
Abstract
A mammalian cell utilizes DNA methylation to modulate gene expression in response to environmental changes during development and differentiation. Aberrant DNA methylation changes as a correlate to diseased states like cancer, neurodegenerative conditions and cardiovascular diseases have been documented. Here we show genome-wide DNA methylation changes in macrophages infected with the pathogen M. tuberculosis. Majority of the affected genomic loci were hypermethylated in M. tuberculosis infected THP1 macrophages. Hotspots of differential DNA methylation were enriched in genes involved in immune response and chromatin reorganization. Importantly, DNA methylation changes were observed predominantly for cytosines present in non-CpG dinucleotide context. This observation was consistent with our previous finding that the mycobacterial DNA methyltransferase, Rv2966c, targets non-CpG dinucleotides in the host DNA during M. tuberculosis infection and reiterates the hypothesis that pathogenic bacteria use non-canonical epigenetic strategies during infection.
Similar content being viewed by others

Introduction
Changes in the transcriptome of a cell closely parallel changes in its epigenome highlighting the pliability of the epigenome towards signals emanating from the environment. Amongst the various epigenetic modifications, DNA methylation is an important component of a mammalian cell’s epigenome1 and several studies have exemplified the dynamic nature of DNA methylation and its contribution in translating an environmental cue to a cellular response2. Interaction with an infectious agent also invokes a response from the infected mammalian cell that manifests as molecular changes including modulation of the epigenome. The susceptibility of a cell’s methylome to manipulation by an infectious agent has been reported. For example, Human papillomavirus protein E7 has been shown to directly associate with DNMT1 leading to aberrant methylation of host DNA3. DNA methylation changes have also been reported in response to the protozoan Leishmania donovani infection of human macrophages4. These epigenetic changes were found to suppress the host immune response, aiding the intracellular survival of the protozoan4.
Mycobacterium tuberculosis, an intracellular pathogen, that infects human macrophages is able to subvert the host defense mechanisms and can lay dormant for years5. Very little is known about the epigenetic changes that accompany infection of macrophages by M. tuberculosis. The mycobacterial protein Rv1988 has been shown to methylate histone H3 at R42 and repress gene expression6 whereas Rv3763 is involved in suppression of IFN-γ induced genes via TLR2, leading to histone hypoacetylation at CIITA promoter7. Previous work from our laboratory has shown direct interaction of Rv2966c with the macrophage epigenome and its ability to effect non-CpG methylation at specific genetic loci8. These studies indicated that M. tuberculosis might be interfering with host epigenetic response to aid in its survival. In the present study, we report global DNA methylation changes acquired upon infection of THP1 macrophages with the virulent Mycobacterium tuberculosis H37Rv. Using MBD (Methyl Binding Protein) protein based methylation-Seq assay, we identified around 23000 differentially methylated regions (DMRs) in infected macrophages. Methylation analysis of randomly selected genetic loci using MeDIP (Methylated DNA Immunoprecipitation) and bisulfite sequencing revealed these changes to be at cytosines present in non-CpG rather than the CpG dinucleotide context. These findings mirror our previous finding where the mycobacterial protein Rv2966c was found to be targeting non-CpG dinucleotides8 and reiterates the hypothesis that bacteria use non-canonical epigenetic strategies during infection9,10,11,12,13.
Results
M. tuberculosis infection causes genome-wide DNA methylation changes in THP1 macrophages
In order to study the DNA methylation dynamics during host-pathogen interaction, genomic DNA was isolated from M. tuberculosis H37Rv infected (0 and 48 hrs) and corresponding uninfected PMA treated THP1 cells (THP1 macrophages). DNA for each time point (in duplicates) was pooled and enriched for methylated regions by MBD protein based affinity pull down. The enriched DNA fragments were sequenced on the Illumina Hi-seq NGS platform (Materials and Methods). THP1 cell line, a monocytic cell line, differentiates into macrophages upon treatment with the mitogen Phorbol myristate acetate ester (PMA)14. In order to eliminate the DNA methylation changes that would have resulted due to differentiation, peaks that were different between uninfected 0 and 48 hrs samples were first removed. The remaining peaks were compared with peaks from infected 48 hrs samples (see flow charts in Supplementary Fig. 1). A total of 23,433 differentially methylated regions (DMRs) were identified, of which 19,506 (~83%) DMRs were hypermethylated and 3,927 (~17%) were hypomethylated in the M. tuberculosis H37Rv infected THP1 macrophages (Fig. 1). This indicated that regions of hypermethylation were five-fold more enriched than hypomethylation in the genome of infected cells.

Pie charts (A) showing the distribution of DMRs (B) with respect to genes within the human genome. Hyper (left panel) and Hypo (right panel) DMRs were categorised as intergenic, promoter-specific and gene body specific transcription end site specific based on their location in the human genome. Hypermethylated DMRs represent genomic regions with gain of methylation while hypomethylated DMRs represent loss of methylation upon infection.
Genomic localization of the identified Differentially Methylated Regions
In order to assess the functional significance of the methylation differences arising due to M. tuberculosis infection, the distribution of these DMRs with reference to genic regions in the genome was examined. Approximately 50% of the hypermethylated DMRs (9830) were found to be associated with gene body (exons and introns), 48% (9308) were intergenic and 1% mapped to the promoter (203) and 1% to the transcription end site (TES, 165) (Fig. 1). In case of hypomethylated DMRs, ~46% (1792) and ~53% (2074) DMRs mapped to gene body and intergenic regions respectively while approximately 1% of DMRs were associated with promoter (34) and TES (27) (Fig. 1).
