
Abstract
Nanotechnology has introduced many manufactured carbon-based nanoparticles (CNPs) into our environment, generating a debate into their risks and benefits. Numerous nanotoxicology investigations have been carried and nanoparticle-induced toxic effects have been reported. However, there remain gaps in our knowledge, primarily regarding mechanism. Herein, we assessed the global alterations induced by CNPs in A549 lung cells using biospectroscopy techniques, including attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy and surface-enhanced Raman spectroscopy (SERS). A549 cells were treated with fullerene (C60), long or short multi-walled carbon nanotubes, or single-walled carbon nanotubes at concentrations of 0.1 mg/L, 0.01 mg/L and 0.001 mg/L. Exposed cells were then analysed by ATR-FTIR spectroscopy and SERS. Spectra were pre-processed via computational analysis and information on biochemical alterations in exposed cells were identified. Additionally, global DNA methylation levels in cells exposed to CNPs at 0.1 mg/L were determined using HPLC-MS and genetic regulators (for DNA methylation) were checked by quantitative real-time RT-PCR. It was found that CNPs exert marked effects in A549 cells and also contribute to increases in global DNA methylation. For the first time, this study highlights that real-world levels of nanoparticles can alter the methylome of exposed cells; this could have enormous implications for their regulatory assessment.
Similar content being viewed by others


Introduction
With rapid developments in nanotechnology, numerous nanomaterials are entering into the environment. Consequently, potential exposures to nanomaterials are likely to have increased dramatically in recent times. Particularly, carbon-based materials (CBMs) are currently widely used in both daily life applications and industry1,2,3,4. Thus, there is an urgent need to understand potential health hazards related to their exposure prior to increasing usage5,6.
CBMs are one of the most attractive nanomaterials due to their unique physicochemical properties depending on different forms, such as fullerenes (C60), single- or multi-walled carbon nanotubes (SWCNTs or MWCNTs) [see Electronic Supporting Information (ESI) Figure S1]. Numerous investigations have been carried out to assess these carbon nanoparticles (CNPs) and toxic effects both in vitro and in vivo have been reported7,8,9,10. Commonly, generation of reactive oxygen species (ROS) is considered a major factor involved in the toxicity of CNPs11. However, there remain knowledge gaps between nanotoxicology and real-world effects of CNPs12. Quite a few conventional toxicology assays appear limited in their applicability towards assessment of nanotoxicity (e.g., nanoparticles may interact with the dye in the MTT or LDH assays; nanoparticles might well interfere with enzymatic immunoassays); thus novel testing methodologies are required13. Such novel testing methodologies could verify existing toxicology assays whilst leveraging additional insights.
Raman or attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy techniques are vibrational spectroscopic approaches capable of deriving biochemical information from biological samples14. Vibrational spectroscopy approaches, in a reagent-free fashion, have the capability to inexpensively analyse biological samples; these methods have been used to investigate cancer and assess toxic effects of environmental pollution15,16,17,18,19,20,21. Infrared (IR) spectroscopy is the measurement of the energy absorption of the chemical bond vibrational or rotational movement occurring at a specific energy level. This generates spectra with peaks representing chemical bonds as well as the vibration/rotations of groups of atoms in a sample. The mid-IR region (1800–900 cm−1), known as the “biochemical fingerprint” region, is where the majority IR absorption occurs for biochemical structures. As a scattering technique, Raman spectroscopy provides a complementary spectrochemical approach. Surface-enhanced Raman spectroscopy (SERS) is a variation of the technique that exhibits high detection sensitivity in generating fingerprint spectra22,23. Its detection sensitivity relies on strong electromagnetic enhancement, typically produced by Ag or Au nanoparticles (NPs); Raman signals of molecules close to these NPs can be enhanced by many orders of magnitude24,25. This makes SERS an ultrasensitive tool, down to the single molecule level26; however, there are limitations associate with this approach relating to a lack of reproducibility and/or consistency in the level of enhancement.
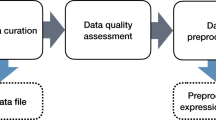
Our objective herein was to perform an assessment of four CNPs (C60, long or short MWCNTs, single-walled CNTs) to induce cellular alterations using biospectroscopic techniques (Fig. 1). Due to their size, such CNPs can readily become airborne, inhaled and induce pulmonary injury. Hence, the human epithelial-like lung A549 cell line was employed. For in vitro or in vivo toxicological tests, dose is a key parameter to understanding real-world effects. Although acute effects are more likely to be observed following high-dose exposures, recording the consequences of low-dose levels over a lifetime is necessary to understand possible risks of CNPs. Thus, cells were exposed to CNPs at ppb levels at a dose range of 0.1 mg/L, 0.01 mg/L and 0.001 mg/L. Following exposure, ATR-FTIR spectroscopy and SERS were employed. Also, global DNA methylation levels in cells exposed to 0.1 mg/L CNPs were determined by HPLC-MS; disruption of methylation in cells is linked to diseases such as cancer27,28,29. This study aims to assess the ability of CNPs to induce effects at ppb levels.

Experimental overview of application of biospectroscopy techniques towards nanotoxicology testing.
