
Abstract
Cross-dehydrogenative couplings of two different C-H bonds have emerged as an attractive goal in organic synthesis. However, achieving regioselective C-H activation is a great challenge because C-H bonds are ubiquitous in organic compounds. Actually, the regioselective couplings promoted by enzymes are a common occurrence in nature. Herein, we have developed simple, efficient and general transition metal-free intramolecular couplings of alphatic and aromatic C-H bonds. The protocol uses readily available aryl triazene as the radical initiator, cheap K2S2O8 as the oxidant and the couplings were performed well with excellent tolerance of functional groups. Interestingly, α-carbon configuration of some amino acid residues in the substrates was kept after the reactions and the couplings for substrates with substituted phenylalanine residues exhibited complete β-carbon diastereoselectivity for induction of the chiral α-carbon. Therefore, the present study should provide a novel strategy for regioselective cross-dehydrogenative couplings of two different C-H bonds.
Similar content being viewed by others
Introduction
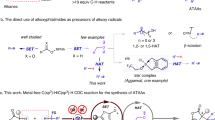
Constructing carbon-carbon and carbon-heteroatom bonds via carbon-hydrogen (C-H) activation has attracted much attention in chemical transformation and this strategy can reduce the number of steps to the target molecule and decrease cost and waste1,2,3,4,5,6,7. However, achieving regioselective C-H activation is a great challenge because C-H bonds are ubiquitous in organic molecules. The most common protocols are referred to as transition metal-catalyzed directed C-H bond functionalization, in which a directing functional group is able to coordinate to metal of the catalyst and the metal inserts a proximal C-H bond to get a thermodynamically stable five- or six-membered metallacyclic intermediate and a subsequent reaction occurs at the ortho position of the directing group (Fig. 1a)8,9,10,11. There has been great progress in the cross-coupling of two different C-H bonds in the absence of directing group12,13,14,15,16,17,18, but the reactions usually occur on adjacent C-H bonds to a heteroatom (Fig. 1b), which is away from the ideal chemical transformation19. Therefore, the cross-coupling of two different C-H bonds that are not in close proximity to a heteroatom is in undeveloped stage in the absence of ortho-site directing group. Actually, the C-H activation is a common occurrence in nature20,21,22 and the regioselective cross-dehydrogenative couplings of two different C-H bonds promoted by enzymes are often found in the biosynthesis of natural products and biologically active molecules23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38. The replication of the enzymatic processes in the laboratory using simple and readily available natural resources is the best procedure39. Therefore, it is highly desirable to develop a chemical regioselective cross-coupling of C-H bonds under the inspiration of the enzymatic reactions. Aryl triazenes that are easily prepared from the corresponding anilines are both stable and adaptable to numerous synthetic transformations40,41,42,43,44. Recently, Baran and co-workers used o-tosyl triazene chloride (TzoCl) as the ‘portable desaturase’ to install on aliphatic amines or alcohols and the desaturation of unactivated aliphatics was performed well when 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) and trifluoroacetic acid (TFA) or trifluoromethanesulfonic acid (TfOH) were added to the system45. Inspired by the previous excellent researches, we initiated our project on intramolecular cross-coupling of two different C-H bonds. As shown in Fig. 1c, our strategy is as follows: Conjugation of A with TzoCl on the amino gives B, B provides the corresponding aryl radical C under treatment of oxidant and H-abstraction of aliphatic C-H bond by the aryl radical leads to alkyl radical D since aryl radicals were confirmed to be highly reactive, very short-lived intermediates46,47. Subsequent intramolecular electrophilic reaction of alkyl radical with aryl ring in D affords the target product E.

Design on transition metal-free intramolecular coupling of unactivated aliphatic and aromatic C-H bonds.
(a) Ortho-site directed C-H activation. (b) Adjacent C-H bond activation to a heteroatom. (c) Description of the concept on our coupling of two C-H bonds.