As 12946 intragenic DMRs (gene body, promoter and TES) were found to be associated with 7573 genes it indicated that multiple DMRs were associated with a single gene. Out of these 7573 genes, 4996 (3684 with hyper and 1312 with hypo-DMRs) were associated with a single DMR, 2555 (2278 with hyper and 277 with hypo-DMRs) with 2–10 DMRs and 22 (only hyper DMRs) with more than 10 DMRs (Table 1). Furthermore, out of the 7573 genes that were associated with a DMR, 2208 genes (listed in Supplementary Table 1) were found to be associated with both hypermethylated and hypomethylated DMRs. The epigenetic circuitry utilises DNA methylation to organize specific genetic loci into specific chromatin conformations15. The presence of both hypermethylated and hypomethylated DMRs within the same genetic loci could suggest multiple chromatin conformations within the same gene.
Analysis of DMRs mapping to the gene body revealed that DMRs were predominantly associated with introns as 93% of these DMRs mapped to the introns while only 4–5% mapped to the exons. The remaining DMRs were associated with 5′ or 3′ UTRs (Table 2). Amongst the DMRs mapping to the introns, 40% of DMRs mapped to the first two introns while ~12% mapped to the last two intron of a gene, irrespective of the gene size (Table 2). This observation was similar to what has been observed for B-cell methylome where MRIs (Methylated Region of Interest) were found frequently at 5′ and 3′ ends of a gene16.
Examination of the intergenic DMRs showed that approximately 60% of the intergenic DMRs were present within 70 Kb of a TSS (52% for hypo and 62% for hyper-DMRs, Fig. 2A). Apart from regulatory elements, non-genic regions are generally associated with repetitive DNA sequences and ncRNA genes17. To examine if some of the DMRs were associated with any specific repetitive element, the percentage of a specific repetitive DNA element associated with the DMRs was compared with the percentage of this repetitive element within the whole genome (Table 3, see Materials and Methods). While SINE elements constitute only around 13% of the total length of the repetitive DNA sequence in the human genome, this percentage was significantly higher at approximately 33% in relation to the DMRs (Table 3). No major difference was observed for any other repetitive DNA elements.

(A) Histogram showing distance of hyper (white bars) and hypo (black bars) DMRs from the transcription start site (TSS) of the nearest gene. Correlation of hyper (left panel) and hypo (right panel) DMRs with size (B), number of genes (C) and gene density (D) for each chromosome was plotted.
ncRNAs including lncRNA, miRNA, piRNA and snoRNA are known to be associated with gene regulation18. Recent reports have also shown differential expression of host miRNA in response to M. tuberculosis infection19,20,21.In our study, approximately 8% (1850) of the DMRs were associated with non-coding RNA genes. Majority (~90%) of these DMRs were found to be intergenic. As many of the ncRNAs are present within or in the vicinity of protein coding genes22, it was no surprise that more than half of these DMRs (954) mapped within other genes. 61% of the ncRNA DMRs were associated with miRNA genes (1124), ~24% with lncRNA (452), ~8% with piRNA (156) and 6.4% with snoRNA (118) genes (Table 4).
Chromosomal distribution of DMRs
In the human genome, distribution of genes on chromosomes has been found to be non-random and often in clusters. Studies have identified clusters of tissue-specific genes across different chromosomes23,24,25 that are co-expressed or co-regulated. To assess if these DMRs were preferentially associated with certain chromosomes and mirrored non-random chromosomal distribution of genes, distribution of the various DMRs on different chromosomes was examined (Supplementary Table 2).
While a positive correlation for both hypermethylated and hypomethylated DMRs was observed when compared to chromosome size as well as the number of genes per chromosome (Supplementary Table 2, Fig. 2B,C), no correlation was observed when gene density per megabase of chromosome was plotted against the number of DMRs, (Supplementary Table 2, Fig. 2D). The density of DMRs across the various chromosomes varied with chromosomes 17, 19 and 22 having high gene densities were found to have least number of DMRs. On the other hand, chromosome 1, 3 and 6 having low gene density had the highest number of DMRs (Fig. 3).

DMR density per Mb for each chromosome was calculated and plotted as histograms using Circos. The innermost circle displays the hypomethylated DMRs and the outermost circle displays the hypermethylated DMRs. Windows with more than 10 hypermethylated DMRs are shaded red and windows with more than 5 hypomethylated DMR are shaded green. DMR hotpsots within a chromosome are bound by a rectangle. The middle circle represents the ideogram. Bands drawn within the ideogram represent cytogenetic bands as are observed by Giemsa staining (data taken from UCSC Genome Browser).
To further investigate the observed enrichment (within chromosomes 1, 3 and 6) and depletion (within chromosomes 17, 19 and 22) of DMRs, we calculated DMR density using a 5 Mb sliding window with a 500 kb slide. A hypermethylation hotspot was defined as any 5 Mb window with average DMR density higher than 38.47 (average DMR density for chromosome 6), the highest average DMR density observed amongst the 24 chromosomes (supplementary Table 3). Based on these criterion, 23 hotspots present on 11 different chromosome were identified (Fig. 3, Table 5). Chromosome 1 had the largest number of hotspots (5 nos.). A region on the long arm of chromosome 6 (6q21-6q27) containing several immunologically important genes (including CCR6, TIAM2, ULBP2, LRP11, GPR126, FYN, NOX3) was found to have the highest DMR density (Table 5, Supplementary Table 4).
Functional classification of genes associated with DMRs
To examine if M. tuberculosis infection induced DNA methylation changes were associated with any specific genes or gene families, the genes associated with DMRs were classified using Gorilla Gene Ontology tool26 and the output was visualized using Revigo27. The gene families that showed significant enrichment (p < 0.05) included genes involved in signaling, cell communication, metabolism, transport, cell cycle, cytoskeleton reorganization, transcriptional regulation and chromatin modification (Fig. 4, Supplementary Table 5). DMR-associated immune response genes included genes corresponding to the HLA complex, cytokines, complement system.