(A) General schematic for brief introduction of nanotoxicity and the basic principle of attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy and surface-enhanced Raman spectroscopy (SERS) for this experimental approach (B) Raw spectra in the “biochemical fingerprint region” from A549 cells following ATR-FTIR spectroscopy; and (C) Raw spectra of A549 cells following SERs.
Results
ATR-FTIR spectral dataset
The spectra derived following ATR-FTIR spectroscopy were inputted into principal component analysis (PCA) following pre-processing. PCA scores plots allow one to visualise clustering among classes (see ESI Figure S2). This allows observation of a dose-response in one-dimensional (1-D) scores plots in PC1 space (Fig. 2); scores in PC1 space contribute most variance in the spectral data. To determine whether the treatment groups were significantly different from the corresponding control group, repeated-measures one-way analysis of variance (ANOVA) with Dunnett’s post-hoc tests were conducted to examine the treated vs. control cells in PC1 space (see ESI Table S4). It shows that in all treatment groups, CNP-induced effects observed differ significantly (P < 0.05) from the control group.

One-dimensional (1-D) PCA scores plots in PC1 space of attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectral dataset derived from A549 cells exposed to carbon nanoparticles at each concentration vs. vehicle control.
Their corresponding loadings plots are on the right. Panels represent: (A, B) C60 fullerene; (C, D) long MWCNTs; (E, F) short MWCNTs; and, (G, H) SWCNTs.
Loadings plots derived from the PC1 space identify the distinguishing wavenumbers corresponding to the most important variances for each CNP treatment. The eight wavenumbers contributing most in each loadings plot are marked. For each CNP exposure, the distinguishing wavenumbers ranked as follows: 1624, 1658, 1589, 1547,1493, 1709, 1396 and 1103 cm−1 in C60 treatment group; 1539, 1501, 1624, 1585, 1662, 1466, 1732 and 1065 cm−1 in long MWCNTs group; 1620, 1547, 1501, 1655, 1585, 1462, 1717 and 1400 cm−1 in short MWCNTs group; and, 1508, 1466, 1678, 1732, 1061, 1582, 1018 and 1115 cm−1 in SWCNTs group, respectively. These wavenumbers contribute most to segregation in PC1 space (for tentative assignments, see ESI Table S1).
We then set out to compare cellular responses when cells were exposed to different CNPs at 0.1 mg/L, i.e., the highest exposure examined in this study. Following PCA, linear discriminant analysis (LDA) was applied; this supervised analysis takes category information into account. Scores in the first three LD spaces were used for visualisation (Fig. 3A,B). Separation between groups was significant. Moreover, cluster vectors plots derived from PCA-LDA highlighted biochemical alterations in cells induced by the different CNPs (see ESI Figure S4). To simplify the identification of the most pronounced wavenumbers responsible for alterations, a peak detector was employed and a cluster vectors peak plot was generated (Fig. 4A; tentative assignments for the first seven wavenumbers are listed in ESI Table S3).

Scores plots by principal component analysis-linear discriminant analysis (PCA-LDA) derived from spectral dataset of the A549 cells exposed to CNPs at 0.1 mg/L vs. vehicle control (90% confidence ellipsoids).
(A) Three-dimensional (3-D); and (B) 2-D PCA-LDA scores plots from the attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectral dataset. (C) 3-D; and (D) 2-D PCA-LDA scores plots from the SERS spectral dataset. C60, C60 fullerene; Long, long MWCNTs; Short, short MWCNTs; and, Single, SWCNTs.

Cluster vectors peak plots following principal component analysis-linear discriminant analysis (PCA-LDA) indicating the wavenumber basis for segregation corresponding to A549 cells exposed to CNPs at 0.1 mg/L.
Each cell population exposed to a different CNP was compared with the vehicle control. The size of the symbol in cluster vectors peak plot is proportional to the height of the corresponding peaks in the cluster vectors plots, which are relative to the extent of biochemical alteration vs. vehicle control. (A) Cluster vectors peak plot derived from attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectral dataset; and (B) Cluster vectors peak plot derived from SERS spectral dataset. C60, C60 fullerene; Long, long MWCNTs; Short, short MWCNTs; and, Single, SWCNTs.
SERS spectral dataset
Following a similar workflow, SERS spectral data was pre-processed prior to further analysis. The PC1 scores plots show significant separation between treatment groups vs. control (Fig. 5; statistical results in ESI Table S4). Similarly, the top eight distinguishing wavenumbers in the loadings plots were: 1454, 1006, 930, 1364, 1621, 1423, 858 and 1487 cm−1 for C60 exposure; 1055, 518, 1586, 463, 761, 1522, 625 and 1335 cm−1 for long MWCNTs exposure; 1006, 1456, 1341, 1372, 765, 946, 1718 and 1623 cm−1 for short MWCNTs exposure; and, 1623, 442, 1179, 802, 938, 1030, 1417 and 728 cm−1 for SWCNTs exposure (tentative assignments are listed in ESI Table S2).

One-dimensional (1-D) principal component analysis (PCA) scores plots in PC1 space of SERS spectral dataset derived from A549 cells exposed to carbon nanoparticles at each concentration vs. vehicle control.
Their corresponding loadings plots are on the right. Panels represent: (A, B) C60 fullerene; (C, D) long MWCNTs; (E, F) short MWCNTs; and, (G, H) SWCNTs.
