
Abstract
Gut microbiome shapes various aspects of a host’s physiology, but these functions in aquatic animal hosts have yet to be fully investigated. The sea cucumber Apostichopus japonicus Selenka is one such example. The large growth gap in their body size has delayed the development of intensive aquaculture, nevertheless the species is in urgent need of conservation. To understand possible contributions of the gut microbiome to its host’s growth, individual fecal microbiome comparisons were performed. High-throughput 16S rRNA sequencing revealed significantly different microbiota in larger and smaller individuals; Rhodobacterales in particular was the most significantly abundant bacterial group in the larger specimens. Further shotgun metagenome of representative samples revealed a significant abundance of microbiome retaining polyhydroxybutyrate (PHB) metabolism genes in the largest individual. The PHB metabolism reads were potentially derived from Rhodobacterales. These results imply a possible link between microbial PHB producers and potential growth promotion in Deuterostomia marine invertebrates.
Similar content being viewed by others

Introduction
Apostichopus japonicus (Selenka) is a temperate sea cucumber species occurring in the western Pacific Ocean, Yellow Sea, Sea of Japan and Sea of Okhotsk1. It ingests organic matter, bacteria, protozoa, diatoms as well as algae and animal detritus and thus plays an important role in benthic nutrients recycling2. A. japonicus is also an important fishery resource in many Asian countries, especially in China3. Wild stocks have drastically declined due to overfishing and in 2013 this species was listed as “Endangered” on the IUCN Red List of Threatened Species4. Wild stocks of sea cucumber species have been sustained by seed production and aquaculture3, however a lack of information concerning ecology, physiology and biochemistry of this species is an obstacle to further success in this field. Extremely large growth gap among the juveniles of A. japonicus (Fig. 1a) is also a common problem in the seed production of sea cucumbers5,6,7. As a result, there is a right-skewed body size distribution among cultured animals even when raised in the same tank under identical conditions (e.g. temperature, light, animal density). This can be seen in Fig. 1a,b in which the largest individual is ca. 50 times larger than the smallest one. Population densities, congenital hosts’ traits and food competition have all been considered as possible causes, however, the cause of this huge growth gap is still unknown5.

Growth gaps observed in the cultured sea cucumbers.
(a) Representatives of the larger and the smaller sea cucumbers cultured under identical conditions are displayed from lower to upper. (b) A histogram for body weight of sea cucumbers showed right-skewed size distribution. These specimens are 20 months old and were raised on artificial diets for 17 months. For the final two months in the Kumaishi farm, the animals were fed with naturally occurred diatoms before being studied. Size distributions of the larger and smaller individuals used in this study were highlighted in green and purple, respectively. (c) Box plot for actual body weights from 10 larger and 10 smaller individuals used in this study. The body weight averages of two groups, “larger” and “smaller”, were statistically significant.
The increasing demand for A. japonicus livestock has led to further research seeking optimal conditions for seeding and farming. Most of the research has focused on i) the effects of abiotic factors on sea cucumbers growth8,9; ii) the effects of different diets and ration composition on growth10,11; iii) the use of stable isotopes to assess carbon turnover, metabolism, growth and absorption of different food sources12,13; iv) the effects of potential probiotics on growth, immunity and resistance to pathogens14,15; and v) the effect of stock density on growth rates16. Holothurians are also used as model animals to study visceral regeneration of digestive systems17,18. Despite its importance, a few studies have assessed A. japonicus microbial diversity using either culture-dependent or independent methods19,20,21.
The relationship between gut microbiota and hosts has been widely studied in many organisms. For instance, microbiome in ruminants and termites are largely responsible for the digestion of indigestible components such as cellulose and hemicellulose22,23 and is also responsible for fatty acid absorption in zebra fish24. Obesity has been associated with mouse gut microbiota25; colonization of germ-free mice with an ‘obese microbiota’ results in a significant increase in total body fat compared to colonization with a ‘lean microbiota’26. Gut microbiota can improve immune system response27 and can also modulate brain development and behavior28,29. These studies indicate a strong interaction between microbiota and host, however, little is known about that of A. japonicus sea cucumber, which is one of the phylogenetically close Deuterostomia species30.
We have observed a growth gap in juvenile sea cucumbers in Hokkaido hatcheries. Most previous studies have assessed the influence of abiotic and biotic factors on sea cucumber growth manipulating the concentrations, composition or intensity of the environmental factors8,9,10,11,13,14,15,16. However, to our knowledge, there have been no studies investigating the possible intrinsic causes of sea cucumber growth. Our hypothesis is that gut microbes play a major role on sea cucumber growth. To explore any possible contributions of the gut microbiome to its host’s growth, individual fecal microbiome comparisons (both taxonomically and functionally) of both large and small individuals of sea cucumber A. japonicus were performed. To reach our goal, we developed a non-destructive (without animals sacrifice) protocol for sea cucumber feces collection and DNA extraction to assess the gut microbial diversity in live specimens. We analyzed the microbial diversity taxonomically by pyrosequencing the V1-V2 region of 16S rRNA gene and functionally by massively parallel sequencing metagenomes. The non-destructive individual analysis we introduce here opens the way to both marine animal conservation and also to studies in the dynamics of host-microbes interaction.
Results
Larger and smaller Apostichopus japonicus have different fecal microbial communities
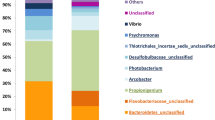
We assessed fecal microbiota of the larger and smaller individual sea cucumbers using tag sequencing of the V1-V2 region of 16S rRNA gene. After trimming, 104,679 qualified reads from the sea cucumber samples (52,996 and 51,683 reads from 10 larger and smaller individual samples, respectively) and 5,241 qualified reads from the seawater sample were used to cluster into OTUs (similarity 97%) (Table S1). After removing Eukarya, mitochondria and chloroplast sequences, we used 63,574 bacterial sequences for taxonomic assignment. Chloroplast sequences, which might be from ingested algae, were more frequently associated with smaller individuals [48.4 ± 16% (mean ± SD)] than larger individuals (37 ± 15%), however, these were not significant (P > 0.05). In total 1,976 OTUs with average Good’s coverage of 93.5 ± 1.8% were obtained. Eukaryotic diversity, consisting of >90% of stramenopiles, was not significantly different between the two groups.