GO analysis of genes associated with gene body DMRs present within promoter, gene body and TES visualized as a REViGO Interactive Graph.
The functional significance of genes found to be associated with regions of differential methylation in M. tuberculosis infected cells was also assessed by examining the interaction networks of the identified proteins. Using the web based PANTHER Gene Ontology tool28, we extracted the molecular function and biological process associated with each gene, followed by manual curation of the genes involved in immune response, chromatin modification, DNA replication and repair. The interaction network for the corresponding proteins was determined by the STRING search web tool29 and the interaction network was generated using clustering coefficient (Fig. 5). Most of the players involved in epigenetic reprogramming, were clustered in one sub-network. Interestingly, except for DNMT1 (a maintenance methyltransferase), all other DNA methyltransferase genes, DNMT3A, DNMT3B, DNMT3L, were found to have M. tuberculosis infection related DMR(s). SIN3A, a protein known to be associated with HDAC mediated repression of MHC class II proteins and found to be associated with HLA-DRα promoter in M. avium infected THP1 cells30, formed an important node connecting immune response with the epigenetic machinery (Fig. 5). Other epigenetic modulators like DNA demethylases TET2 and TET3, histone variant genes, INO80 complex, PRC2 complex genes, EED and SUZ12 were also associated with DNA methylation changes upon infection (Fig. 5).

Network analysis was performed on the set of genes found to be in the vicinity of DMRs identified in the MBD-seq. The interaction between proteins was generated using STRING (high confidence) and output was visualized using Cytoscape (Bottom left panel). Prominent nodes with their first neighbors are highlighted as a circular layout. The prominent node identified were SIN3A, ATM, PRKCA, IFNG, VAV3. Zoomed images represent circular layouts depicting genes involved in signaling (orange and purple), immunity (red), DNA repair (yellow) and chromatin organization (blue).
We next examined the function of genes present within the top five hotspots to assess whether any particular gene(s) or gene family associated more prominently with the infection-induced differential DNA methylation (Table 5). It was interesting to note that several immunologically important genes were present within the 5 hotpots, (especially in hotspot 1, 3 & 4, Fig. 6, supplementary Table 4). Hotspot 1 present on chromosome 6 included immunologically important genes like CCR6, TIAM2, ULBP2, LRP11, GPR126, FYN and NOX3 (Fig. 6). Hotspot 3 on chromosome 1 included ADAR, FCRL2, IL20, CD46, CD55, TNFSF4, IGFN1, IL19, TRAF5, etc, whereas hotspot 4 on chromosome 6 contained the HLA genes that are involved in antigen presentation apart from several other immunologically important genes (Fig. 6). The association of DMRs with histone genes (H2A gene cluster) was also noticeable in hotspot 3 on chromosome 1 (Fig. 6).

Each DMR is plotted as a tile. The track immediately inside the ideogram represents hypermethylated DMRs, and the inner most track depicts hypomethylated DMRs. Hypermethylated and hypomethylated DMRs associated with the conserved sequence motif are colored red and green respectively. Regions within the chromosome identified as DMR hotspots are shaded red in the ideogram. Immune system related genes are labelled in red, ncRNA in blue and chromatin related genes in green. Hotspot cluster with histone genes is highlighted in green whereas the hotspots associated with HLA cluster on chromosome 6 and genes involved in immune response on chromosome 1 are highlighted in red. Histone and HLA genes that were not part of a hotspot are highlighted in grey. The heat map circle (second from inside) shows the gene density across the respective chromosome with regions of very high gene density as red, high as yellow, medium as green and low as grey.
DMRs are associated with a conserved motif
To examine if a common motif was associated with the identified DMRs, the sequences corresponding to hypermethylated DMRs were tested for the presence of any conserved motif using MEME tool31. Since several immune response genes were present in the top 5 hotspots the analysis was initially performed on all the immune response genes associated DMRs. A 28 base pair motif with a conserved ‘GCCTCC’ core was identified in these DMRs using MEME (Fig. 7A). Based on this observation, the complete set of 23,433 DMRs were scanned for the presence of this motif. This motif was found to occur 8646 times in 6646 out of the 19,506 hypermethylated and 1455 times in 1178 out of the 3,927 hypomethylated DMRs (Supplementary Tables 6 and 7) with some DMRs showing the presence of this motif at multiple positions. In 515 genes this motif was present in both hypermethylated and hypomethylated DMRs (Supplementary Table 8). The enrichment of this motif in the human genome was also calculated. Taking the number of bases covered into consideration, this particular motif was more than 100 fold enriched in the DMR data set as compared to the whole genome. Scanning of individual chromosomes revealed that chromosome 19 had the maximum density of this motif. However, no significant enrichment of this motif was observed with any DMR across the different chromosomes (Supplementary Table 9). To further examine the correlation of this motif with DMRs, the distance of this motif from the peak maxima was calculated (Supplementary Table 10). In majority of the DMRs, the motif was found to occur within 150 bp from the peak maxima (Fig. 7B) and in 16% (1037) of hypermethylated and 20% of hypomethylated DMRs, the peak maxima and the motif overlapped (distance of 14 bp from the center of the 28 bp motif to peak maxima).

(A) The 28 bp motif identified in the DMRs and represented as a logo. The conserved ‘GCCTCC’ core has been underlined. The motif was generated using MEME tool. (B) Histogram showing the distance of the motifs, present within individual DMRs, from the peak maxima. Note that the maximum distance from the peak maxima for most of the DMRs was equivalent to the length of DNA wrapped around one nucleosome. Hyper DMRs are denoted by white bars and hypo DMRs with black bars.