PCA-LDA can give rise to visualisation in either 2-D or 3-D scores plots (Fig. 3C,D). Significant segregation was observed between CNPs treatment groups vs. control. Furthermore, a cluster vectors peak plot displayed the main biochemical alterations induced by different CNPs (Fig. 4B; tentative assignments of the first seven wavenumbers are listed in ESI Table S3).
Global DNA methylation levels determined by HPLC-MS
Global methylation levels in A549 cells were observed to increase following CNPs (0.1 mg/L) exposure. Mean levels of global genomic methylation in the control group was 0.88%, while mean values increased to 0.9967%, 0.9867%, 1.003% and 0.93% following exposure to C60, long or short MWCNTs and SWCNTs, respectively (Fig. 6). Global DNA methylation level was significantly elevated by C60 (P < 0.05) or short MWCNTs (P < 0.05) treatment (see ESI Table S5).

Global DNA methylation levels in A549 cells exposed to CNPs at 0.1 mg/L or without treatment, i.e., control.
Values are expressed as mean ± SD (n = 3). C60, C60 fullerene; Long, long MWCNTs; Short, short MWCNTs; and, Single, SWCNTs.
Quantitative real-time RT-PCR
Quantitative analysis was used to assess gene expression levels of DNMTs in A549 cells exposed to CNPs (0.1 mg/L). Post-exposure, DNMT1 expression levels were determined as 1.001- (C60), 0.8905- (long MWCNTs), 0.9297- (short MWCNTs) and 0.9284-fold (single-walled CNTs) of the control group. Similarly, DNMT3a expression was determined as 1.036- (C60), 0.7928- (long MWCNTs), 0.9018- (short MWCNTs) and 0.9606-fold (single-walled CNTs) of the control group, whilst DNMT3b levels decreased to 0.9849- (C60), 0.8198- (long MWCNTs), 0.8171- (short MWCNTs) and 0.7553-fold (single-walled CNTs) of the control group (Fig. 7). Generally, a down-regulating tendency in DNMT transcription in A549 cells following CNPs exposure was observed except for C60. However, significance (P < 0.05) was only observed in the down-regulation of DNMT3b expression following SWCNTs treatment (see ESI Table S6).

Quantitative real-time RT-PCR analysis of gene expression in A549 cells exposed to CNPs at 0.1 mg/L - candidate genes were: (A) DNMT1 (B) DNMT3a; and, (C) DNMT3b.
Total RNA was isolated, reverse transcribed and amplified with the specific primers. Relative quantification of each gene expression level was normalized according to GAPDH expression. The data of exposure groups were calibrated to the control values (control = 1); values are expressed as mean ± SD (n = 3). C60, C60 fullerene; Long, long MWCNTs; Short, short MWCNTs; and, Single, SWCNTs.
Discussion
When A549 cells are exposed to CNPs, alterations in IR spectra indicate that the dose-response is non-linear except in cells treated with long MWCNTs (Fig. 2). Similarly, spectral datasets from SERS indicate non-linear responses in cells following such exposures (Fig. 5). These non-linear responses are often associated with low-dose effects, which is significant in eco-toxicological assessment as it is more typical of environmental contamination. Biospectroscopy exhibits an ability to detect non-linear responses or low-dose effects in cells17,18.
However, more pronounced segregation between exposure groups vs. control is observed in the SERS-derived scores plot compared to IR (Fig. 5). The IR spectrum is a measurement of absorption after an IR beam is transmitted into the cells. However, the dominant contributor to most SERS processes is the electromagnetic enhancement mechanism; the maximum SERS enhancing region decreases dramatically with distance (r−10 for spheres) and the largest enhancement is found only a few nanometre from the Au nanoparticle surface24. This suggests that most of the signal collected employing SERS is derived from entities in close proximity to the surface of the Au nanoparticle, likely the cell membrane. Therefore, SERS is likely to only generate enhanced localised information of cellular components in an information rich manner. However, it is unknown how reproducible or consistent such enhancement of the Raman signal is. Consequently, ATR-FTIR and Raman spectroscopy techniques are mutually complementary.
In order to determine how CNPs alter cells, loadings plots post-PCA of the spectral dataset were employed to highlight the important wavenumbers related to alterations in exposed cells vs. control. Both loadings plots derived from the PC1 (Figs 2 and 5) and PC2 (see ESI) space were used, since loadings in these two PC factors explain most of the variance contributing to segregation among groups in 2-D scores plots (see ESI). In each loadings plot, the top eight distinguishing peaks were highlighted as corresponding to the main biomarkers for the dose-response in CNP-treated cells (all wavenumbers and their tentative assignments are listed in ESI). The loadings plots for both IR and SERS spectra suggest that the four different CNPs shared a similar mode of action in altering cells. Datasets derived from IR spectra highlight significant alterations induced by CNPs in Amide I (~1650 cm−1), Amide II (~1500 cm−1), lipid (~1750 cm−1) and protein (~1400 cm−1), while small changes in the region of DNA/RNA (vasPO2−, ~1225 cm−1; vsPO2−, ~1080 cm−1) are observed as well (Fig. 2), suggesting that CNPs may have some genotoxicity. Although signals collected following SERS only generate partial information in the cells, significant alterations are observed in lipid and Amide I regions [v(C = C), ~1640 cm−1; CH2 bending, ~1440 cm−1], phenylalanine (~1001 cm−1) and DNA (~720 cm−1) (Fig. 3). All spectral profiles indicate that alterations induced by CNPs are mostly located in the outer region of cells21,30. It is believed that generation of ROS and subsequent oxidative stress (OS) is the predominant mechanism of nanotoxicity31,32. Thus, CNPs could directly or indirectly cause genomic damage inside the cell with no necessity for them reaching the nucleus33,34.