The fecal microbiota of the larger and the smaller individuals were clustered separately from one another and separated from the seawater microbiota from the rearing tank (Fig. 2a). We found two major taxa in the fecal microbiota. The most abundant phylum was Proteobacteria; 57.4 ± 4.4% and 52.7 ± 4.9% of reads in the larger and the smaller individuals, respectively. Proteobacteria accounted for 77.3% of reads in the seawater sample. Bacteroidetes was the second most abundant phylum in sea cucumber samples (34.0 ± 4.8% and 37.8 ± 6.3% in larger and smaller individuals, respectively). Only 11.4% of this phylum was observed in the seawater sample. Comparing classes among Proteobacteria in the fecal microbiota, the relative abundances of Alphaproteobacteria (Larger, 15.3 ± 1.8%; Smaller, 11.5 ± 2.8%) and Deltaproteobacteria (Larger, 2.3 ± 0.7%; Smaller, 1.1 ± 0.8%) were significantly different between the larger and smaller individuals (P < 0.05, q < 0.05). The relative abundances of minor phyla (Actinobacteria, Firmicutes, Fusobacteria and Spirochaetes) were significantly different between the two groups (P < 0.05, q < 0.05, see Table S2).

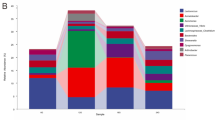
The microbiota of larger and smaller sea cucumbers feces are different.
(a) The larger and the smaller sea cucumbers clustered individually and seawater sample differed from sea cucumber samples. (b) Ranking of the order level abundances in fecal microbiota of the larger (green) and the smaller (purple) individuals. Only significant taxa were shown in this bar plot; Rhodobacterales, Desulfobacterales and Oceanospirillales were significantly more abundant in the larger individuals, Marinicellales, Acidimicrobiales were more abundant in the smaller individuals.
The most abundant order was Flavobacteriales (Larger, 29.0% ± 5.2%; Smaller, 33.5% ± 6.4%), followed by Alteromonadales (Larger, 23.9% ± 5.0%; Smaller, 25.1% ± 3.2%), but the data was not significantly different between the two groups (P > 0.05). Rhodobacterales among Alphaproteobacteria was the third most abundant (Larger, 14.8 ± 2.0% and Smaller, 11.1 ± 2.8%) and the relative abundances between larger and smaller individuals were significantly different (Fig. 2b and Table S2 (P < 0.05, q < 0.05)). Desulfobacterales (Larger, 1.1% ± 0.5%; Smaller, 0.2% ± 0.1%) (Deltaproteobacteria) and Oceanospirillales (Larger, 0.9% ± 0.7%; Smaller, 0.3% ± 0.4%), were more abundant in larger individuals (P < 0.05, q < 0.05), on the other hand Marinicellales (Larger, 2.8% ± 0.6%; Smaller, 3.5% ± 0.7%) and Acidimicrobiales (Larger, 0.9% ± 0.3%; Smaller, 1.3% ± 0.4%) were more abundant in smaller individuals (P < 0.05, q < 0.05). Relative abundances of some other orders, for instance Fusobacterales, Rhizobiales and Actinomycetales, were also significantly different between the two groups (P < 0.05, q < 0.05), but these were quite low occupation (<1%) (Fig. 2b).
Species richness and diversity were not statistically significant between the two groups of microbiota (Fig. 3a–c), on the contrary, the unweighted UniFrac analysis demonstrated clear clustering of the larger and smaller individuals microbiota (Fig. 3d).

Microbial diversity comparisons between larger and smaller Apostichopus japonicus individuals.
(a) Species richness, (b) Shannon index, (c) evenness and (d) unweighted UniFrac-based 2D PCoA plot was based on all OTUs from the larger, the smaller sea cucumbers and seawater sample. Percent variation expected were PC1 15.69%, PC2 10.99%. Samples indicated with arrows were used for functional metagenomic analysis. The larger sea cucumbers, the smaller ones and seawater sample are indicated in green, purple and blue, respectively.
Core fecal microbiota of Apostichopus japonicus juveniles and OTUs correlated to hosts’ body weight
We hypothesized that larger and smaller sea cucumbers could possess unique microbiota. Comparisons of individual fecal microbiota of A. japonicus demonstrated that 30, 12 and 35 OTUs were defined as larger individual core (LIC), smaller individual core (SIC) and all individual core (AIC) microbiota, respectively (Fig. S1a). The OTUs from LIC and SIC microbiota were further affiliated to 10 and 6 major bacterial orders, respectively. Rhodobacterales, Alteromonadales, Cytophagales, HTCC2188 (Gammaproteobacteria) and Myxococcales were absent in the SIC and Marinicellales was not observed in the LIC microbiota (Fig. S1b). Those of the AIC microbiota were classified into nine orders (e.g. Rhodobacterales, Flavobacteriales and Alteromonadales). A total of 1,728 OTUs were also defined as a pan fecal microbiota of the cultured juvenile A. japonicus.
We further identified that 15 OTUs had strong positive correlation with sea cucumber body weight (r > 0.7) with statistical significance (corrected P < 0.05), respectively (Table S3). Two and six OTUs were affiliated to LIC and larger only, respectively. OTU584 (Legionellaceae) showed the highest positive correlation. OTU1150 (Alteromonadales OM60) was found both in all A. japonicus individuals used in this study and the seawater.