Results and Discussion
Development of a method
At first, 2-N-Tzo-amino-N,3-dimethyl-N-phenylbutanamide (1a) was chosen as the model substrate to optimize the reaction conditions including catalysts, oxidants, additives, solvents and temperature under nitrogen atmosphere. When TEMPO was used as the oxidant, TFA or TfOH as the additive referencing to Baran’s conditions45, the desired product (2a) was observed in only 6% and 7% yields using CH3NO2 as the solvent (Table 1, entries 1 and 2). Interestingly, use of two equiv of K2S2O8 as the oxidant led to a 50% yield in the absence of additive at 100 °C (Table 1, entry 3). Other solvents, CH3CN, 1,2-dichloroethane (DCE), toluene, dioxane, DMF and DMSO, were attempted (Table 1, entries 4–9) and CH3CN gave the highest yield (53%) (Table 1, entry 4). In order to confirm whether trace amount of transition metals in the system mediate this reaction, the solvent in the resulting solution of entry 4 was removed by a rotary evaporator and the residue was determined by ICP mass spectrometry. Mn, Pd, Rh, Cu, Fe, Ni, Pt, Au, Ag, Co and Cr almost were not observed (Data determined by ICP mass spectrometry on the residue after the solvent in the resulting solution of entry 4 was removed by a rotary evaporator: Mn (0.1 ppm), Pd (<0.5 ppm), Rh (<0.5 ppm), Cu (44.1 ppm), Fe (72.1 ppm), Ni (4.1 ppm), Pt (<0.5 ppm), Au (8.3 ppm), Ag (11.6 ppm), Co (0.12 ppm) and Cr (2.11 ppm)). Structure of 2a containing L-valine residue was identified by NMR and the result showed that configuration of the chiral α-carbon in 2a was retentive and no racemization was observed. Other oxidants were tested (Table 1, entries 10–17) and they were inferior to K2S2O8 (Table 1, entry 4). Yield obviously dropped when temperature was decreased to 60 °C (Table 1, entry 18). Reducing amount of K2S2O8 to one equivalent led to a lower yield (Table 1, entry 19) and increasing its amount to three equivalents gave a similar yield to entry 4 (see Table 1, entry 20). Three common transition-metal catalysts, Pd(OAc)2, CuBr2 and AgNO3 (Table 1, entries 21–23), were added to the reaction system, respectively and the results showed that addition of transition-metal catalysts did not improve efficiency of the reaction, which exhibits that the present reaction is a transition metal-free process. Reaction in air provided a lower yield (Table 1, entry 24). Therefore, the optimal conditions for the intramolecular coupling of aliphatic and aromatic C-H bonds are as follows: two equiv of K2S2O8 as the oxidant, CH3CN as the solvent at 100 °C for 2 h under nitrogen atmosphere.
Couplings of aliphatic tertiary C-H and aromatic C-H bonds
We investigated the scope of substrates containing L- and D-amino acid residues for intramolecular couplings of aliphatic tertiary C-H and aromatic C-H bonds (Fig. 2). When R2 in the substrate was ethyl in stead of methyl in 1a, 2b was obtained in 65% yield. Various substitutes for R1 were attempted. As shown in Fig. 2a, the substrate with p-Me relative to NR2 on the aromatic ring provided higher yield than that with o-Me relative to NR2 because of steric hindrance (compare 2c and 2d in Fig. 2a). Existence of piperidine in 2e was favor for the intramolcular coupling of aliphatic tertiary C-H and aromatic C-H bonds because N-Tzo-valine and aromatic C-H bond were on the same side of tetrahydroquinoline. NMR data exhibited that α-carbon configuration in the chiral amino acid residues is kept. In order to further ascertain structures of the newly synthesized products (2), 2k was made from the substrate with D-valine residue, its single crystal was prepared (see Supporting Information for details) and X-ray diffraction analysis showed that α-carbon configuration of D-valine residue in 2k was remained (Fig. 2b). The reactions showed good tolerance of functional groups including ether (see 2f in Fig. 2a), C-F bond (see 2g in Fig. 2a), C-Cl bond (see 2h in Fig. 2a), C-Br bond (see 2i in Fig. 2a), trifluoromethyl (see 2j in Fig. 2a), ester (see 2k in Fig. 2a). We also attempted substrates containing different R3 and R4 and they afforded the reasonable yields (see 2l and 2m in Fig. 2a).

Substrate scope for couplings of aliphatic tertiary C-H and aromatic C-H bonds.