To compare the effects induced by different CNPs, a parallel experiment was carried out in which A549 cells were exposed to a concentration of 0.1 mg/L. Cells were then collected for biospectroscopic analysis, as well as further global DNA methylation assessment. Following application of PCA-LDA, both 3-D and 2-D scores plots from IR and SERS spectra were derived (Fig. 3). IR spectra indicate that long MWCNTs induce the most pronounced alterations in A549 cells, while C60 seem to be less harmful; the C60-treated group exhibit a relatively high overlap with the control (Fig. 3A,B). Moreover, the 2-D scores plot derived from IR spectra also shows that short MWCNTs and SWCNTs exert marked effects to a similar extent, but possibly through different underlying mechanisms due to significant segregation between these categories (Fig. 5B). This result is consistent with our previous investigations showing that larger-sized CNPs may induce more pronounced alterations35,36. In contrast to IR spectra, SERS spectra indicate that short MWCNTs induce the most marked responses in cells, followed by SWCNTs and then long MWCNTs, with large overlap between the C60-exposed group and the control, again suggesting that C60 NPs have the least effect (Fig. 3D). Both spectral datasets suggest that C60 is the least toxic of the four CNPs37. In general, cluster vectors derived from IR spectra show that most alterations are located in the lipid, Amide I and protein regions, while the DNA/RNA region is slightly affected. Both IR and SERS spectra highlighted that the most pronounced biochemical alteration in cells after C60 exposure is in the lipid region, which could be a result of its spherical shape and its relatively lipophilic properties. Moreover, SERS detected marked alterations in the DNA/RNA region (intensity ~760 cm−1) induced by the three types of CNTs.
Beyond the global biochemical information generated by biospectroscopy, global DNA methylation levels in cells treated with CNPs at 0.1 mg/L were measured using HPLC-MS38. These results show that global DNA methylation levels were significantly elevated by C60 and short MWCNTs treatment, with marked increases also observed following long MWCNTs or SWCNTs exposure (Fig. 6; see ESI Figure S8). Additionally, utilization of quantitative real-time RT-PCR determined how CNPs impact on DNA intracellular methylation processing. As target genes, DNMTs are positive regulators, which can mediate DNA methylation by catalysing the transfer of a methyl group to DNA. DNMT1 mainly contributes to maintaining the pre-existing methylation pattern during replication, while DNMT3a and DNMT3b are mostly involved in de novo methylation39, which is considered to be implicated in cell growth and differentiation and also in abnormal methylation in tumorigenesis40. Generally, a down-regulating tendency in DNMTs transcription in A549 cells after CNPs exposure is observed. Only the transcription level of DNMT3b was decreased significantly by SWCNTs, in contrast to the observation of increased global DNA methylation levels.
Conclusion
Herein, biospectroscopy, both ATR-FTIR spectroscopy and SERS, is presented as a novel approach for nanotoxicity assessment, which also provides biochemical information underlying CNPs-induced cellular alterations. Both IR and SERS spectra show that C60, long or short MWCNTs and SWCNTs exert effects in A549 cells and induce alterations in lipid, protein and even genomic regions. Additionally, it was determined that CNPs at 0.1 mg/L reduce DNMT expression, but contribute to global genomic hypermethylation; to our knowledge, this is the first demonstration of a CNP-induced epigenomic effect29,41. Further study is required to determine real-world effects of CNPs for risk assessment.
Methods
Chemicals and carbon nanoparticles
All CNPs were purchased from Sigma. Short MWCNTs were >90% pure being 10–15 nm in diameter and 0.1–10 μm in length. Long MWCNTs were >90% pure and were 110–170 nm in diameter and 5–9 μm in length. C60 had a purity >99.5% and particle size of 1 nm. SWCNTs were described as CarboLex AP-grade (the purity of AP-grade products ranges from 50% to 70% by volume); major impurities are carbon nanospheres and carbon-encapsulated catalyst nanoparticles - the diameter was 1.2–1.5 nm. All CNPs were analysed by Raman spectroscopy (Renishaw PLC, Gloucestershire, UK) with a 785 nm laser and determined to be of high purity. Additionally, images of CNPs were taken using a scanning electron microscope (SEM) [JSM 5600 (JEOL)] (see ESI Figure S1). Bovine serum albumin (BSA) obtained from Sigma was ≥98%. CNPs were dispersed in 1% BSA solution with a 15-min ultrasonication bathed in ice water and stock solutions were made at concentrations of 100 mg/L. CNT solutions were stable and well-dispersed, while C60 appeared to agglomerate.
Gold nanoparticles (Au NPs) for SERS were synthesized using trisodium citrate as the reductants (see ESI Figure S5)42,43; these were prepared and characterised by Dr Li Cui (at Institute of Urban Environment, Xiamen, China).