Comparative metagenome analysis of fecal microbiome
Metagenome sequencing using the HiSeq platform was performed on the largest and the smallest specimens (except specimens containing insufficient fecal DNA samples) as representatives of larger and smaller individuals used in the above analyses. From the largest and the smallest individuals, 22,386,506 reads (2.2 Gb) and 22,803,396 reads (2.3 Gb) were obtained, respectively. After quality filtering, 18,582,516 and 18,156,817 reads from the largest and the smallest individual, respectively, were used for MG-RAST annotation. The total number of annotated reads was 6,049,986 and 4,634,631 for the largest and the smallest individual, respectively. Bacterial reads occupied 92.6% and 87.6%, eukaryotic reads occupied 7.2% and 12.2% and archaeal reads occupied 0.16% and 0.15% in the largest and the smallest library, respectively. Bacterial community based on the metagenomic reads was similar to those obtained by the 16S rRNA gene based microbiota analysis (Fig. S4).
The metagenomic analysis demonstrated that 25 functional features on subsystem category (Level 1) were significantly more abundant in one of the samples. Microbial reads annotated to ‘carbohydrates’, ‘amino acids and derivatives’, ‘cofactors, vitamins, prosthetic groups, pigments’, ‘fatty acids, lipids and isoprenoids’, ‘cell wall and capsule’ and others were significantly more abundant in the largest individual and ‘protein metabolism’, ‘RNA metabolism’, ‘respiration’, ‘virulence, disease and defense’ and others were significantly more abundant in the smallest individual (Fig. S2a). In more detail, the reads affiliated to genes responsible for polyhydroxybutyrate (PHB) metabolism, n-phenylalkanoic acid degradation, fatty acid metabolism cluster, acetyl-CoA fermentation to butyrate, fatty acid degradation regulons, serine-glyoxylate cycle and butyrate metabolism in Level 3 subsystem category were significantly more abundant in the largest individual. Among those, the largest positive effect size (more abundant in the largest individual) was observed in reads affiliated to PHB metabolism (Fig. 4). Interestingly further KEGG mapping of the annotated reads into the PHB metabolism to validate which metabolic pathways are more prominent revealed phaA (acetyl-CoA C-acetyltransferase), phaB (acetoacetyl-CoA reductase) and phaC (PHB synthase), which are essential in PHB synthesis from acetyl-CoA to PHB, were more abundant in the largest individual’s metagenomic library (Fig. S3a). In particular, the abundance of phaA gene (EC 2.3.1.9) reads impacted not only mapping into PHB metabolism but also that of other subsystem categories (Fig. S3b–g). The abundance of enoyl-CoA hydratase (EC 4.2.1.17), which is involved in butyrate metabolism, lysine and tryptophan synthesis and PHB degradation, also affected to positive effective size of most of the subsystem categories (Fig. S3a–e,g). Notably, 67% in average of total prokaryotic reads annotated to the phaABC genes were affiliated to Rhodobacterales, in accordance with the results obtained by 16S rRNA typing, where higher abundance of Rhodobacterales in the larger individuals’ microbiota was found.

An extended error bar plot showing more abundant features in the largest sea cucumber.
Green bars indicate the largest sea cucumber and purple indicate the smallest sea cucumber. Proportion (left side) means a possible abundance of microbes possessing each functional feature and difference between proportions (=effect sizes) for each feature is indicated by a green dot. For this analysis, features were filtered by q value (0.05) and effect size (0.05). Relative abundances of metagenome reads related to polyhydroxybutyrate (PHB) metabolism and butyrate metabolism were more abundant in the largest individual.
Reads affiliated to essential cellular functions in subsystem Level 3 categories (e.g. bacterial ribosome LSU or SSU, bacterial RNA polymerase, F0F1-type ATPase) were significantly more abundant in the metagenomic library from the smallest animal (Fig. S2b). Validation of the taxonomic affiliation showed, for example, bacterial RNA polymerase reads were consisted of diverse array of the sea cucumber gut microbiome.
Discussion
A. japonicus sea cucumber is a good candidate to study the evolution, dynamics and functions of gut microbiome in Deuterostomia invertebrates due to their unique ecophysiology17,18 and it is in urgent need of both conservation and development of intensive aquaculture3,4. To our knowledge, this is the first individual fecal microbiome analysis of A. japonicus to assess the possible interactions between the microbiome and the host animals’ growth. Our analyses showed that the observed differences in fecal microbiota of larger and smaller individuals might be associated with PHB synthetic metabolism. Interestingly, both high-throughput sequencing approaches suggest that Rhodobacterales may play a key role in sea cucumber growth. Feeding trials assessing direct effect and causality of these gut microorganisms on their hosts’ growth are necessary to support this idea, but our results provide a new insight into a link between PHB producing microbes and their potential contribution to sea cucumber growth. Only a few studies have investigated the gut microbiota of holothurians sea cucumbers, all of them using dissection techniques. These studies have assessed gut bacterial diversity and concluded selective feeding and proposed some potential probiotic bacteria19,31. None of these previous studies have investigated the roles of bacterial communities on the host’s health or growth. Using the non-destructive methodology presented here, live specimens can be used to monitor gut microbial dynamics and host-microbes interaction; for example, the dynamics of gut microbiome before and after evisceration and after probiotic treatment could be monitored. Again, we would like to emphasize that sea cucumber is one of the best model animals to study in terms of organ regeneration among Deuterostome animals in both molecular and genetic basis. In fact, studies have revealed numbers of novel genes differentially expressed during the gut regeneration process17. It is interesting to understand how the gut microbiome, including PHB producers, is affected such examples of the regeneration process using the non-destructive fecal microbiome methodology. The strategy also aids to the “endangered” species conservation in the wild. Without unnecessary mortality, on site fecal collections of a Stichopodidae sea cucumber and further analyses are in progress.