M. tuberculosis infection induces methylation of non-CpG cytosines in host DNA
To validate the MBD-seq data and confirm DNA methylation changes observed in THP1 macrophages upon infection with Mycobacterium tuberculosis, we performed bisulfite sequencing on few of the DMRs. These DMRs were chosen either randomly or based on the role of the DMR associated gene in host defense mechanism against M. tuberculosis infection. Bisulfite analysis of these regions revealed negligible difference in methylation of cytosines present in CpG dinucleotide context. In fact, majority of the CpGs were found to be methylated in both uninfected and infected THP1 macrophages for the regions analysed (Fig. 8A, Supplementary Fig. 2). Surprisingly, a significant difference was obtained in the methylation of cytosines in non-CpG context for all the regions that we tested (Fig. 8A, B). This was true for both gain and loss of DNA methylation and the combined non-CpG methylation difference between uninfected and infected THP1 macrophages was also statistically significant (Fig. 8C). Examination of the bisulfite data also indicated that while change in the levels of cytosine methylation was observed in all the non-CpG dinucleotides, CpT methylation change was the most prominent (Fig. 8B,C).

(A) DNA methylation analysis by bisulfite sequencing. Cytosine methylation profile for some of the hypermethylated (HR) and hypomethylated (HO) DMRs was examined by bisulfite sequencing of genomic DNA from uninfected (U) and M. tuberculosis infected (I) THP1 macrophages. Red circles represent CpG and squares represent non CpG dinucleotides. Filled symbols (grey or black) represent methylated cytosine. 10 or more clones per sample were analysed. (B) Statistical analysis of methylated non-CpG dinucleotides in hypermethylated (HR) and hypomethylated (HO) DMRs. Ratio of mC to total C per sample was plotted on Y-axis. HO2 - chr.3:28247576-28247639; HO4 - chr7:97993741-97993898; HR1 - chr6:167283490-167283654; HR4 - chr.3:162931192-162931429; HR10 - chr.5:39213638-39213737; HR11 - chr.1:164394535-164394664. (C) Percentage methylation of non-CpG dinucleotides. Proportion of methylated CpA, CpT and CpC dinucleotides was calculated with respect to total cytosines. The regions analysed are listed on the X-axis. Coordinates for each region are provided in Materials and Methods.
To confirm the genuineness of the observed non-CpG differential methylation by bisulfite analysis, methylation analysis was performed by MeDIP analysis on some of the DMRs examined by bisulfite sequencing. For this analysis, DNA was isolated from three different sets of THP1 macrophages infected with M. tuberculosis H37Rv. MeDIP results confirmed that the non-CpG methylation difference observed by bisulfite sequencing reflected the methylation difference that were observed initially by MBD-seq (Fig. 9A).

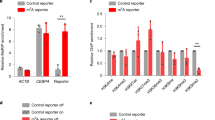
(D) MeDIP validation of bisulfite sequencing based cytosine methylation enrichment of selected DMRs. The enrichment is represented as percentage input for each region. (E) Expression analysis by quantitative RT-PCR for the genes listed below the X-Axis. The level of expression in uninfected THP1 macrophages is shown by a horizontal line (UI level). The experiment was done at least thrice in duplicates. The error bars represent standard deviation (SD). * Indicate significant difference (Student’s t-test, *P < 0.05).
Altered DNA methylation associated with gene expression changes
In order to study the effect of infection-induced DNA methylation changes on the expression levels of DMR-associated genes upon infection, expression of 28 genes associated with one or more DMRs was examined by Real Time RT-PCR. RNA for this analysis was isolated from three different sets of M. tuberculosis H37Rv infected or uninfected THP1 macrophages. 19 out of the 28 genes tested showed change in gene expression. 7 of them (ADORA3, CCR6, DNMT3B, HDAC9, NEDD4L, PBX1, SOX5) showed a decrease in expression, 12 genes showed increase in expression (BAIAP2L1, BCL2L1, CXCR4, FYN, NOTCH2, PARP1, PRKCD, PRMT3, RNASET2, RPS6KA2, ZCWPW2, ZNF148) while for 9 genes expression levels did not change (ANKS6, CMC1, COL15A1, DNMT3A, FOXP2, GABBR2, HRH2, PIK3R1, VAV3) 48 hours post-infection (Fig. 9B).
Discussion
The dynamic nature of the epigenome allows a cell to translate environmental cues and mount appropriate response by modulating its transcriptional machinery. Here we show genome-wide DNA methylation changes upon infection of a macrophage by Mycobacterium tuberculosis. Importantly, the DNA methylation changes were non-canonical and observed only for cytosines present in non-CpG dinucleotide context.
Changes in DNA methylation upon M. tuberculosis infection was observed across the whole THP1 macrophage genome, on every chromosome and in proximity of 7573 genes. Although a few studies have shown correlation of gene body DNA methylation with gene regulation, most studies show a strong association of promoter DNA methylation with gene transcription32,33. It was, therefore, surprising to find that most of the DNA methylation changes upon M. tuberculosis infection were associated with non-promoter, non-exonic regions of the genome. Studies have shown that tissue-specific-methylated non-promoter regions are usually cis-regulatory elements like enhancers, transcriptional activators or repressors34. Therefore, the observation that DNA methylation predominantly at non-promoter regions was altered would indicate that regulatory regions were targeted by M. tuberculosis during infection of THP1 macrophages. This hypothesis is also supported by our finding that several ncRNA genes, which have been implicated in the regulation of gene expression35, were the target of infection-induced DNA methylation changes.