Cell culture and CNPs treatment
Human lung epithelial A549 cells were routinely maintained in RPMI 1640 medium at pH 7.2, supplemented with 10% inactivated foetal bovine serum and 1% penicillin/streptomycin and grown in humidified atmosphere supplied with 5% CO2 in air at 37 °C. A549 cells were cultured in 60 mm dishes prior to incorporation into experiments. Following this, cells were disaggregated with trypsin (0.05%)/EDTA (0.02%) solution and were immediately re-suspended in complete medium and seeded in 30 mm dishes. Then cells were grown for 24-h to attach and followed by a further 24-h treatment or without test agents. The four CNPs were introduced into the treatment at concentrations of 0.1 mg/L, 0.01 mg/L or 0.001 mg/L. Each control and each exposure group was conducted in triplicate.
Post-exposure, A549 cells were washed three times with cold PBS, scraped and centrifuged at 1000 g for 3 min. The resulting cell pellets were fixed with 4% formalin in PBS for 30 min. The fixed cells were washed using distilled water and then added to Low-E glass slides (Kevley Technologies, Chesterland, OH, USA), air-dried and stored in desiccator prior to ATR-FTIR spectroscopy. For SERS, the fixed and washed cells were mixed with Au NPs. After vortexing, an aliquot of 10 μL of the mixture were dropped onto a glass slides for SERS measurement.
Spectrochemical analysis
All A549 cell samples on Low-E slides were interrogated using a Bruker TENSOR 27 FTIR spectrometer (Bruker Optics Ltd., Coventry, UK) equipped with a Helios ATR attachment containing a diamond internal reflection element (IRE). Instrument parameters were set at 32 scans and 8 cm−1 resolution. For each slide, 10 IR spectra were acquired at different points across the sample. Prior to starting a new slide, the ATR crystal was cleaned with deionized water and a background was then taken.
SERS spectra were acquired by using a LabRAM Aramis (HORIBA JobinYvon) confocal micro-Raman system equipped with a 1200 g/mm grating, He-Ne 632.8 nm laser (laser power ≤70 mW prior to lens). The system calibration was carried out using a silicon calibration source for wavenumber shift. A 50× objective (Olympus) with a numerical aperture of 0.55 was used to focus the laser beam and collect Raman signal with a working distance of about 8 mm. In order to reduce any laser damage to cells, DuoScan in the micromapping mode with a scanning area of 30 μm × 30 μm was applied with an acquisition time of 5 sec. For each sample, 5 spectra were acquired.
Computational analysis of spectral data
Spectral data processing, acquired from both ATR-FTIR and Raman spectroscopy, were performed using IRootLab toolbox ( http://irootlab.googlecode.com) running on MATLAB r2010a (The MathWorks, Inc., US)44. IR spectra were pre-processed as followings: cut to 1800–900 cm−1 (the biochemical fingerprint range), 2nd differentiation and vector normalisation. SERS spectra were initially processed in LabSpec 6 (software provided with the Raman system) for cosmic ray removal and background correction (see ESI Figure S6). The spectral data were then inputted into MATLAB and pre-processed following wavelet de-noising, cut to 400–1800 cm−1, 2nd differentiation and vector normalisation45. The differentiation is employed both as a means of baseline correction and to resolve overlapped bands45. The wavelet de-noising utilizes non-linear filtering implemented through multi-scale decomposition and thresholding for de-noising, especially for the spectra containing sharp peaks. Computational analysis using multivariate techniques including PCA and LDA can efficiently analyse such large spectral datasets46,47. The main difference between PCA and LDA is that the former is an unsupervised method, whilst the latter is supervised. PCA looks for projections to maximize variance and LDA looks for projections that maximize the ratio of between-category to within-category scatter48. Following pre-processing, PCA was applied to the dataset. PCA reduces the dimensions of the data. Undoubtedly PCA is capable of identifying important information in spectral data, but has less discriminatory power. Often, in order to interpret complex biochemical information with labelled classes, further analysis using supervised procedures like LDA is required. The output data derived from PCA or PCA-LDA (the first ten PC factors of PCA are used for LDA since >99% of variance is captured) can then be visualized as 1-D, 2-D or 3-D scatterplots (“scores plots”). In scores plots, nearness between two categories implies similarity, while distance indicates dissimilarity. To reveal the biochemical alterations associated with each category in the dataset, both loadings plots and cluster vectors16 were developed. To simplify the identification of the main biochemical alterations distinguishing each category, peak detector was used to indicate the first few highest peaks in the loadings plots or cluster vectors plots.
Global DNA methylation determined by HPLC-MS
A549 cells exposed to four CNPs at 0.1 mg/L were collected and stored in PBS at −20 °C prior to quantitative real-time RT-PCR or DNA methylation analysis. DNA was extracted from A549 cells using the DNeasy Cell Kit (Qiagen, Germany) following the manufacturer’s instructions. RNase A was added to columns in the kit to remove RNA residue. DNA hydrolysis was conducted using a mixture degradase kit (DNA Degradase Plus, Zymo Research, USA) following the manufacturer’s protocol. To confirm complete hydrolysis of DNA, agarose gel electrophoresis was employed to test the result of the hydrolysis (see ESI Figure S7). The DNA hydrolysis mixtures were then stored at −20 °C for mass spectrometric analysis.