We found a similar number of bacterial OTUs and diversity (Shannon index) in A. japonicus gut microbiota to other studies using similar DNA extraction and pyrosequencing strategies19. The diversity indices of the A. japonicus fecal microbiota are similar to those observed in Hydra microbiota but lower than those of fish and mammals32. The fecal microbiota of juvenile A. japonicus is predominantly in Proteobacteria and Bacteroidetes, whereas Bacteroidetes was less dominant in maricultured adult specimens19. It is well known that gut microbiota is influenced by food intake, by environmental factors and by developmental stages of hosts and their genetics32,33. The observed differences in microbiota along with sea cucumber’s body weight may be caused by one of (or synergy among) the above-mentioned factors. Similar to the previous report on host habitat selection of the gut microbiota of zebrafish and mouse34, 8 and 10 phyla detected in the sea cucumber microbiota were shared with those of mice/human and zebrafish, however the abundance and diversity below phylum level differed. The juvenile A. japonicus individuals also showed microbiota signatures observed in both fish guts (Proteobacteria rich) and the human gut (Bacteroidetes rich)32. In the previous report on gut microbiota transplantation experiments between zebrafish and mouse, a restoration tendency to the original composition of gut microbiota is observed; abundance of Proteobacteria increases, but that of Bacteroidetes drops to below the detectable limit in a mouse-microbiota transplanted zebrafish34. A hybrid type of sea cucumber microbiota observed here might be affected by not only their conserved status of these hosts in surviving in aquatic environments but also their host species specific selective pressure. Archaeal sequences were found to be rare in the metagenomic libraries. Similar results were found in a previous study35, therefore it is not necessary to discuss their roles in the gut microbiota here.
Rhodobacterales, Desulfobacterales and Oceanospirillales (in more detail Rhodobacteraceae and Desulfobulbaceae; see family table in TableS2) were significantly abundant in the larger individuals and Marinicellaes and Acidimicrobiales were similarly abundant in the smaller. Rhodobacteraceae is one of the most diverse aquatic bacterial groups (>100 genera) consisting of aerobic photo- and chemoheterotrophs and a diverse array of polyhydroxyalkanoates (PHA), commonly PHB, producers isolated from marine environments36,37,38. Rhodobacteraceae sequences and isolates have frequently been observed in marine environments37 as well as in the gut microbiota of wild and maricultured A. japonicus19,35. More recently, dietary β-glucan supplementation for A. japonicus triggered Rhodobacteraceae abundance in the gut microbiota and host NF-κB signaling pathway and then it was speculated that the activation of the signaling pathway was caused by the increased mass of the bacterial group20. Rhodobacteraceae can be involved in sulfur and carbon biogeochemical cycling in water columns and on the photic sea floor due to their physiological traits. This bacterial group is being considered as transient fractions in the gut microbiota from the environments35. Interestingly, the metagenomic comparisons described here reveal a further possible hypothesis that Rhodobacterales affects the host growth of A. japonicus as a PHB producer. PHB is known to be accumulated in commonly nutrient-limited bacterial cells, especially under increased carbon to nitrogen ratio conditions39,40. There are three types of PHB, 1) high molecular weight storage PHB consisting of >1,000 3-hydroxybutyrate (3HB) residues, 2) low molecular weight PHB which has ca. 100 to 200 3HB units and 3) conjugated PHB (cPHB) consisting of low numbers of 3HB units (<ca. 30) which are covalently linked to proteins40. PHB-like granules have recently been observed in human cells41. PHB degradation is also catalyzed by PHB/PHA depolymerase in the first step of biodegradation and the degradation process can serve as energy sources for the host cells. PHB has been tested as feed additives promoting host growth in fish and crustacean aquaculture42,43,44. The PHB also conferred protection to an Artemia host against a pathogenic vibrio by modulating the Hsp70-triggered innate immune response45. A PHB depolymerase gene (phaZ) was also retrieved from an EST library from the intestinal tissues of A. japonicus (GenBank JK730361.1). It is possible to consider that the bacterial PHB, especially that produced by Rhodobacterales, might also serve as an energy source for a host sea cucumber, causing the observed higher growth in the larger individuals. PHB producing bacteria has also been found in termite guts, but its proper role has not been fully elucidated yet46,47. This hypothesis further gives us a novel ecological insight into carbon cycling as a bridge between storage granule producing bacteria and extra-nutrition for benthic marine invertebrates. Considering our results, we suggest the use of PHB producers as probiotics in sea cucumber aquaculture. Future bioassays studies using isolated PHB producing bacteria or their PHB are necessary to confirm their probiotics potential in promoting sea cucumber growth.
Another group of bacteria more abundant in larger individuals was Desulfobulbaceae, which is a strictly anaerobic sulfate-reducing bacteria (SRB)48,49. The presence of Desulfobulbaceae sequences suggests that anoxygenic environments are formed in the gut of A. japonicus juveniles. In a previous study, Desulfosarcina, a member of Desulfobacterales, was also predominant in the hindgut contents of the maricultured A. japonicus19. The roles of SRB are presumed to be related to acetate production and nitrogen fixation19. SRB colonizes ca. 50% of human guts and are frequently detected in healthy adults fecal microbiota50. SRB are known to be the most efficient hydrogenotrophs, they can utilize H2 and some fermentation products (e.g. lactate, formate) produced in the guts as electron donors49. Thus, SRB contribute towards maintaining redox balance and maximizing microbial energy production. The higher abundance of SRB sequences observed in larger individuals’ feces suggests higher redox states in their gut. Such conditions are more likely to be suitable for fermentation and PHB accumulation, which might affect the host’s growth.
The lack of information on detection and/or isolation of Oceanospirillales, Marinicellales and Acidimicrobiales in the growth of the host sea cucumber resulted the delays in discovering details in the structure-function relationships. However, Halomonas meridiana and Marinomonas pontica (Oceanospirillales) have been isolated from sea sediment of the host ’s habitat21 and the guts of A. japonicus35. In particular, the Halomonas strain showed polysaccharide degrading abilities (starch and carboxymethyl cellulose sodium salt)21, which expects the roles in supporting roles in digestion of ingested detritus by the host animals. Marinicellales reads were detected from hind gut of Apostichopus japonicus using 16S rRNA gene sequencing analysis19 and Acidimicrobiales strains were isolated from the abdominal epidermis of a sea cucumber Holothuria edulis51. Both groups of bacteria actually might have associations with the sea cucumber, further ecophysiological and ecogenomics studies could infer the host effects on the unique host animals.
