed by others
Introduction
Chicken, Gallus gallus, is valuable not only as a food source but also as a model organism for scientific research1. In the past thousands of years, hundreds of chicken breeds have diverged under natural and artificial selection in a wide variety of circumstances. As a result, chickens have undergone significant phenotypic differentiation in body size, plumage, egg color, and flying ability2.
The Gyeongbuk Araucana (GA) domestic chicken is a hybrid breed developed in Gyeongbuk, Korea by crossing the Golden Duckwing Araucana and the White Leghorn, two breeds with very distinct characteristics. The White Leghorn, a small breed with a rump, is renowned for its prolific egg-laying as well as a good feed-to-egg conversion ratio. Meanwhile, the Golden Duckwing Araucana is a similarly sized small rumpless and tufted breed that produces blue-shelled eggs at a relatively slow rate. These two breeds were crossed to produce a Korean chicken variety which possesses qualities favorable to commercial poultry production from both parent breeds, including the blue shell tint of the eggs they lay and high egg production rate. Although both the Golden Duckwing Araucana and White Leghorn are small breeds, GA resulting from the cross are extremely large and can fly well. Additionally, GA chickens display a combination of phenotypic traits from both parents: a rump but no ear tufts (Fig. 1). Although GA is a relatively new breed, it has already been registered in the Domestic Animal Diversity Information System (DAD-IS) of the FAO (Food and Agriculture Organization).

(A) blue egg shell of Gyeongbuk Araucana. (B) general appearance of Gyeongbuk Araucana chicken breed.
Analysis of genetic information and patterns can be useful for discovering the origin of specific breed or detecting specific traits within breeds. Recent phylogenetic analyses using various genetic information revealed the origin of the domesticated chicken3 and the Korean native chicken (KNC)4. Additionally, genetic variants present between several chicken breeds have been utilized to support the characterization of specific traits in chicken breeds5,6,7. In this way, a wide range of genomic studies on domestic animals, and in chickens specifically, have been conducted to investigate the genetic architecture of these species. However, no such study has been performed in GA. For this reason, we performed whole genome sequencing on GA chickens and additionally performed whole genome sequencing of Leghorn (LH) and KNC. The whole genome paired-end reads for other 12 chicken breeds were also obtained from the sequence read archive (SRA) in EMBL-EBI database. Using genomic information from 28 chickens, we identified candidate genomic characteristics which may be related to Gyeongbuk Araucana’s phenotypic traits. Our results also confirmed the results of previous studies related to blue egg formation8,9. This study is the first of its kind to report a comprehensive view of the GA chicken breed at a genomic level.
Results and Discussion
Short read alignment and variant calling
The whole genomes of 9 chicken samples (3 GA, 3 LH, and 3 KNC) were sequenced to an average depth of 11.4 fold , with 9,422,388,891 bp in a total of 1,095,144,380 reads. Short sequencing reads of each breed were aligned to the chicken reference genome (Galgal 4.75) from the Ensembl database with an overall alignment rate of 97.81%. The average depth for the overall dataset of 28 Chickens was 17.2 fold, and the mapping rate in different breeds varied from 95.89% to 98.42%. (Table S1). We then identified 16,342,621 single nucleotide variants (SNVs) and 2,254,900 Indels in all 28 samples after removing PCR duplicates and recalibrating base quality of sequencing reads. To obtain variants for population analyses, we removed the variants supposing missing data present in any of the 28 individuals or in non-chromosomal regions. Although 7,122,335 SNVs (43.58%) had been identified previously (ftp://ftp.ensembl.org/pub/release-83/variation/vcf/gallus_gallus/Gallus_gallus.vcf.gz), we defined 9,220,286 SNVs as candidates for novel variants (56.42%). The detailed results of variant calling and functional annotation are shown in Figures S1 and S2, Tables 1 and S2.