During different developmental stages or during cell differentiation, regulation of gene expression through DNA methylation is achieved both by gain and loss of DNA methylation at specific loci36,37. On a random basis, it is expected that the number of genetic loci gaining or losing DNA methylation should be equal. During M. tuberculosis infection of THP1 macrophages more than 90% of the differentially methylated regions showed gain of DNA methylation. The basis of DNA methylation gain could either be due to overexpression of host DNA methyltransferases like DNMT3A, DNMT3B or DNMT1 or down regulation of host DNA demethylases like TET proteins. Our preliminary data suggests that the expression levels of the known host DNA methyltransferases was not altered (Fig. 9B). Since changes in histone modifications are more dynamic than DNA methylation and the differentiated cells normally utilize the changes in histone modifications to initially respond to environmental cues38,39, it is possible that observed DNA methylation changes are secondary effect of changes in associated histone modifications. On the other hand, it is possible that the increase in DNA methylation was due to a mycobacterial protein that was secreted into the host by the pathogen. We have previously reported that the secretory mycobacterial protein Rv2966c can methylate the host genome8. This would indicate that the hypermethylation observed in macrophages upon mycobacterial infection could be due to the action of Rv2966c. Further work that dissects out the contribution of Rv2966c vis-a-vis a host DNA methyltransferase would help in understanding the reason for the bias towards hypermethylation during M. tuberculosis infection.
Mammalian DNA methyltransferases are known to methylate cytosines predominantly in CpG dinucleotides and the level of non-CpG methylation has been found to be very low or negligible in most differentiated cells40. Therefore, it was surprising to find that the cytosine methylation during M. tuberculosis infection of macrophages was predominantly at non-CpG dinucleotides. At some of the loci, while all CpG dinucleotides were fully methylated in both uninfected and M. tuberculosis infected macrophages, differential methylation between the two samples was solely due to methylation of cytosines in non-CpG dinucleotides. The mycobacterial protein Rv2966c has been shown to be a cytosine methyltransferase that predominantly targets non-CpG dinucleotides8. Therefore, our observation indicating non-CpG DNA methylation underscores the role of the mycobacterial protein Rv2966c in host DNA methylation upon M. tuberculosis infection. Non-CpG DNA methylation has been hypothesized to be removed only during replication41. Since macrophages, like other differentiated cells, do not undergo cell division, the acquired non-CpG DNA methylation and the associated chromatin changes at specific loci would be maintained in the M. tuberculosis infected macrophages. This would indicate that success of M. tuberculosis as a pathogen could partly be due to the non-canonical epigenetic mechanisms that it uses to modulate host gene expression.
While the differential DNA methylation was observed within or in the vicinity of genes belonging to diverse gene families we observed significant enrichment for a few gene families including immune response, chromatin modification, DNA replication and repair. Macrophages are antigen presenting cells and the prime target of infection by M. tuberculosis. Concordant with this fact, HLA genes, involved in antigen presentation and hence amplification of immune response, were found to be located within the DMR hotspot on chromosome six. Also targeted were the cytokine genes (IL16, IL17) and chemokine genes (CCL17, CCL18) that are secreted by activated macrophages and are essential for downstream regulation of T cell dependent immune response42,43,44,45. As indicated before, several micro RNAs, known to regulate gene expression, and lncRNA, known to be associated with chromatin modifying complexes like PRC2 and MLL, were found to be target of differential methylation. Our findings are in agreement with recent studies that have shown changes in the miRNA and lincRNA profile of TB patients and indicate that ncRNA along with important epigenetic effector molecules play an important role in the regulation of immunity-related genes during M. tuberculosis infection20,21,46.
In summary, our study shows that during M. tuberculosis infection, macrophages undergo genome-wide non-canonical DNA methylation changes that have the potential to modulate the host gene expression. However, further work is needed to understand the mechanism that underlie these DNA methylation changes and dissect out the role of specific mycobacteria proteins working alone or in concert with host epigenetic effectors in modulating host gene expression.
Materials and Methods
Infection of THP1 cells with M. tuberculosis H37Rv
PMA treated THP1 cells (ATCC) were infected with M. tuberculosis H37Rv as described elsewhere8 at an MOI of 1:1 or 1:10 (cells:bacteria) for 4 hrs followed by treatment with Gentamycin for 2 hrs. Cells were harvested after 0, 24 and 48 hours of infection and DNA & RNA were isolated using Qiagen All Prep Kit. For examining the efficiency of infection, dilution plating of mycobacteria harvested from infected THP1 cells, with and without gentamycin treatment, was done and colony forming units were counted at each time point (supplementary Fig. 3A). In order to assess the viability of infected THP1 macrophages, MTT assay was performed after 0, 24 and 48 hours of infection (Supplementary Fig. 3B). In addition, THP1 macrophages were also infected with PKH67 (Sigma) labeled M. tuberculosis H37Rv cells. Briefly 2 × 108 mycobacterial cells were washed with PBS and resuspended in diluent C (Sigma). Cell suspension were then added to 2X dye and incubated at room temperature for 5 min. with periodic mixing. Reaction was quenched by addition of equal volume of serum, followed by three washes with PBS. Labeled cells were finally resuspended in 1 ml of RPMI and used for infection of THP1 macrophages. Infected cells were fixed after 0 and 24 hrs post-infection. The infected cells were visualized under confocal microscope (supplementary Fig. 3C).
MTT assay was performed as per manufacturer’s instructions. Briefly, cells were washed once with phenol free RPMI, followed by incubation with 1 mg/ml MTT for an hour at 37 °C. The cells were solubilized by addition of DMSO, formazan crystal was dissolved by titrating and absorbance measured at a wavelength of 540 nm. For CFU estimation of internalized bacteria, THP1 cells were lysed by addition of 0.05% SDS to release intracellular mycobacteria. 10 fold dilutions were plated on plain 7H11 plates and colonies were enumerated after 3–4 weeks of incubation.