We found a strong positive correlation between OTUs affiliated to Legionellaceae and Alteromonadales OM60 to the body weight of A. japonicus juveniles. Legionella spp. are related to PHB production52 and Alteromonadales OM60 (such as Congregibacter spp.53) is related to polyphosphate (polyP) production. Recently, in mammalian cells, cPHB has been found in a wide variety of tissues and in atherosclerotic plaques and polyP has been linked to a variety of functions, including blood coagulation, cell proliferation, apoptosis and mitochondrial ion transport and energy metabolism54. Those bacterial polymers might trigger unique biological functions in host Deuterostomia invertebrates. Once the specific functions are unveiled, these bacteria could also be candidates for probiotics in sea cucumber aquaculture.
In conclusion, we firstly assessed possible links between gut microbiome and host growth in cultured A. japonicus juveniles. We suggest that Rhodobacterales bacteria retaining PHB metabolism genes might contribute to the production of larger individuals. Our data also revealed a healthy microbiome in larger individuals composed of SRB, commonly observed in human healthy adults, in balancing the redox state of gut environments. These results imply a link between microbial PHB producers and potential growth promotion in marine invertebrates. We need further studies using gut microbiota transplant from large to small sea cucumber gnotobiotic individuals to assess the direct effects of gut microbes on sea cucumbers’ growth.
Methods
Sea cucumbers sampling
Cultured sea cucumber A. japonicus was used for this study. The animals were fertilized and grown in Hokkaido Fisheries Station, Muroran, Japan. The juveniles were fed with cultured diatoms and an artificial diet (Norwegian Ascophyllum nodsum powder) for 18 months. These animals were then moved to a farm in Kumaishi, Japan, in June 2014 and then were acclimated with naturally occurring diatoms for two months in a 500 ton tank and reared in sand-filtered coastal seawater pumped up from 10 m depth. Seawater temperature in the tank ranged from 8.5 °C to 13 °C.
In order to verify the sea cucumber body weight distribution, 816 individuals from one tank were weighed (Fig. 1b). The mean body weight and the median were 0.41 g and 0.14 g, respectively. We categorized two groups of these individuals from the top 20% because the smaller individuals within this criterion were too small for feces sampling. We classified large individuals as weighing >2.0 g as ‘larger sea cucumbers’ and small individuals as weighing <1.9 g as ‘smaller sea cucumbers’.
Collection of individual sea cucumber feces and seawater
For bacterial DNA preparation, we collected feces of individual sea cucumber to avoid sacrificing animals and seawater in a rearing tank under semi-aseptic conditions on site. All sampling procedures were performed inside an instant clean booth, illuminated by ultraviolet light for 15 minutes (GL-15, Panasonic, Osaka, Japan). For feces collection, we selected 10 individuals from each group (larger and smaller) (Fig. 1b,c). Sea cucumber individuals were cleaned using filter-sterilized seawater and moved individually into sterile beakers with 300 mL of filter-sterilized seawater until feces was released (~20 min). The filter-sterilized seawater was prepared as follows; natural seawater pumped 10 m depth was added in a pressure tank and filtered through a 0.22 μm Sterivex filter (Sterivex™-GV Sterile Vented Filter Unit 0.22 μm, EMD Millipore, Billerica, USA) by positive pressure using filtered (0.22 μm) high purity N2 gas. Feces were collected into a 1.5 mL tube using an adopted 5 mL tip and preserved at −80 °C until DNA extraction.
Five liters of the same seawater used in the sea cucumber rearing tank was filtered through a 0.22 μm Sterivex filter by positive pressure using filtered (0.22 μm) N2 gas. The Sterivex filter was filled with SET buffer (sucrose 20%, EDTA 50 mM, Tris-HCl 50 mM) and preserved at −80 °C until DNA extraction.
Microbial DNA extraction
Microbial DNA extraction from sea cucumber feces was performed using the NucleoSpin Soil Kit (MACHEREY-NAGEL, Düren, Germany), according to the manufacture’s protocol. Microbial DNA extraction from seawater was performed using the NucleoSpin Tissue kit (MACHEREY-NAGEL), according to the modified manufacture’s protocol. We used SDS (20%) and proteinase K (20 mg mL−1) for pre-lysis instead of buffer T1 and proteinase K. Also, we used 1 mL buffer B3 instead of 200 μL buffer B3.
16S rRNA genes pyrosequencing
The hypervariable V1-V2 region of the 16S rRNA gene was amplified by PCR with barcoded 27Fmod and 338R primers55. PCR was performed in 50 μL of 1 μL Ex Taq PCR buffer composed of 10 mM Tris–HCl (pH 8.3), 50 mM KCl and 1.5 mM MgCl2 in the presence of 250 mM dNTP, 1 U Ex Taq polymerase (Takara Bio, Ootsu, Shiga), forward and reverse primers (0.2 μM) and ~20 ng template DNA. Thermal cycling consisted of initial denaturation at 96 °C for 2 min, followed by 25 cycles of denaturation at 96 °C for 30 s, annealing at 55 °C for 45 s and extension at 72 °C for 1 min and final extension at 72 °C on a 9700 PCR system (Life Technologies Japan, Tokyo, Japan). Negative controls were treated similarly, except that no template DNA was added to the PCR reactions. PCR products of ~370 bp were visualized using electrophoresis on 2% agarose gels, while negative controls failed to produce visible PCR products and were excluded from further analysis. PCR amplicons were purified using AMPure XP magnetic purification beads (Beckman Coulter, Brea, CA, USA) and quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies Japan). Equal amounts of each PCR amplicon were mixed and then sequenced using either 454 GS FLX Titanium or 454 GS JUNIOR (Roche, Basel, Switzerland). Based on sample specific barcodes, reads were assigned to each sample followed by the removal of reads lacking both forward and reverse primer sequences. Data was further qualified by removal of reads with average quality values < 25. Filter-passed reads were filed as FASTA for downstream analyses, after trimming off both primer sequences.