Population differentiation of 28 chickens
To understand the population differentiation, structure, and relationship between various breeds more deeply, we performed several analyses based on the high quality variants. First, principle component analysis (PCA) was performed to distinguish 15 different breeds using two main components explaining the dispersion of samples (Fig. 2A). GA, LH, and KNC were well divided by breed using Eigenvector 1 (12.85% of the total variance) and Eigenvector 2 (11.08% of the total variance) for PCA. Results revealed clear structural differences between populations. We further measured kinship coefficient, also known as coefficient of coancestry, and IBS (identical by state) to measure the pairwise relationship between each sample (Figure S3). The relationships were clear in GA chickens but not in one of the LH samples and not in most of the KNC samples. From the results of three analyses, it has been clearly demonstrated that GA genetic profile is overall significantly distinct from that of other chicken breeds.

(A) Results of principal component analysis (PCA) of 15 chicken breeds. PC1 (x-axis) versus PC2 (y-axis). PC1 and PC2 represent 12.85% and 11.08% of the total variance in the PCA, respectively. (B) Linkage disequilibrium (LD) pattern of GA, KNC, and LH.
Gene prediction using unmapped reads
To identify breed-specific genes in GA, we assembled unmapped reads into contigs. However, they were inadequate for detection of gene regions given their short length. Thus, we performed whole genome assembly using all short read sequences (Table S3) and then matched the unmapped assembled contigs to the assembled whole genome contigs using BLAST. 424, 459, and 375 putative peptides from the matched contigs of 3 GA samples (GA1, GA2, and GA3, respectively) were annotated. Of the total putative genes, 61 were shared between all GA samples. These genes were predominantly related to growth (such as ‘epithelium development (4 genes, p-value = 0.01)’, ‘cell cycle (6 genes, p-value = 0.03)’, and ‘structural molecule activity (5 genes, p-value = 0.03)’). Additionally, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis was performed. Results revealed that several genes were involved in the Notch signaling pathway that is responsible for functions relating to cardiac valve homeostasis10, stabilization of angiogenesis11, and neuronal function and development12. Given GA’s large body size, it makes sense that genes related to growth were predominantly identified. In addition, as the process of gene prediction was based on sequence similarity between assembled contigs and reference genes, the predicted genes are not regarded as totally novel genes. On the other hand, alignments of unmapped reads to the regions with genes implicates that the genes in these particular breeds have considerable variants, so sequenced reads couldn’t be mapped to the reference genome.
Selective sweep signal in GA
Compared to other breeds, GA display a consistently high level of linkage disequilibrium over varying distances of the genome (Fig. 2B). This may reflect the fact that only a small number of the Golden Duckwing Araucana, one of the GA’s parent breeds, was imported to Korea causing GA chickens to have a low level of genetic diversity.
To find distinctly selected genomic regions in GA, we measured genome-wide variation between GA and KNC (Fig. 3A). KNC is a well-suited breed for comparison since it lives geographically close to and is genetically different from GA. In order to overcome the limitation of small sample size, we detected candidate genomic regions using the overlap of top 5% regions of pairwise nucleotide variation (log10 (θπ ratio)) and genetic differentiation (Weir and Cockerham’s FST) as significant regions following protocol used in previous studies13. The FST statistic is less sensitive to small sample size if sufficient variant loci are examined14,15. From the chicken genome, 46,354 windows were used to detect selective sweep since these windows contain more than 50 SNPs (92.9% of the genome). We identified 223 genes as a positive signature of the each statistic (log10 (θπ ratio) >3.378, FST > 0.387). Although the Z chromosome is a highly conserved region (Figure S2), most of the selected regions were located on the Z chromosome (17.4%). Meanwhile, the selected genes were predominantly related to cardiac development and morphogenesis (Fig. 3B). In avian species, flying has been found to associate with the cardiovascular system; due to the physiological strain resulting from flying, most flying birds have strong cardiac muscles16. MYLK2, identified as significant from our analyses, has previously been established as important in cardiac muscle tissue morphogenesis as well as fast-twitch skeletal muscle fiber contraction, both of which are required for quick bursts of activity such as flight17. Similarly, we identified several genes (BCL2L1, GSS, SCARA3, ERBB4) associated with response to oxidative stress18,19. As chickens only fly at high intensity in short bursts, it is particularly important for them to have a mechanism to protect their body from the damage of free radicals.

Manhattan plot of distribution of pairwise nucleotide variation (log2 (θπ ratio)) and genetic differentiation (Weir and Cockerham’s FST).