To perform chromatographic separation, a Kinetex C18 column (100 mm × 4.6 mm, 2.6 μm; Phenomenex, USA) was employed in a HPLC system (LC-20AD, Shimadzu, Japan). The injection volume was 20 μL. The mobile phase consisted of water (A) and methanol (B). A gradient elution protocol was as follows: 0–0.01 min, 3% B; 0.01–5.00 min, 5% B; 3.00–12.00 min, 50% B; 12.00–15.00 min, 100% B; 15.00–25.00 min, 3% B at a flow rate of 0.5 mL.min−1. For the mass spectrometric analysis, an electrospray ionization tandem mass spectrometry (LCMS-8030, Shimadzu, Japan) system was used, operating in positive ionization mode and conditioned at a capillary temperature of 400 °C and medium N2 curtain gas. Optimized multiple reaction monitoring (MRM) conditions were set to evaluate dC from m/z 228.1 to 111.9 and 5 mdC from m/z 242.1 to 126.0. Data acquisition and processing were performed via Analyst software. The global DNA methylation ratio (MR) was determined by MR = [5 − mdC]/([5 − mdC] + [dC]).
Quantitative real-time RT-PCR
Total RNAs were extracted from cells using the RNeasy@ Mini Kit (Qiagen, Venlo, Netherlands). Subsequently, reverse transcription of cDNA synthesis was performed with 1 μg total RNA using PrimeScript@ RT reagent kit with gDNA Eraser cDNA synthesis kits (Takara, Otsu, Shiga, Japan). Real-time PCR was carried out in a 20 μL reaction mixture in triplicate, using SYBR Green Master Mix reagents (Roche, Basel, Switzerland) on a Roche LightCycle@ 480 ll real-time PCR system following the manufacturer’s protocol (95 °C for 10 min followed by 40 cycles at 95 °C for 15 sec and 60 °C for 30 sec). The primer sequences specific for DNMT1, DNMT3a, DNMT3b and GAPDH were designed using the primer5 software (see ESI Table S7). Gene expression levels were normalised to GAPDH expression. The relative levels of each target mRNA transcripts to the control GAPDH were analysed by 2−ΔΔCt method and expressed as fold change.
Statistical analysis
The data are expressed as the mean ± SD. Significant differences among multiple groups were determined using a one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc tests. Probabilities of P < 0.05 were considered as statistically significant. All these tests were conducted in GraphPad Prism 4 (GraphPad Software, USA).
Additional Information
How to cite this article: Li, J. et al. Low-dose carbon-based nanoparticle-induced effects in A549 lung cells determined by biospectroscopy are associated with increases in genomic methylation. Sci. Rep. 6, 20207; doi: 10.1038/srep20207 (2016).
References
Jensen, A. W., Wilson, S. R. & Schuster, D. I. Biological applications of fullerenes. Biorg. Med. Chem. 4, 767–779, doi: 10.1016/0968-0896(96)00081-8 (1996).
Baughman, R. H., Zakhidov, A. A. & de Heer, W. A. Carbon nanotubes–the route toward applications. Science 297, 787–792, doi: 10.1126/science.1060928 (2002).
Bianco, A., Kostarelos, K. & Prato, M. Applications of carbon nanotubes in drug delivery. Curr. Opin. Chem. Biol. 9, 674–679, doi: 10.1016/j.cbpa.2005.10.005 (2005).
Gooding, J. J. Nanostructuring electrodes with carbon nanotubes: A review on electrochemistry and applications for sensing. Electrochim. Acta 50, 3049–3060, doi: 10.1016/j.electacta.2004.08.052 (2005).
Nel, A., Xia, T., Mädler, L. & Li, N. Toxic potential of materials at the nanolevel. Science 311, 622–627, doi: 10.1126/science.1114397 (2006).
Oberdörster, G., Oberdörster, E. & Oberdörster, J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ. Health Perspect. 823–839 (2005).
Warheit, D. B. et al. Comparative pulmonary toxicity assessment of single-wall carbon nanotubes in rats. Toxicol. Sci. 77, 117–125, doi: 10.1093/toxsci/kfg228 (2004).
Muller, J. et al. Respiratory toxicity of multi-wall carbon nanotubes. Toxicol. Appl. Pharmacol. 207, 221–231, doi: 10.1016/j.taap.2005.01.008 (2005).
Davoren, M. et al. In vitro toxicity evaluation of single walled carbon nanotubes on human A549 lung cells. Toxicol. In Vitro 21, 438–448, doi: 10.1016/j.tiv.2006.10.007 (2007).
Smith, C. J., Shaw, B. J. & Handy, R. D. Toxicity of single walled carbon nanotubes to rainbow trout, (Oncorhynchus mykiss): respiratory toxicity, organ pathologies and other physiological effects. Aquat. Toxicol. 82, 94–109, doi: 10.1016/j.aquatox.2007.02.003 (2007).
Fu, P. P., Xia, Q., Hwang, H.-M., Ray, P. C. & Yu, H. Mechanisms of nanotoxicity: generation of reactive oxygen species. J. Food Drug Anal. 22, 64–75, doi: 10.1016/j.jfda.2014.01.005 (2014).
Kahru, A. & Dubourguier, H.-C. From ecotoxicology to nanoecotoxicology. Toxicology 269, 105–119, doi: 10.1016/j.tox.2009.08.016 (2010).