Taxonomic assignment and microbial diversity analyses
Sequences of Eukarya, chloroplasts and mitochondria were removed from the dataset using Metaxa software56. Sequence analysis was performed using Quantitative Insights Into Microbial Ecology (QIIME) 1.8 software package57 as shown below; i) Sequences were clustered into OTUs (similarity 97%) using uclust algorithm58. ii) Representative sequences of each OTU were assigned taxonomically using uclust consensus taxonomy assigner with default parameters (minimum percent similarity 90%) and Greengenes reference version 13.8. iii) Sequences were aligned to Greengenes Core reference using PyNAST algorithm59 and phylogenic trees were generated using FastTree for tree based analyses. iv) Alpha diversity estimates, observed species, Faith’s phylogenic diversity [PD] and Chao1 were calculated and compared between the larger and the smaller individuals using nonparametric test based on 999 iterations using a rarefaction of 1,835 reads from each sample. v) For beta diversity, even sampling of 1,835 reads per sample was used and we performed unweighted UniFrac analysis60 and visualized in PCoA plots61. Beta diversity was compared in a pairwise fashion (larger vs smaller sea cucumbers, larger vs seawater, smaller vs seawater) using unweighted UniFrac distance matrixes, using permutational multivariate analysis of variance (PERMANOVA) with 999 permutations to determine statistical significance. vi) Taxonomic relative abundances of fecal microbial communities between larger and smaller sea cucumbers were compared using Welch’s t test with Storey’s FDR (false discovery rate) for multiple test correction. Significance was assumed to be P < 0.05 and q < 0.05. Taxonomic affiliations of 16S rRNA gene reads below genus level could not be fully achieved due to the lack of marine microbial reference sequences even in this affiliation setting using QIIME, family table was the lowest hierarchy analyzed in this study (Table S2).
OTUs relevant to body weight of sea cucumbers
To verify if specific OTUs abundance correlated to sea cucumber body weight, we performed multiple Pearson correlations and P values were adjusted using the Holm method. We searched for highly (r > 0.7) and significantly (corrected P < 0.05) correlated OTUs against the body weight of the sea cucumbers. OTUs that occurred in only one sample were removed from this analysis. OTUs present in all larger individuals, in all smaller individuals and all individuals tested were defined as ‘lager individuals core (LIC)’, ‘smaller individuals core (SIC)’ and ‘all individuals core (AIC)’, respectively.
Metagenomic sequencing
Each representative DNA samples of the largest and the smallest body weight sea cucumbers, from which sufficient volumes were available after 16S typing, were used for metagenome sequencing. Metagenomic sequencing was performed on HiSeq platform by Hokkaido System Science, Co. Ltd., Sapporo, Japan. The DNA quality and quantity were estimated using Nanodrop, Qubit Fluorometer and Agilent 2200 TapeStation System. By using TruSeq Nano DNA LT Sample Prep Kit, genomic DNA was fragment and insert DNA of 350 bp was selected and connected adapter sequences. Sixty nanograms of each DNA sample was amplified with nine cycling. Subsequently, 100 bp paired-end sequencing of two samples was performed on HiSeq platform. Using Chastity, sequences showing low fluorescence purity were removed.
Pre-processing and data annotation by MG-RAST
Sequencing data set in FASTQ format of samples (the largest and the smallest) was uploaded to MG-RAST server version 3.5. For quality control, firstly, dereplication (removing artificial replicate sequences) was performed using DRISEE (duplicate read inferred sequencing error estimation). Secondly, the removal of low quality sequences was performed by a modified DynamicTrim. After filtering, data gene calling was performed using FragGeneScan algorithm to predict protein or rRNA coding region. Clusters of proteins (similarity 90% or greater) were generated using uclust by BLAT analysis. The sequences assignments were conducted using the MG-RAST server and the following cut-off parameters: expected value less than 1 × 10−5, 60% minimum identity and 15 base pair or amino acid minimum alignment. Taxonomic annotation was performed using the National Center for Biotechnology Information (NCBI) GenBank database and functional annotation was completed using the SEED database62.
Metagenomic analysis
Annotated metagenomes were analyzed using the MG-RAST analysis tools and STAMP software v2.0.963. To test if the abundance of each functional feature of the largest and the smallest sea cucumbers were different, Fisher’s exact test with Newcombe-Wilson confidence interval calculation method and Storey’s FDR for multiple test correction method were used in STAMP software. P-values of <0.05 were considered statistically significant. To visualize metagenomic results, profile bar plots and extended error bar plots were generated at Subsystem Level 1 and 3. To plot data, results were filtered by 0.05 of q-value and 0.05 of effect size64.
To investigate the taxonomic affiliation of the significantly different functional features between the largest and the smallest sea cucumbers, we used the MG-RAST workbench tool implementing KEGG Orthology and GenBank databases to annotate with the functional features.
All sequences generated in this study are deposited in BaMBa65 under data package identification number pmeirelles.19.1 and DDBJ/GenBank/EMBL database under BioProject number PRJDB4366.
Additional Information
How to cite this article: Yamazaki, Y. et al. Individual Apostichopus japonicus fecal microbiome reveals a link with polyhydroxybutyrate producers in host growth gaps. Sci. Rep. 6, 21631; doi: 10.1038/srep21631 (2016).
References
Purcell, S., Samyn, Y. & Conand, C. Commercially important sea cucumbers of the world. FAO Species Cat. Fish. Purp. 6 (2012).
Choo, P. Population status, fisheries and trade of sea cucumbers in Asia. FAO Fish. Tech. Pap. 516, 81–188 (2008).
Lovatelli, A. et al. Advances in sea cucumber aquaculture and management. FAO Fish. Tech. Pap. 463, 1–425 (2004).
Hamel, J. F. & Mercier, A. Apostichopus japonicus. The IUCN Red List of Threatened species (2013). Available at: http://www.iucnredlist.org/details/full/180424/0. (Accessed: 26th November 2015).
Watanabe, S., Sumbing, J. G. & Hazel, M. J. Growth pattern of the tropical sea cucumber, Holothuria scabra, under captivity. JARQ 48, 457–464 (2014).