Using the same procedure, we compared LH and KNC, using KNC as a reference, in order to identify positively selected regions in LH. From the results, we identified positive selection regions which overlap between LH and GA; identification of genomic regions responsible for egg production in the LH breed may help decipher the genetic basis of GA’s superior egg laying performance. We found that all overlapped windows were located on the Z chromosome. Among the genomic area, a window at 32.1 ~ 32.2 Mbp was found to be significantly related with egg production in the chicken QTL region20. It was also involved within the previously reported QTL region related to maternal age at the first egg production21. Results of our analyses provide important insight into the genomic features of a breed which has not been investigated previously. However, as a relatively small sample size was used in the present study, future genomic studies on this breed using a larger sample size may help further elucidate the underlying genetic factors behind this novel and unique breed.
Identification of transposable element variants (TEV)
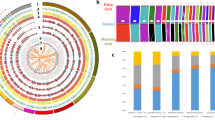
Using Retroseq software22, we obtained a total of 412,208 candidate TEVs from 9 genomic samples (GA, LH, KNC) and 19 public genome sequences. Most of the TEVs (~250,000) were located in the intergenic region, while only 149 TEVs were found in the exon region (Table S4). From GA, we identified 22,033 candidate TEVs which differed from the chicken reference genome (Galgal 4.75). Most of the TEVs were annotated as intergenic sequences and only 11 TEVs as exon sequence (Table 2 and Fig. 4A). As found in previous studies23, the majority of TEVs belonged to the CR1 families of LINEs (CR1 : 10,524, LINE : 10,676, LTR : 8,587, DNA : 2,611, SINE : 159), which is consistent in other non-GA chicken breeds as well.

(A) A circular plot for distribution of TEVs on the genome of GA. Four types of TE are represented by different colors (LTR : green, LINE : red, SINE: orange, and DNA: blue). (B) A clustering tree based on LTR presence patterns for 28 chicken genomes. Three groups for LH, GA, and KNC are well-defined. The neighbor-joining tree using whole genome variants and other TE types (LINE, SINE, and DNA) are shown in Figure S4.
Clustering analysis based on TE variants identified from 28 chicken genomic samples
Using TEV information from 9 chicken genome along with 19 public genome sequences, we performed clustering analysis based on the pattern of TE presence in 28 chicken genomes. Several other studies have constructed phylogenetic trees based on the TE presence pattern in primates24,25,26,27. These studies on the relationship between species showed that incongruent TE insertion sites can be caused by several factors, including incomplete lineage sorting and hybridization between species. Considering these factors within species and mating-free environment, we estimated that these factors would considerably impact TE presence patterns and consequently cause a confounding result. As predicted, we obtained results similar to those estimated for TE types like LINE, SINE, and DNA transposons (Figure S4). However, for LTR, the constructed tree was similar to that of SNP based-clustering, which roughly segregates whole individuals by breed (Figs 4B and S4A). This result indicates that LTR polymorphism might be used as a marker for revealing the relationship of relatively close organisms and is consistent with those from a previous study on the effects of retroviral insertions on phenotypic traits of breeds28 as well as blue egg shell formation10,27.
The clustering results based on the LTR presence pattern are shown in Fig. 4B. Like the SNP-based tree, GA and LH were clustered into one group and close to KNC, which was expected given the the origin of GA as a result of hybridization between the Golden Duckwing Araucana and White leghorn.
Candidate retroviral insertions specific to GA chickens
The genetic determination of blue egg shell coloration has been identified in Araucana chickens; it has been revealed that EAV-HP insertion promotes the expression of SLCO1B3 gene in the uterus of the oviduct in Araucana chickens, which causes blue egg shell formation9,29. Our results identified retroviral insertions from three GA genomes in SLCO1B3 gene equally. Dong Xiang (DX) chickens are another blue-egg laying breed29; EAV-HP insertion was also identified in the DX genome sequence adjacent to the insertion of the SLCO1B3 gene in GA, which indicates that GA and DX share genetic characteristics with other breeds that lay blue eggs. All of these results are consistent with previous reports9,29.