Kroll, A., Pillukat, M. H., Hahn, D. & Schnekenburger, J. Current in vitro methods in nanoparticle risk assessment: limitations and challenges. Eur. J. Pharm. Biopharm. 72, 370–377, doi: 10.1016/j.ejpb.2008.08.009 (2009).
Kelly, J. G. et al. Biospectroscopy to metabolically profile biomolecular structure: a multistage approach linking computational analysis with biomarkers. J. Proteome Res. 10, 1437–1448, doi: 10.1021/pr101067u (2011).
Llabjani, V. et al. Polybrominated diphenyl ether-associated alterations in cell biochemistry as determined by attenuated total reflection Fourier-transform infrared spectroscopy: a comparison with DNA-reactive and/or endocrine-disrupting agents. Environ. Sci. Technol. 43, 3356–3364, doi: 10.1021/es8036127 (2009).
Llabjani, V., Trevisan, J., Jones, K. C., Shore, R. F. & Martin, F. L. Binary mixture effects by PBDE congeners (47, 153, 183, or 209) and PCB congeners (126 or 153) in MCF-7 cells: biochemical alterations assessed by IR spectroscopy and multivariate analysis. Environ. Sci. Technol. 44, 3992–3998, doi: 10.1021/es100206f (2010).
Llabjani, V. et al. Differential effects in mammalian cells induced by chemical mixtures in environmental biota as profiled using infrared spectroscopy. Environ. Sci. Technol. 45, 10706–10712, doi: 10.1021/es202574b (2011).
Llabjani, V., Trevisan, J., Jones, K. C., Shore, R. F. & Martin, F. L. Derivation by infrared spectroscopy with multivariate analysis of bimodal contaminant-induced dose-response effects in MCF-7 cells. Environ. Sci. Technol. 45, 6129–6135, doi: 10.1021/es200383a (2011).
Patel, I. I. et al. High contrast images of uterine tissue derived using Raman microspectroscopy with the empty modelling approach of multivariate curve resolution-alternating least squares. Analyst 136, 4950–4959, doi: 10.1039/c1an15717e (2011).
Pang, W. et al. Identification of benzo[a]pyrene-induced cell cycle-associated alterations in MCF-7 cells using infrared spectroscopy with computational analysis. Toxicology 298, 24–29, doi: 10.1016/j.tox.2012.04.009 (2012).
Riding, M. J. et al. Concentration-dependent effects of carbon nanoparticles in gram-negative bacteria determined by infrared spectroscopy with multivariate analysis. Environ. Pollut. 163, 226–234, doi: 10.1016/j.envpol.2011.12.027 (2012).
Hering, K. et al. SERS: a versatile tool in chemical and biochemical diagnostics. Anal. Bioanal. Chem. 390, 113–124, doi: 10.1007/s00216-007-1667-3 (2008).
McNay, G., Eustace, D., Smith, W. E., Faulds, K. & Graham, D. Surface-enhanced Raman scattering (SERS) and surface-enhanced resonance Raman scattering (SERRS): a review of applications. Appl. Spectrosc. 65, 825–837, doi: 10.1366/11-06365 (2011).
Stiles, P. L., Dieringer, J. A., Shah, N. C. & Van Duyne, R. P. Surface-Enhanced Raman Spectroscopy. Annu. Rev. Anal. Chem. 1, 601–626, doi: 10.1146/annurev.anchem.1.031207.112814 (2008).
Sharma, B., Frontiera, R. R., Henry, A.-I., Ringe, E. & Van Duyne, R. P. SERS: Materials, applications and the future. Mater. Today 15, 16–25, doi: 10.1016/S1369-7021(12)70017-2 (2012).
Kneipp, K. et al. Single molecule detection using surface-enhanced Raman scattering (SERS). Phys. Rev. Lett. 78, 1667 (1997).
Ehrlich, M. DNA methylation in cancer: too much, but also too little. Oncogene 21, 5400–5413 (2002).
Esteller, M. & Herman, J. G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. 196, 1–7, doi: 10.1002/path.1024 (2002).
Das, P. M. & Singal, R. DNA Methylation and Cancer. J. Clin. Oncol. 22, 4632–4642, doi: 10.1200/jco.2004.07.151 (2004).
Riding, M. J. et al. Mechanistic insights into nanotoxicity determined by synchrotron radiation-based Fourier-transform infrared imaging and multivariate analysis. Environ. Int. 50, 56–65, doi: 10.1016/j.envint.2012.09.009 (2012).
Nel, A., Xia, T., Mädler, L. & Li, N. Toxic potential of materials at the nanolevel. Science 311, 622 (2006).
Shvedova, A. A., Pietroiusti, A., Fadeel, B. & Kagan, V. E. Mechanisms of carbon nanotube-induced toxicity: focus on oxidative stress. Toxicol. Appl. Pharmacol. 261, 121–133, doi: 10.1016/j.taap.2012.03.023 (2012).
Schuster, G. B. Long-range charge transfer in DNA: transient structural distortions control the distance dependence. Acc. Chem. Res. 33, 253–260, doi: 10.1021/ar980059z (2000).
Elsaesser, A. & Howard, C. V. Toxicology of nanoparticles. Adv. Drug Del. Rev. 64, 129–137, doi: 10.1016/j.addr.2011.09.001 (2012).