Battaglene, S. C., Evizel Seymour, J. & Ramofafia, C. Survival and growth of cultured juvenile sea cucumbers, Holothuria scabra. Aquaculture 178, 293–322 (1999).
Ramofafia, C., Foyle, T. P. & Bell, J. D. Growth of juvenile Actinopyga mauritiana (Holothuroidea) in captivity. Aquaculture 152, 119–128 (1997).
Dong, Y., Dong, S., Tian, X., Wang, F. & Zhang, M. Effects of diel temperature fluctuations on growth, oxygen consumption and proximate body composition in the sea cucumber Apostichopus japonicus Selenka. Aquaculture 255, 514–521 (2006).
Dong, Y., Dong, S. & Ji, T. Effect of different thermal regimes on growth and physiological performance of the sea cucumber Apostichopus japonicus Selenka. Aquaculture 275, 329–334 (2008).
Xia, S. et al. Effects of different seaweed diets on growth, digestibility and ammonia-nitrogen production of the sea cucumber Apostichopus japonicus (Selenka). Aquaculture 338–341, 304–308 (2012).
Shi, C. et al. Effects of diatom concentration in prepared feeds on growth and energy budget of the sea cucumber Apostichopus japonicus (Selenka). Aquac. Res. 46, 609–617 (2013).
Sun, Z. L., Gao, Q. F., Dong, S. L., Shin, P. K. S. & Wang, F. Estimates of carbon turnover rates in the sea cucumber Apostichopus japonicus (Selenka) using stable isotope analysis: The role of metabolism and growth. Mar. Ecol. Prog. Ser. 457, 101–112 (2012).
Gao, Q. F., Wang, Y., Dong, S., Sun, Z. & Wang, F. Absorption of different food sources by sea cucumber Apostichopus japonicus (Selenka) (Echinodermata: Holothuroidea): Evidence from carbon stable isotope. Aquaculture 319, 272–276 (2011).
Zhao, Y. et al. Effects of potential probiotic Bacillus subtilis T13 on growth, immunity and disease resistance against Vibrio splendidus infection in juvenile sea cucumber Apostichopus japonicus. Fish Shellfish Immunol. 32, 750–755 (2012).
Yang, Z. P., Sun, J. M., Xu, Z., Zhang, C. C. & Zhou, Q. Beneficial effects of Metschnikowia sp. C14 on growth and intestinal digestive enzymes of juvenile sea cucumber Apostichopus japonicus. Anim. Feed Sci. Technol. 197, 142–147 (2014).
Dong, S. et al. Intra-specific effects of sea cucumber (Apostichopus japonicus) with reference to stocking density and body size. Aquac. Res. 41, 1170–1178 (2010).
Ortiz-Pineda, P. A. et al. Gene expression profiling of intestinal regeneration in the sea cucumber. BMC Genomics 10, 262 (2009).
Mashanov, V. S. & García-Arrarás, J. E. Gut regeneration in holothurians: A snapshot of recent developments. Biol. Bull. 221, 93–109 (2011).
Gao, F., Li, F., Tan, J., Yan, J. & Sun, H. Bacterial community composition in the gut content and ambient sediment of sea cucumber Apostichopus japonicus revealed by 16S rRNA gene pyrosequencing. PLoS One 9, 1–10 (2014).
Yang, G., Xu, Z., Tian, X., Dong, S. & Peng, M. Intestinal microbiota and immune related genes in sea cucumber (Apostichopus japonicus) response to dietary β-glucan supplementation. Biochem. Biophys. Res. Commun. 458, 98–103 (2015).
Zhang, X. et al. Physiological characterization of aerobic culturable bacteria in the intestine of the sea cucumber Apostichopus japonicus. 10 (2013).
Russell, J. B. & Rychlik, J. L. Factors that alter rumen microbial ecology. Science 292, 1119–1122 (2001).
Warnecke, F. et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450, 560–565 (2007).
Semova, I. et al. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12, 277–288 (2012).
Ley, R. E. et al. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 102, 11070–11075 (2005).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013).
Collins, S. M. & Bercik, P. The relationship between intestinal microbiota and the central nervous system in normal gastrointestinal function and disease. Gastroenterology 136, 2003–2014 (2009).
Dunn, C. W. et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452, 745–749 (2008).
Plotieau, T., Lavitra, T., Gillan, D. C. & Eeckhaut, I. Bacterial diversity of the sediments transiting through the gut of Holothuria scabra (Holothuroidea; Echinodermata). Mar. Biol. 160, 3087–3101 (2013).
Hacquard, S. et al. Microbiota and host nutrition across plant and animal kingdoms. Cell Host Microbe 17, 603–616 (2015).
Benson, A. K. et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 107, 18933–18938 (2010).
Rawls, J. F., Mahowald, M. A., Ley, R. E. & Gordon, J. I. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127, 423–433 (2006).
Enomoto, M., Nakagawa, S. & Sawabe, T. Microbial communities associated with holothurians: presence of unique bacteria in the coelomic fluid. Microbes Environ. 27, 300–305 (2012).
Pujalte, M. J., Lucena, T., Ruvira, M. A., Arahal, D. R. & CarmenMacian, M. Rhodobacteraceae In The Prokaryotes 4th edn 545–577 (2013).
Cho, J. C. & Giovannoni, S. J. Oceanicola granulosus gen. nov., sp. nov. and Oceanicola batsensis sp. nov., poly-β-hydroxybutyrate-producing marine bacteria in the order ‘Rhodobacterales’. Int. J. Syst. Evol. Microbiol. 54, 1129–1136 (2004).
Zheng, Q. et al. Paracoccus beibuensis sp. nov., Isolated from the South China Sea. Curr. Microbiol. 62, 710–714 (2011).
Madison, L. L. & Huisman, G. W. Metabolic engineering of poly (3-Hydroxyalkanoates): from DNA to plastic. Microbiol. Mol. Biol. Rev. 63, 21–53 (1999).
Jendrossek, D. & Pfeiffer, D. New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly(3-hydroxybutyrate). Environ. Microbiol. 16, 2357–2373 (2014).