As we detected the presence of TE insertions using multiple TE probe sequences at the breakpoint, it was necessary to verify that the sequences of TE insertions are identical. In order to validate the presence of EAV-HP sequence, we carried out local de novo assembly with Velvet30 using the reads mapped within 1000 upstream and downstream of the candidate breakpoint. From this process, we obtained several contigs and aligned these with the TE probes we used for TE identification. Next, we conducted multiple sequence alignment over sequences recovered from the above process of several individuals, and retrieved partial conserved TE sequences (142 bp) from 3 GA and DX genomes. Collectively, the commonly identified insertion of EAV-HP in three GA chickens was identical to that of previous experimental studies9,29.
Retroviral insertion can influence the transcription of genes in many ways. Several studies on mice found that the effects of intronic retrovirus insertion on the transcription of the resident gene result in an alteration of the ratios of the splice variants by premature transcription, either by providing a cryptic promoter or by altering splicing. In the case of blue egg shell formation, upstream insertion of resident genes can influence expression of the gene31. Consequently, these transcriptional effects can modify phenotypic traits, which has been shown in chickens28 as well as mice32,33.
Here, we propose candidate TEVs specific to GA chickens. We identified one LTR insertion specific to GA within SUCLG1 gene. This insertion was located in the 3′ UTR region of the gene and we retrieved the partial sequence of 188 bp conserved in all three GA genomes. SUCLG1 gene has been investigated for its role as an α subunit of succinate-CoA ligase-encoding gene, which forms a complex with nucleoside diphosphate kinase and plays an important role in the salvage of deoxyribonucleotides for mitochondrial DNA synthesis34. SCN5A also contained a LTR insertion identified only in GA within the intron region (136 bp conserved sequence). SCN5A is a gene encoding cardiac-specific voltage-gated sodium channel and known to be related to many cardiovascular diseases35,36. This result was consistent with identification of heart and muscle development-related genes within selection signal.
Conclusions
In this study, we conducted whole genome sequencing and analyzed genomic variants in order to reveal genetic characteristics of the GA breed. As a novel chicken breed, GA breed displays population structure clearly separated from other chicken breeds based on overall genomic variants and TE insertion information. Furthermore, we identified candidate genes which could provide insight into the association between genetic traits and phenotypic distinctions such as large body size, flight ability, egg production, and blue egg shell formation using various measures. This information and the sequencing data used in this study could be invaluable for understanding genomic features of a novel breed which has not been previously studied, as well as provide a basis for selective breeding of traits crucial to the poultry production industry.
Methods
Ethics statement
The experiment and all its procedures were approved by the regional Ethical Committee (Gyeong Buk Animal Bioethics committee permit number: 2012-0049), and the methods were carried out in accordance with the approved guidelines.
DNA sequencing and sample collection
We obtained blood samples from male GA, LH, and KNC chickens. Samples were collected from the Livestock Research Institute, Yeongju, Korea. To prevent clotting, blood drawn from the carotid artery was treated with heparin. To generate inserts of ~300 bp fragments, 3 μg of genomic DNA was randomly sheared using Covaris System. The TruSeq Dna Sample Prep. Kit (Illumina, San Diego, CA) was used for library construction following manufacturer guidelines. Whole genome sequencing was performed using the Illumina HiSeq 2000 platform. Additionally, we downloaded genomic data of 14 chicken samples from the EMBL-EBI database, which included 2 Silkies (one from China and one from Taiwan), 1 Taiwanese native chicken (TNC), 1 Leghorn, 1 Tibetan chicken (TB), 1 Shouguang (SG), 1 Wenchang (WC), 1 Beijing You (BY), 1 White Plymouth Rock (WPR), 1 Dong Xiang (DX), 1 Cornish (CN), 1 Luxi Game (LG), 1 Rhode Island Red (RIR), and 1 Red Jungle Fowl (RJF). Also, data from 5 Korean Native Chickens, used in a previous study, were included to increase the quality of variants calling and following analyses (Table S5). A quality check on raw sequence data was performed using fastQC37 software, and potential adapter sequences were removed prior to sequence alignment using Trimmomatic-0.3238.