(a) Structures of the obtained products (2). (b) Crystal structure of compounds 2k. Reaction condition: under nitrogen atmosphere, 1 (0.1 mmol), K2S2O8 (0.2 mmol), CH3CN (2.0 mL), reaction temperature (100 °C), reaction time (2 h) in a sealed Schlenk tube. *Isolated yield.
Couplings of benzylic C-H and aromatic C-H bonds
Inspired by the excellent results above, we extended the substrate scope using substituted phenylalanine residues in 3 in stead of the above amino acid residues containing tertiary C-H bond in 1. As shown in Fig. 3a, the substrates with phenylalanine residue provided the reasonable yields under the standard conditions (see 4a–e in Fig. 3a) and those with electron-withdrawing groups at para-site of NMe on the aromatic ring exhibited higher reactivity (see 4d and 4e in Fig. 3a). We attempted other substrates with different substituted phenylalanine residues and they also gave good results. The substrates containing electron-withdrawing groups on aromatic ring of phenylalanine residue (see 4l–q in Fig. 3a) afforded higher yields because of higher acidity of benzylic C-H than those containing electron-donating groups (see 4g and 4h in Fig. 3a). Similarly to 2k, single crystal of 4b was prepared and its structure was identified by X-ray diffraction analysis (Fig. 3b) (see Supporting Information for details). The result showed that the couplings displayed complete β-carbon diastereoselectivity because of effect of ortho-site α-chiral carbon in the amino acid residues and they all were cis-form configuration. The couplings of benzylic C-H and aromatic C-H bonds exhibited excellent tolerance of functional groups including C-F bond (see 4i and 4q in Fig. 3a), C-Cl bond (see 4b in Fig. 3a), C-Br bond (see 4c and 4j in Fig. 3a), C-I bond (see 4k in Fig. 3a), trifluoromethyl (see 4d in Fig. 3a), esters (see 4e and 4h in Fig. 3a), ether (see 4g in Fig. 3a), nitrile (see 4l in Fig. 3a), nitro (see 4m–q in Fig. 3a).

Substrate scope for couplings of benzylic C-H and aromatic C-H bonds.
(a) Structures of the obtained products (4). (b) Crystal structure of compounds 4b. Reaction condition: under nitrogen atmosphere, 3 (0.1 mmol), K2S2O8 (0.2 mmol), CH3CN (2.0 mL), reaction temperature (100 °C), reaction time (2 h) in a sealed Schlenk tube. *Isolated yield. Structures of diastereomers were identified by 1H NMR analysis and X-ray diffraction analysis.
Couplings of α-C-H bond of carbonyl and aromatic C-H bonds
We further investigated intramolecular couplings of α-C-H bond of carbonyl and aromatic C-H bond for the substrates (5) containing aspartic acid derivatives. As shown in Fig. 4a, the examined substrates provided moderate to good yields. Unfortunately, the reactions led to racemization of α-carbon in aspartic acid residue because of unknown factors and cis- and trans-forms were observed through coupling constants between α- and β-C-H. Interestingly, cis- and trans-isomers were isolated by silicon gel column chromatography and their structures were identified by 1H NMR analysis. Single crystal of cis-isomers in 6m was prepared using similar procedures to 2k and X-ray diffraction analysis exhibited that two cis-forms were observed (Fig. 4b) (see Supporting Information for details). In addition, for substrates containing electron-withdrawing groups R1 such as Cl, Br, CF3 and ester, trans-form diastereomers were major products (see 6c–f in Fig. 4a) and that containing the bigger amide afforded major cis-form diastereomer (see 6l in Fig. 4a). The method also displayed good tolerance of functional groups including esters (see 6a–i in Fig. 4a), amides (all the substrates in Fig. 4a), C-Cl bond (see 6c in Fig. 4a), C-Br bond (see 6d in Fig. 4a) and CF3 (see 6e in Fig. 4a).

Substrate scope for couplings of α-C-H bond of carbonyl and aromatic C-H bond.
(a) Structures of the obtained products (6). (b) Crystal structure of compounds 6m. Reaction condition: under nitrogen atmosphere, 5 (0.1 mmol), K2S2O8 (0.2 mmol), CH3CN (2.0 mL), reaction temperature (100 °C), reaction time (2 h) in a sealed Schlenk tube. *Isolated yield (cis- and trans-isomers were separated by silica gel column chromatography and their structures were identified by 1H NMR analysis).