The M. tuberculosis work was either performed in the P3 facility of National Institute of Immunology, Delhi, India or in the BSL-II type-II negative pressure facility within the laboratory of Dr. Sharmistha Banerjee, Department of Biochemistry, University of Hyderabad, Hyderabad, India as per approved IBSC guidelines.
Methylated DNA pull-down using Methyl Miner kit
Genomic DNA isolated from uninfected and M. tuberculosis H37Rv infected THP1 macrophages was estimated using Qubit and DNA quality was checked by resolving on 0.8% agarose. Equal amount of DNA from two biological replicates was pooled and 3 μg of the pooled genomic DNA was fragmented using Covaris 2.0 in the range 150–180 bp. MBD-based methylated DNA pull down was performed on the fragmented DNA using Methyl Miner kit (Invitrogen) as per the manufacturer’s instructions. Enriched DNA was eluted twice using high salt buffer and precipitated. The efficiency of pull down was checked by end point PCR with GAPDH as negative control and a reported methylated region on Chr.22 as positive control (supplementary Fig. 4A,B). The eluted fractions were estimated by Qubit and fragment size checked by Bioanalyser (supplementary Fig. 4C).
Library Preparation and Sequencing
Library Preparation was performed at Genotypic Technology’s Genomics facility (Bangalore, India) as follows. Libraries for multiplex ChIP Sequencing were constructed using NEXTflex™ ChIP-Seq Sample Preparation Kit protocol outlined in “Preparing Samples for ChIP Sequencing of DNA” (BIOO Scientific# IP-5143-01). Briefly, DNA was subjected to a series of enzymatic reactions that repair frayed ends and phosphorylated the fragments. The end-repaired fragments were adenylated with a single nucleotide ‘A’ overhang (BIOOScientific) followed by adapter ligation (NEXT Flex adapters). The fragments with ligated adapters were subjected to pre-size selection PCR for 5 cycles followed by size selection on 2% Low Melting Agarose. The size selected samples were enriched with 13 cycles of PCR. The prepared libraries were quantified using Nanodrop Spectrophotometer and Qubit followed by quality validation using High Sensitivity Bioanalyzer Chip (Agilent) (supplementary Table 11).
Sequence Analysis and Peak calling
MBD libraries were sequenced on Illumina HiSeq platform at a depth of ~30 million paired end reads (100 t × 2). Only those reads which passed the Q30 filter were retained. We analyzed the quality of the sequences using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and mapped the short reads to the reference human genome (build hg19) using Bowtie247, with a maximum mismatch of 2 bases. The alignment rate was >95% for all samples. We identified regions of significant enrichment using PeakSeq v1.148 using Input as background control, tag extension of 200 bp, and a target false discovery rate (q value) of 0.01. The number of peaks obtained for each sample are outlined in Supplementary Table 1.
Identification of Differentially Methylated Regions (DMRs)
To identify regions that showed a difference in their methylation levels upon M. tuberculosis H37Rv infection, a coverage based approach was followed. Firstly, for identification of regions hypermethylated upon infection, we took the peaks that were identified in I48 sample and calculated the coverage of these regions in all other samples using bedtools49. The coverage in each sample was normalized to its sequencing depth. Following this, all peaks that were smaller than 50 bp were discarded. To remove peaks that were significantly different between 0 and 48 hrs samples (probably due to cell culture) irrespective of infection, all regions that showed >25% difference in the coverage between U0 and U48 were removed. From the remaining regions, all peaks which had a coverage of less than 10 reads in I48 sample were removed to ensure that the coverage was significant to be called methylated. In addition, all regions that showed less than 50% increase in coverage in I48 with respect to U48 were also removed. To identify hypomethylated DMRs, the same approach was followed, except that we started with the peak-set obtained in U48, and the comparisons were done with respect to coverage of U48 instead of I48.
Identification of hotspots
To identify potential hotspots of hypermethylated DMRs, the increase in the density (normalized for sequencing depth) for each DMR was calculated by subtracting the normalized density of a DMR in U48 from that of I48. The total increase for 500 kb sliding windows of 5 MB each was calculated for all chromosomes. A given 5 MB window was considered a hotspot if the DMR density in this window (per MB) was greater than 38.47, the average DMR density for chromosome 6, the highest average DMR density observed amongst the 24 chromosomes.
Annotation of Peaks
For annotation, a non-redundant human gene database containing 28517 genes was created by retaining only the largest transcript of each official gene symbol. Each peak was annotated by mapping it to the nearest gene using bedtools, and further categorized the peaks into 4 groups – TSS (−1 kb to +0.5 kb of Transcription Start Site), TES (−0.5 kb to +0.5 kb of Transcription End Site), Gene Body (peaks present within the gene body but not TSS or TES, and intergenic peaks (all other peaks). Genic peaks were further annotated by mapping their peak maximas to 5′ UTR, CDS, Intron or 3′ UTRs.
MeDIP
MeDIP was performed using Auto MeDIP Kit on automated platform SX-8G IP–Star Compact (Diagenode). Briefly, 1.5 microgram of DNA from uninfected and infected THP1 cells was sheared using Bioruptor to 200–500 bp. As per the manufacturer’s protocol, the sheared DNA was immunoprecipitated with 5-methylcytosine antibody, a portion of sheared DNA (10%) was kept as input and remaining immunoprecipitated DNA, bound to magnetic beads was isolated. qPCR for selected genomic locus using the primers listed below was performed and efficiency was calculated as %(me-DNA-IP/Total Input).

Bisulfite Sequencing
Genomic DNA from uninfected and M. tuberculosis infected (48 hrs post-infection) THP1 macrophages was subjected to bisulfite conversion using Epitect Bisulfite Kit (Qiagen). PCR was performed on converted DNA using strand-specific modified primers. The PCR product was then cloned into XcmI digested pBSK-TA vector. At least 10 clones were analyzed for each sample.