Short reads alignment and variants calling
Paired-end sequence reads were mapped to the chicken reference genome (Galgal 4.75) from the Ensembl database using Bowtie239 with default settings. For downstream processing and variant calling, we used several open-source software packages: Picard tools (http://picard.sourceforge.net), SAMtools40, and Genome Analysis Toolkit (GATK)41. “CreateSequenceDictionary” and “MarkDuplicates” Picard command-line tools were used to read reference FASTA sequence for writing bam file with only sequence dictionary, and to filter potential PCR duplicates, respectively. Using SAMtools, we created index files for the reference and bam files. We then performed local realignment of sequence reads to correct misalignment due to the presence of small insertion and deletion using GATK “RealignerTargetCreator” and “IndelRealigner” arguments. Base quality score recalibration was performed to get accurate quality scores and to correct the variation in quality with machine cycle and sequence context. For variant calling, GATK “UnifiedGenotyper” and “SelectVariants” arguments were used with the following filtering criteria: all variants with 1) a Phred-scaled quality score of less than 30; 2) read depth less than 5; 3) MQ0 (total count across all samples of mapping quality zero reads) >4; or a 4) Phred-scaled P-value using Fisher’s exact test more than 200 were filtered out to reduce false positive calls due to the strand bias. Annotation of the calling variants were performed using Snpeff42 with default settings.
Short reads assembly and functional annotation
We used the “Error correction” module of Allpaths-LG43 with default settings to eliminate possible sequencing errors. Error-corrected paired-end reads were merged into FASTA format using “Fq2fa” module from IDBA v1.1.1 software which stands for iterative De Bruijn graph De novo assembler for short reads sequencing data with highly uneven sequencing depth. We assembled error corrected paired-end reads using IDBA_UD from IDBA package44 with the following parameters: 1) Perform pre-correction before assembly (“–pre_correction”), and 2) minimum k value should be more than 30 (–mink 30). Using Gapcloser45, we filled predicted gaps in the assembled sequences using default settings. RepeatMasker46 was used to screens DNA sequences for interspersed repeats and low complexity DNA sequences before gene prediction for the candidate contigs. We matched the unmapped assembled contigs to the whole genome assembled contigs using BLAST, and potential coding regions in the contigs sequence were predicted using AUGUSTUS47. Coding regions were aligned to chicken reference peptide sequences (Galgal 4.75) from the Ensembl database using BLASTP for functional annotation. A detailed schematization of this process is shown in Figure S5.
Population stratification and selective sweep analysis
We used Genome-Wide Complex Trait Analysis (GCTA)48 to calculate eigenvectors, which are equivalent to those estimated by the EIGENSTRAT software tool for principal component analysis (PCA). Autosomal genotype data was converted to PLINK format, the input format required for GCTA, using VCFtools49.
To investigate linkage disequilibrium patterns, the coefficient of determination (r2) between any two SNVs was calculated using Haploview50. The average of pairwise r2 was calculated according to the distance between two SNVs in a 500 kb window and averaged for the whole genome.
Pairwise relatedness and inbreeding coefficient were measured using the KING program51 command with parameter ‘–kinship –ibs’. VCFtools 4.049 was used to compute nucleotide variation (θπ) and genetic differentiation (FST) by applying a sliding window approach (bin size 100 kb, step size 20 kb).
Transposable element (TE) probes
As the majority of TEs in the chicken genome are of the retrotransposon type52, we focused on four types of retrotransposons in this study: LTR, LINE, SINE, and DNA transposons. Thus, we obtained all possible TE probe sequences from Repeatmasker Genomic datasets (http://www.repeatmasker.org/) and used these as the alignment subjects for TE identification. The coordinates of TEs from the Gallus gallus reference genome (from Repeat library 4, 20140131) were also used TE identification.
Transposable element variants (TEV) identification
We identified TE variants (TEV) across 28 chicken genomes using Retroseq software22. Retroseq employs discordantly or solely mapping reads to seek candidate TE insertion sites, called breakpoints. In the alignment step, it was necessary to determine the appropriate insert size of paired reads to obtain pure discordantly mapping reads induced by TE insertion not by the mapping distance. We set the minimum insert size (1000 bp) to guarantee this requisite.