Mechanistic investigations
In order to explore mechanism on the couplings of aliphatic and aromatic C-H bonds, the following control experiments were performed. As shown in Fig. 5a, treatment of 2-N-Tzo-amino-N,3-dimethyl-N-phenylbutanamide (1a) with K2S2O8 was performed in the presence of one equiv of 2,2,6,6-tetramethylpiperidinyl-1-oxyl (TEMPO) under the standard conditions and only 9%-yielded product (2a) was obtained. The result showed that the reaction could undergo a radical intermediate process. Deuterium-labelling phenylalanine was prepared according to the previous procedure48 and intramolecular coupling of substrate 7 was carried out under the standard conditions (Fig. 5b). Pleasedly, product 8 was obtained in 47% yield, which implied transfer of deuterium from alphatic β-C-D bond to aromatic C-D bond. Therefore, a possible reaction mechanism for couplings of alphatic and aromatic C-H bonds is proposed in Fig. 5c. First, homolysis of K2S2O8 yields radical F under heating condition and treatment of substrate B with F provides radical cation H freeing anion G. Deprotonation of H by G gives radical J leaving I and subsequent desorption of N-ethylideneethanamine (K) and N2 leads to highly reactive aryl radical C. Intramolecular 1,6-H abstraction from alphatic C-H bond to aromatic C-H bond donates alphatic alkyl radical D and cyclization of D affords radical L. Treatment of L with F produces cation M leaving G and deprotonation of M in the presence of G provides the target product (E).

Mechanistic investigations.
(a) Evidence for the in situ formation of an intermediate aryl radical. (b) Deuterium-labelling study to support a H-abstraction event during coupling of alphatic and aromatic C-H bonds. (c) Proposed reaction mechanism for couplings of alphatic and aromatic C-H bonds.
Application of the methods
In order to explore affect of position for hydrogen-transfer from alphatic C-H to aromatic C-H, compounds 9 was prepared and treated under the standard conditions. Pleasedly, product 10 was obtained in 40% yield (Fig. 6a). The result showed that 1,7-H abstraction is also feasible for the coupling of alphatic and aromatic C-H bonds. We attempted oxidation of 4k and 6k to lead to quinolinones. As shown in Fig. 6b, treatment of 4k or 6k with ten equiv of activated MnO2 was performed in 1,2-dichloroethane (DCE) at 80 °C for 24 h and the corresponding quinolinones 11 or 12 was obtained in 71% and 64% yields, respectively. Therefore, the present study is effective for synthesis of quinolinone derivatives.

Application of the methods.
(a) Treatment of compound 9 under the standard conditions. (b) Oxidation of compounds 4k and 6k with activated MnO2.
Conclusion
We have developed simple, efficient and general transition metal-free intramolecular regioselective cross-dehydrogenative couplings of alphatic and aromatic C-H bonds. The protocol uses readily available aryl triazene as the radical initiator, cheap K2S2O8 as the oxidant and the couplings were performed well under mild conditions with excellent tolerance towards various functional groups. Interestingly, α-configuration of some amino acid residues in the substrates was kept after the reactions and the couplings for substrates containing substituted phenylalanine residues exhibited complete β-carbon diastereoselectivity because of effect of ortho-site α-chiral carbon. Although the reactions for substrates containing aspartic acid derivatives gave cis- and trans-form racemates, the cis- and trans-isomers could be easily separated by silica gel column chromatography. The reaction mechanism indicated that initiation of reactions began in formation of aryl radicals from treatment of aryl triazenes with K2S2O8, in which aryl triazene seems to act as a radical initiator and the reactions immediately process once K2S2O8 starts. Therefore, the present method should provide a new strategy for intramolecular regioselective cross-dehydrogenative couplings of two different C-H bonds.