Hypomethylated

Expression Analysis by Real Time PCR of M. tuberculosis infected THP1 macrophages
THP1 cells were infected with M. tuberculosis H37Rv as described above at an MOI of 10:1 for 4 hrs followed by treatment with gentamycin for 2 hrs. Cells were harvested at 0 hrs, 24 hrs and 48 hrs of infection and DNA-RNA were isolated using Qiagen All Prep Kit. 1 μg of RNA was converted to cDNA using SuperscriptIII (Invitogen). The change in expression upon infection of M. tuberculosis H37Rv in THP1 cells was evaluated by Real Time PCR using Mesa Green qPCR Mastermix Plus (Eurogentec) in ABI Prism SDS 7500 system. GAPDH was used as internal control. Ct values were normalized for GAPDH and fold change in infected sample with respect to uninfected samples was plotted.

Identification of motif using MEME tool
A text file containing chromosome number and the peak coordinates for DMRs associated with immune response genes was uploaded to Galaxy server to extract the sequences in fasta format. The same file was then uploaded to MEME suite to extract any conserved motif. The motif matrix obtained from the MEME output was then used to search for occurrence of that motif in the hypermethylated and hypomethylated DMRs. Occurrence of motif in DMRs was identified using FIMO.
Identification and categorization of repeats in the DMRs
The association of DMRs with repeats was checked using a custom python script. Firstly, the repeat-masked sequence of all DMRs was extracted from the hg19 reference genome. For each DMR, the number of bases that were repeat-masked was calculated, and expressed as ratio to the total size of DMR. To further check if any given category of repeats was specifically associated with DMRs, the list of all repeat elements was downloaded using UCSC table browser50. Using this list, the number of bases of a DMR associated with a given repeat category was calculated. Specific enrichment of a repeat category was checked by comparing the fraction of DMRs associated with each category with the fraction of the entire genome associated with that category.
Gene Ontology
The list of official gene symbol for genes associated with non-intergenic DMRs was submitted to GORILLA tools and functional overrepresentation test was performed to identify the enriched gene ontology (GO) categories26. The output was submitted to ReviGO27 and interactive graph was generated for selected GO terms. Using the web based PANTHER Gene Ontology tool (www.pantherdb.org), the molecular function associated with each gene was extracted and the genes were manually curated into the enriched GO categories.
The proteins involved in immune response, chromatin modification, DNA replication and repair were submitted to STRING (www.string-db.org). The interaction network so generated was extracted as a text file and submitted to Cytoscape (www.cytoscape.org) and clustering coefficient based circular layout algorithm was used to generate the network.
Additional Information
How to cite this article: Sharma, G. et al. Genome-wide non-CpG methylation of the host genome during M. tuberculosis infection. Sci. Rep. 6, 25006; doi: 10.1038/srep25006 (2016).
References
Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 (2002).
Meaney, M. J. & Szyf, M. Environmental programming of stress responses through DNA methylation, life at the interface between a dynamic environment and a fixed genome. Dialogues Clin. Neurosci. 7, 103–123 (2005).
Laurson, J., Khan, S., Chung, R., Cross, K. & Raj, K. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 31, 918–26 (2010).
Marr, A. K. et al. Leishmania donovani infection causes distinct epigenetic DNA methylation changes in host macrophages. PLos Pathog. 10, e1004419 (2014).
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998).
Yaseen, I., Kaur, P., Nandicoori, V. K. & Khosla, S. Mycobacteria modulate host epigenetic machinery by Rv1988 methylation of a non-tail arginine of histone H3. Nat. Commun. 6, 8922 doi: 10.1038/ncomms9922 (2015).
Pennini, M. E., Pai, R. K., Schultz, D. C., Boom, W. H. & Harding, C. V. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J. Immunol. 176, 4323–4330 (2006).
Sharma, G., Upadhyay, S., Srilalitha, M., Nandicoori, V. K. & Khosla, S. The interaction of mycobacterial protein Rv2966c with host chromatin is mediated through non-CpG methylation and histone H3/H4 binding. Nucleic Acids Res. 43, 3922–3937 (2015).
Bierne, H., Hamon, M. & Cossart, P. Epigenetics and bacterial infections. Cold Spring Harb. Perspect. Med. 2, a010272 (2012).
Masaki, T. et al. Reprogramming adult Schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell 152, 51–67 (2013).
Mujtaba, S. et al. Anthrax SET protein, a potential virulence determinant that epigenetically represses NF-κB activation in infected macrophages. J. Biol. Chem. 288, 23458–23472 (2013).
Rolando, M. et al. Legionella pneumophila effector RomA uniquely modifies host chromatin to repress gene expression and promote intracellular bacterial replication. Cell Host Microbe 13, 395–405 (2013).
Harouz, H. et al. Shigella flexneri targets the HP1γ subcode through the phosphothreonine lyase OspF . EMBO J. 33, 2606–2622 (2014).
Tsuchiya, S. et al. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 42, 1530–1536 (1982).
Hashimshony, T., Zhang, J., Keshet, I., Bustin, M. & Cedar, H. The role of DNA methylation in setting up chromatin structure during development. Nat. Genet. 34, 187–192 (2003).
Rauch, T. A., Wu, X., Zhong, X., Riggs, A. D. & Pfeifer, G. P. A human B cell methylome at 100-base pair resolution. Proc. Natl. Acad. Sci. USA 106, 671–678 (2009).
de Koning, A. P., Gu, W., Castoe, T. A., Batzer, M. A. & Pollock D. D. Repetitive elements may comprise over two-thirds of the human genome. PLos Genet. 7, e1002384 (2011).