Due to the difference in depth coverage across the paired-end reads data from various data production processes, it was necessary to adjust the parameters for each group for each different data production process. Additionally, for the confidence of breakpoint, we filtered the raw calls of TEVs and only recovered the calls tagged as “FL = 8” which met all breakpoint criteria. Final calls of TEVs were classified into four groups (LTR, LINE, SINE, and DNA transposons) based on the nomenclature and classification used in Repeatmasker. For population analyses, TEV calls within 100 bps were clustered and regarded as a single TEV call; using these cluster positions, we identified breed-specific breakpoint and related genes.
Clustering analysis based on TE presence polymorphism
Using cluster position, presence of a TE insertion was coded as “1” or “2” according to genotypes, and absence of TE insertion was coded as “0” for each individual. To improve the reliability of each TE insertion loci, loci present in less than 3 individual genomes were excluded. This data matrix was used as an input for calculation of p-distance matrix calculated by the hamming distance metric. We implemented the neighbor-joining method and bootstrapped the data using the phangorn53 package within R to cluster samples.
Additional Information
Accessions codes: The samples that were sequenced were archived at the Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra/) under the accession SRP051746.
How to cite this article: Jeong, H. et al. Whole genome sequencing of Gyeongbuk Araucana, a newly developed blue-egg laying chicken breed, reveals its origin and genetic characteristics. Sci. Rep. 6, 26484; doi: 10.1038/srep26484 (2016).
References
Darwin, C. & Bynum, W. F. The origin of species by means of natural selection: or, the preservation of favored races in the struggle for life. (AL Burt, 2009).
West, B. & Zhou, B.-x. Did chickens go north? New evidence for domestication. Worlds Poult Sci J 45, 205–218 (1989).
Eriksson, J. et al. Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. Plos. Genet 4, e1000010 (2008).
Kwak, W. et al. Uncovering Genomic Features and Maternal Origin of Korean Native Chicken by Whole Genome Sequencing. Plos. ONE 9, e114763 (2014).
Chang, C. et al. A global analysis of molecular markers and phenotypic traits in local chicken breeds in Taiwan. Anim Genet 43, 172–182 (2012).
Dorshorst, B., Okimoto, R. & Ashwell, C. Genomic regions associated with dermal hyperpigmentation, polydactyly and other morphological traits in the Silkie chicken. J. Hered. 101, 339–350 (2010).
Freese, N. H., Lam, B. A., Staton, M., Scott, A. & Chapman, S. C. A Novel Gain-Of-Function Mutation of the Proneural IRX1 and IRX2 Genes Disrupts Axis Elongation in the Araucana Rumpless Chicken. Plos. ONE 9, e112364 (2014).
Wang, Z. et al. An EAV-HP insertion in 5′ flanking region of SLCO1B3 causes blue eggshell in the chicken. Plos. Genet 9, e1003183 (2013).
Wragg, D. et al. Endogenous retrovirus EAV-HP linked to blue egg phenotype in Mapuche fowl. Plos. ONE 8, e71393 (2013).
Iso, T., Hamamori, Y. & Kedes, L. Notch signaling in vascular development. Arterioscler Thromb Vasc Biol 23, 543–553 (2003).
Liu, Z.-J. et al. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol Cell Biol 23, 14–25 (2003).
Gaiano, N. & Fishell, G. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci 25, 471–490 (2002).
Li, M. et al. Whole-genome sequencing of Berkshire (European native pig) provides insights into its origin and domestication. Sci Rep 4 (2014).
Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. 70, 3321–3323 (1973).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution, 1358–1370 (1984).
Guglielmo, C. G., Haunerland, N. H., Hochachka, P. W. & Williams, T. D. Seasonal dynamics of flight muscle fatty acid binding protein and catabolic enzymes in a migratory shorebird. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 282, R1405–R1413 (2002).
Al Madhoun, A. S. et al. Skeletal myosin light chain kinase regulates skeletal myogenesis by phosphorylation of MEF2C. EMBO. J. 30, 2477–2489 (2011).
Spielberg, S. P., Boxer, L. A., Oliver, J. M., Allen, J. M. & Schulman, J. D. Oxidative damage to neutrophils in glutathione synthetase deficiency. Br J Haematol 42, 215–223 (1979).
Martindale, J. L. & Holbrook, N. J. Cellular response to oxidative stress: Signaling for suicide and survival*. J Cell Physiol 192, 1–15 (2002).