Li, J. et al. Dose-related alterations of carbon nanoparticles in mammalian cells detected using biospectroscopy: potential for real-world effects. Environ. Sci. Technol. 47, 10005–10011, doi: 10.1021/es4017848 (2013).
Li, J., Ying, G.-G., Jones, K. C. & Martin, F. L. Real-world carbon nanoparticle exposures induce brain and gonadal alterations in zebrafish (Danio rerio) as determined by biospectroscopy techniques. Analyst 140, 2687–2695, doi: 10.1039/c4an02227k (2015).
Jia, G. et al. Cytotoxicity of carbon nanomaterials: single-wall nanotube, multi-wall nanotube and fullerene. Environ. Sci. Technol. 39, 1378–1383, doi: 10.1021/es048729l (2005).
Peng, S.-Y., Zhang, J., Tian, M.-P., Wang, Z.-L. & Shen, H.-Q. Determination of global DNA methylation in biological samples by liquid chromatography-tandem mass spectrometry. Chinese J. Anal. Chem. 40, 1201–1206, doi: 10.1016/S1872-2040(11)60566-5 (2012).
Girault, I., Tozlu, S., Lidereau, R. & Bièche, I. Expression analysis of DNA methyltransferases 1, 3A and 3B in sporadic breast carcinomas. Clin. Cancer Res. 9, 4415–4422 (2003).
Tian, M. et al. Perfluorooctanoic acid induces gene promoter hypermethylation of glutathione-S-transferase Pi in human liver L02 cells. Toxicology 296, 48–55, doi: 10.1016/j.tox.2012.03.003 (2012).
Robertson, K. D. DNA methylation and human disease. Nat Rev Genet 6, 597–610, doi: 10.1038/nrg1655 (2005).
Cui, L., Yao, M., Ren, B. & Zhang, K.-S. Sensitive and versatile detection of the fouling process and fouling propensity of proteins on polyvinylidene fluoride membranes via surface-enhanced Raman spectroscopy. Anal. Chem. 83, 1709–1716, doi: 10.1021/ac102891g (2011).
Cui, L. et al. In situ study of the antibacterial activity and mechanism of action of silver nanoparticles by surface-enhanced Raman spectroscopy. Anal. Chem. 85, 5436–5443, doi: 10.1021/ac400245j (2013).
Trevisan, J., Angelov, P. P., Scott, A. D., Carmichael, P. L. & Martin, F. L. IRootLab: a free and open-source MATLAB toolbox for vibrational biospectroscopy data analysis. Bioinformatics 29, 1095–1097, doi: 10.1093/bioinformatics/btt084 (2013).
Trevisan, J., Angelov, P. P., Carmichael, P. L., Scott, A. D. & Martin, F. L. Extracting biological information with computational analysis of Fourier-transform infrared (FTIR) biospectroscopy datasets: current practices to future perspectives. Analyst 137, 3202–3215, doi: 10.1039/c2an16300d (2012).
Martin, F. L. et al. Identifying variables responsible for clustering in discriminant analysis of data from infrared microspectroscopy of a biological sample. J. Comput. Biol. 14, 1176–1184, doi: 10.1089/cmb.2007.0057 (2007).
Martin, F. L. et al. Distinguishing cell types or populations based on the computational analysis of their infrared spectra. Nat. Protoc. 5, 1748–1760, doi: 10.1038/nprot.2010.133 (2010).
Gautam, R., Vanga, S., Ariese, F. & Umapathy, S. Review of multidimensional data processing approaches for Raman and infrared spectroscopy. EPJ Techn. Instrum. 2, 1–38, doi: 10.1140/epjti/s40485-015-0018-6 (2015).
Acknowledgements
Funding from the Chinese Academy of Sciences and the Engineering and Physical Sciences Research Council (EPSRC; Grant no. EP/K023349/1) is greatly acknowledged.
Author information
Authors and Affiliations
Contributions
J.L. conducted the majority of experiments and data analysis. M.T. assisted with global DNA methylation analysis by HPLC-MS, gene expression analysis by quantitative real-time RT PCR and data analysis. L.C. assisted on SERS analysis and synthesised nanoparticles. H.S. advised on experimental design and provided laboratory support. N.J.F. conducted the electron microscopy. J.D. re-analysed samples independently in order to validate findings. F.L.M. conceived the idea for the study. J.L. wrote the initial manuscript and F.L.M. finalised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, J., Tian, M., Cui, L. et al. Low-dose carbon-based nanoparticle-induced effects in A549 lung cells determined by biospectroscopy are associated with increases in genomic methylation. Sci Rep 6, 20207 (2016). https://doi.org/10.1038/srep20207
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20207
This article is cited by
-
Individual and combined toxicity of carboxylic acid functionalized multi-walled carbon nanotubes and benzo a pyrene in lung adenocarcinoma cells
Environmental Science and Pollution Research (2019)
-
Differences in MWCNT- and SWCNT-induced DNA methylation alterations in association with the nuclear deposition
Particle and Fibre Toxicology (2018)
-
Arsenic-containing hydrocarbons: effects on gene expression, epigenetics, and biotransformation in HepG2 cells
Archives of Toxicology (2018)
-
Fine particulate matter 2.5 exerted its toxicological effect by regulating a new layer, long non-coding RNA
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