Mattick, J. S., Makunin, I. V. & Non-coding R. N. A. Hum. Mol. Genet. 15, R17–29 (2006).
Fu, Y. et al. Circulating microRNAs in patients with active pulmonary tuberculosis. J. Clin. Microbiol. 49, 4246–4251 (2011).
Qi, Y. et al. Altered serum microRNAs as biomarkers for the early diagnosis of pulmonary tuberculosis infection. BMC Infect. Dis. 12, 384 (2012).
Das, K., Saikolappan, S. & Dhandayuthapani, S. Differential expression of miRNAs by macrophages infected with virulent and avirulent Mycobacterium tuberculosis. Tuberculosis (Edinb) 93, Suppl: S47–50 (2013).
Mattick, J.S. Non-coding RNAs, the architects of eukaryotic complexity. EMBO Rep. 2, 986–991 (2001).
Bortoluzzi, S. et al. A comprehensive, high-resolution genomic transcript map of human skeletal muscle. Genome Res. 8, 817–825 (1998).
Cohen, B. A., Mitra, R. D., Hughes, J. D. & Church, G. M. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat. Genet. 26, 183–186 (2000).
Gabrielsson, B. L., Carlsson, B. & Carlsson, L. M. Partial genome scale analysis of gene expression in human adipose tissue using DNA array. Obes Res. 8, 374–384 (2000).
Eden, E., Navon, R., Steinfeld, I., Lipson, D. & Yakhini, Z. GOrilla, a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 (2009).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLos One 6, e21800 (2011).
Thomas, P. D. et al. Applications for protein sequence-function evolution data, mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 34, W645–650 (2006).
Jensen, L. J. et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–416 (2009).
Wang, Y., Curry, H. M., Zwilling, B. S. & Lafuse, W. P. Mycobacteria inhibition of IFN-gamma induced HLA-DR gene expression by up-regulating histone deacetylation at the promoter region in human THP-1 monocytic cells. J. Immunol. 174, 5687–5694 (2005).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 (1994).
Bird, A. P. CpG-rich islands and the function of DNA methylation. Nature 321, 209–213 (1986).
Jones, P. A. Functions of DNA methylation, islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012).
Maston, G. A., Evans, S. K. & Green, M. R. Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 7, 29–59 (2006).
Kaikkonen, M. U., Lam, M. T. & Glass, C. K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 90, 430–440 (2011).
Monk, M., Boubelik, M. & Lehnert, S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 99, 371–382 (1987).
Ji, H. et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 467, 338–342 (2010).
Bonasio, R., Tu, S. & Reinberg, D. Molecular signals of epigenetic states. Science 330, 612–616 (2010).
Barter, M. J., Bui, C. & Young, D. A. Epigenetic mechanisms in cartilage and osteoarthritis, DNA methylation, histone modifications and microRNAs. Osteoarthritis Cartilage 20, 339–349 (2012).
Lister, R. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009).
Ziller, M. J. et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLos Genet. 7, e1002389 (2011).
Ritter, M. et al. Elevated expression of TARC (CCL17) and MDC (CCL22) in models of cigarette smoke-induced pulmonary inflammation. Biochem. Biophys. Res. Commun. 334, 254–262 (2005).
Schutyser, E., Richmond, A. & Van Damme, J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J Leukoc Biol. 78, 14–26 (2005).
Song, C. et al. IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. J. Immunol. 181, 6117–6124 (2008).
Ghigo, E. et al. IL-16 promotes T. whipplei replication by inhibiting phagosome conversion and modulating macrophage activation. PLos One 5, e13561 (2010).
Wang, Y. et al. Long noncoding RNA derived from CD244 signaling epigenetically controls CD8+ T-cell immune responses in tuberculosis infection. Proc. Natl. Acad. Sci. USA 112, E3883–E3892 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Rozowsky, J. et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat. Biotechnol. 27, 66–75 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools, a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Karolchik, D. et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–496 (2004).
Acknowledgements
G.S., R.G., M.Z.K. and S.U. are recipients of Senior Research Fellowship of the Council of Scientific and Industrial Research (CSIR), India towards the pursuit of a PhD degree of the Manipal University, Manipal; University of Hyderabad, Hyderabad and JNU, New Delhi, respectively. This study was supported by funds from CDFD and a grant from Department of Biotechnology, Government of India to S.K., V.K.N. and S.B.
Author information
Authors and Affiliations
Contributions
S.K. and G.S. designed the experiments. G.S. and P.S. performed the experiments. R.G., S.U. helped G.S. and P.S. perform the M. tuberculosis infection experiments in the laboratories of V.K.N. and S.B.; C.F.U. and M.T.T. data was generated by M.Z.K. in V.K.N. laboratory and D.T.S. performed all the bioinformatics analysis. S.K., G.S. and D.T.S. wrote the manuscript with inputs from V.K.N. and S.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sharma, G., Sowpati, D., Singh, P. et al. Genome-wide non-CpG methylation of the host genome during M. tuberculosis infection. Sci Rep 6, 25006 (2016). https://doi.org/10.1038/srep25006
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25006
This article is cited by
-
The spectrum of tuberculosis described as differential DNA methylation patterns in alveolar macrophages and alveolar T cells
Clinical Epigenetics (2022)
-
A differential DNA methylome signature of pulmonary immune cells from individuals converting to latent tuberculosis infection
Scientific Reports (2021)
-
Epigenetic interaction of microbes with their mammalian hosts
Journal of Biosciences (2021)
-
The bovine alveolar macrophage DNA methylome is resilient to infection with Mycobacterium bovis
Scientific Reports (2019)
-
Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species
Epigenetics & Chromatin (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