Tuiskula-Haavisto, M. et al. Mapping of quantitative trait loci affecting quality and production traits in egg layers. Poult. Sci 81, 919–927 (2002).
Sasaki, O. et al. Genetic mapping of quantitative trait loci affecting body weight, egg character and egg production in F2 intercross chickens. Anim Genet 35, 188–194 (2004).
Keane, T. M., Wong, K. & Adams, D. J. RetroSeq: transposable element discovery from next-generation sequencing data. Bioinformatics 29, 389–390 (2013).
Abrusán, G., Krambeck, H.-J., Junier, T., Giordano, J. & Warburton, P. E. Biased distributions and decay of long interspersed nuclear elements in the chicken genome. Genetics 178, 573–581 (2008).
Xing, J. et al. A mobile element-based evolutionary history of guenons (tribe Cercopithecini). BMC biology 5, 5 (2007).
Xing, J. et al. A mobile element based phylogeny of Old World monkeys. Molecular phylogenetics and evolution 37, 872–880 (2005).
Li, J. et al. Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene 448, 242–249 (2009).
Meyer, T. J. et al. An Alu-based phylogeny of gibbons (Hylobatidae). Molecular biology and evolution 29, 3441–3450 (2012).
Chang, C.-M. et al. Complete association between a retroviral insertion in the tyrosinase gene and the recessive white mutation in chickens. BMC Genomics 7, 19 (2006).
Wang, Z. et al. An EAV-HP Insertion in 5′ Flanking Region of SLCO1B3 Causes Blue Eggshell in the Chicken. Plos genetics 9, e1003183 (2013).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829 (2008).
Isbel, L. & Whitelaw, E. Endogenous retroviruses in mammals: an emerging picture of how ERVs modify expression of adjacent genes. Bioessays 34, 734–738 (2012).
Vasicek, T. J. et al. Two dominant mutations in the mouse fused gene are the result of transposon insertions. Genetics 147, 777–786 (1997).
Perry, W. L., Copeland, N. G. & Jenkins, N. A. The molecular basis for dominant yellow agouti coat color mutations. Bioessays 16, 705–707 (1994).
Ostergaard, E. et al. A novel missense mutation in SUCLG1 associated with mitochondrial DNA depletion, encephalomyopathic form, with methylmalonic aciduria. Eur J Pediatr 169, 201–205 (2010).
Schott, J.-J. et al. Cardiac conduction defects associate with mutations in SCN5A. Nat Genet 23, 20–21 (1999).
Remme, C. A., Wilde, A. A. & Bezzina, C. R. Cardiac Sodium Channel Overlap Syndromes: Different Faces of SCN5A Mutations. Trends Cardiovasc Med 18, 78–87 (2008).
Bioinformatics, B. FastQC A quality control tool for high throughput sequence data. Cambridge, UK: Babraham Institute (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, btu170 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92 (2012).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. 108, 1513–1518 (2011).
Peng, Y., Leung, H. C., Yiu, S.-M. & Chin, F. Y. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinfo., 4.10. 11–14.10. 14 (2009).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Hillier, L. W. et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004).
Schliep, K. P. phangorn: Phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011).
Acknowledgements
This research was supported by a Grant from the Golden Seed Project (PJ00993001201401), Ministry of Agriculture, Food and Rural Affairs (MAFRA), Ministry of Oceans and Fisheries (MOF), Rural Development Administration (RDA) and Korea Forest Service (KFS), Republic of Korea.
Author information
Authors and Affiliations
Contributions
H.J. and K.K. carried out in-silico analysis and drafted the manuscript. B.K., J.Y. and J.H. carried out biological experiments. K.C. and H.K. contributed to writing and biological interpretation. S.C. and D.O. managed the whole project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jeong, H., Kim, K., Caetano-Anollés, K. et al. Whole genome sequencing of Gyeongbuk Araucana, a newly developed blue-egg laying chicken breed, reveals its origin and genetic characteristics. Sci Rep 6, 26484 (2016). https://doi.org/10.1038/srep26484
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26484
This article is cited by
-
Managing genomic diversity in conservation programs of Chinese domestic chickens
Genetics Selection Evolution (2023)
-
Genetic and phenotypic characterization of the novel mouse substrain C57BL/6N Korl with increased body weight
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