Elustondo, P., Zakharian, E. & Pavlov, E. Identification of the polyhydroxybutyrate granules in mammalian cultured cells. Chem. Biodivers. 9, 2597–2604 (2012).
De Schryver, P. et al. Poly-β-hydroxybutyrate (PHB) increases growth performance and intestinal bacterial range-weighted richness in juvenile European sea bass, Dicentrarchus labrax. Appl. Microbiol. Biotechnol. 86, 1535–1541 (2010).
Najdegerami, E. H. et al. Effects of poly-β-hydroxybutyrate (PHB) on Siberian sturgeon (Acipenser baerii) fingerlings performance and its gastrointestinal tract microbial community. FEMS Microbiol. Ecol. 79, 25–33 (2012).
Nhan, D. T. et al. The effect of poly-β-hydroxybutyrate on larviculture of the giant freshwater prawn Macrobrachium rosenbergii. Aquaculture 302, 76–81 (2010).
Baruah, K. et al. Probing the protective mechanism of poly-β-hydroxybutyrate against vibriosis by using gnotobiotic Artemia franciscana and Vibrio campbellii as host-pathogen model. Sci. Rep. 5, 9427 (2015).
Sudesh, K., Tay, B. & Lee, C. Occurrence of poly (hydroxyalkanoate) in the gut homogenate of a phylogenetically higher termite: Macrotermes carbonarius. Can. J. Chem. 86, 512–515 (2008).
Tay, B. Y., Lokesh, B. E., Lee, C. Y. & Sudesh, K. Polyhydroxyalkanoate (PHA) accumulating bacteria from the gut of higher termite Macrotermes carbonarius (Blattodea: Termitidae). World J. Microbiol. Biotechnol. 26, 1015–1024 (2010).
Kuever, J., Rainey, A. F. & Widdel, F. Desulfobacterales in Bergey’s Manual of systematic bacteriology (ed. George, G. ) 959–1005 (2005).
Kuever, J. Desulfobulbaceae in The Prokaryotes 75–86 (2014).
Rey, F. E. et al. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. USA 110, 13582–7 (2013).
Kurahashi, M., Fukunaga, Y., Sakiyama, Y., Harayama, S. & Yokota, A. Iamia majanohamensis gen. nov., sp. nov., an actinobacterium isolated from sea cucumber Holothuria edulis and proposal of Iamiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 59, 869–73 (2009).
James, B. W., Mauchline, W. S., Dennis, P. J., Keevil, C. W. & Wait, R. Poly-3-hydroxybutyrate in Legionella pneumophila, an energy source for survival in low-nutrient environments. Appl. Environ. Microbiol. 65, 822–827 (1999).
Spring, S., Lünsdorf, H., Fuchs, B. M. & Tindall, B. J. The photosynthetic apparatus and its regulation in the aerobic gammaproteobacterium Congregibacter litoralis gen. nov., sp. nov. PLoS One 4 (2009).
Dedkova, E. N. & Blatter, L. A. Role of β-hydroxybutyrate, its polymer poly-β-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Front. Physiol. 5, 1–22 (2014).
Kim, S. W. et al. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 20, 241–253 (2013).
Bengtsson, J. et al. Metaxa: A software tool for automated detection and discrimination among ribosomal small subunit (12S/16S/18S) sequences of archaea, bacteria, eukaryotes, mitochondria and chloroplasts in metagenomes and environmental sequencing datasets. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol. 100, 471–475 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Caporaso, J. G. et al. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267 (2010).
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J. & Knight, R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172 (2011).
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2, 16 (2013).
Overbeek, R. et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702 (2005).
Parks, D. H., Tyson, G. W., Hugenholtz, P. & Beiko, R. G. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124 (2014).
Yadav, T. C., Pal, R. R., Shastri, S., Jadeja, N. B. & Kapley, A. Comparative metagenomics demonstrating different degradative capacity of activated biomass treating hydrocarbon contaminated wastewater. Bioresour. Technol. 188, 24–32 (2015).
Meirelles, P. M. et al. BaMBa: towards the integrated management of Brazilian marine environmental data. Database 2015 (2015).
Acknowledgements
PMM thanks CAPES for the PhD scholarships (4848-14-9). We thank The Towa Foundation for Food Science and Research (Tokyo, Japan) for supporting this project. We thank for Mr. Azumi at Kumaishi Farm. We thank E. Iioka, M. Kiuchi, R. Kurokawa, K. Komiya, N. Yamashita, C. Shindo and Y. Hattori (The University of Tokyo) for 16S sequencing.
Author information
Authors and Affiliations
Contributions
Project conceived by: Tomoo Sawabe. Experiments designed by: Y.Y. and Tomoo Sawabe. Experiments performed by: Y.Y., P.M.M., W.S., K.O. and Y.S. Data analyzed by: Y.Y., P.M.M., S.M., W.S., M.H., F.L.T. and Toko Sawabe. Paper written by: Y.Y., P.M.M. and Tomoo Sawabe. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yamazaki, Y., Meirelles, P., Mino, S. et al. Individual Apostichopus japonicus fecal microbiome reveals a link with polyhydroxybutyrate producers in host growth gaps. Sci Rep 6, 21631 (2016). https://doi.org/10.1038/srep21631
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21631
This article is cited by
-
Seasonal changes in gut microbiota of sea cucumber over natural aestivation cycle
Journal of Oceanology and Limnology (2024)
-
Unveiling the early life core microbiome of the sea cucumber Apostichopus japonicus and the unexpected abundance of the growth-promoting Sulfitobacter
Animal Microbiome (2023)
-
Recent progress in the use of purple non-sulfur bacteria as probiotics in aquaculture
World Journal of Microbiology and Biotechnology (2023)
-
The digestive tract sections of the sea cucumber Isostichopus badionotus reveal differences in composition, diversity, and functionality of the gut microbiota
Archives of Microbiology (2022)
-
Temporal changes in the gut microbiota in farmed Atlantic cod (Gadus morhua) outweigh the response to diet supplementation with macroalgae
Animal Microbiome (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