Methods
To a 25 mL Schlenk tube charged with a magnetic stirrer, 1, 3 or 5 (0.1 mmol), K2S2O8 (0.2 mmol, 54 mg) and anhydrous MeCN (2.0 mL) were added. The tube was evacuated and back-filled with nitrogen for three cycles and then sealed. It was placed in a preheated oil bath at 100 °C and the reaction was allowed to proceed for 2 hours. After completion of the reaction, the resulting mixture was filtered and the filtrate was concentrated by a rotary evaporator. The residue was dissolved with EtOAc (3 mL) and the solution was washed with water (2 × 3 mL) and brine (2 × 3 mL), dried over MgSO4, filtered and concentrated by a rotary evaporation. The residue was purified with preparative TLC (silica gel, petroleum ether/EtOAc or dichloromethane/MeOH) to provide the target product (2, 4 or 6).
Additional Information
How to cite this article: Tian, H. et al. Transition metal-free intramolecular regioselective couplings of aliphatic and aromatic C-H bonds. Sci. Rep. 6, 19931; doi: 10.1038/srep19931 (2016).
References
Alberico, D., Scott, M. E. & Lautens, M. Aryl-aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 107, 174–238 (2007).
Seregin, I. V. & Gevorgyan, V. Direct transition metal-catalyzed functionalization of heteroaromatic compounds. Chem. Soc. Rev. 36, 1173–1193 (2007).
Daugulis, O., Do, H.-Q. & Shabashov, D. Palladium- and copper-catalyzed arylation of carbon-hydrogen bonds. Acc. Chem. Res. 42, 1074–1086 (2009).
Chen, X., Engle, K. M., Wang, D.-H. & Yu, J.-Q. Palladium(II)-catalyzed C-H activation/C-C cross-coupling reactions: versatility and practicality. Angew. Chem. Int. Ed. 48, 5094–5115 (2009).
Colby, D. A., Bergman, R. G. & Ellman, J. A. Rhodium-catalyzed C-C bond formation via heteroatom-directed C-H bond activation. Chem. Rev. 110, 624–655 (2010).
Hartwig, J. Borylation and silylation of C–H Bonds: A platform for diverse C–H bond functionalizations. Acc. Chem. Res. 45, 864–873 (2012).
Neufeldt, S. R. & Sanford, M. S. Controlling site selectivity in palladium-catalyzed C–H bond functionalization. Acc. Chem. Res. 45, 936–946 (2012).
Kakiuchi, F. & Murai, S. Catalytic C−H/olefin coupling. Acc. Chem. Res. 35, 826–834 (2002).
Lyons, T. W. & Sanford, M. S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010).
Engle, K. M., Mei, T.-S., Wasa, M. & Yu, J.-Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 45, 788–802 (2012).
Colby, D. A., Tsai, A. S., Bergman, R. G. & Ellman, J. A. Rhodium catalyzed chelation-assisted C–H bond functionalization reactions. Acc. Chem. Res. 45, 814–825 (2012).
Li, C.-J. Cross-dehydrogenative coupling (CDC): Exploring C−C bond formations beyond functional group transformations. Acc. Chem. Res. 42, 335–344 (2009).
Girard, S. A., Knauber, T. & Li, C.-J. The Cross-Dehydrogenative Coupling of Csp3-H Bonds: A Versatile Strategy for C-C Bond Formations. Angew. Chem. Int. Ed. 52, 2–29 (2013).
Yeung, C. S. & Dong, V. M. Catalytic dehydrogenative cross-coupling: forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 111, 1215–1292 (2011).
Sun, C.-L., Li, B.-J. & Shi, Z.-J. Direct C-H transformation via iron catalysis. Chem. Rev. 111, 1293–1314 (2011).
Liu, C., Zhang, H., Shi, W. & Lei, A. Bond formations between two nucleophiles: transition metal catalyzed oxidative cross-coupling reactions. Chem. Rev. 111, 1780–1824 (2011).
Cho, S. H., Kim, J. Y., Kwak, J. & Chang, S. Recent advances in the transition metal-catalyzed twofold oxidative C-H bond activation strategy for C-C and C-N bond formation. Chem. Soc. Rev. 40, 5068–5083 (2011).
Kozhushkov, S. I. & Ackermann, L. Ruthenium-catalyzed direct oxidative alkenylation of arenes through twofold C-H bond functionalization. Chem. Sci. 4, 886–896 (2013).
Newhouse, T., Baran, P. S. & Hoffmann, R. W. The economies of synthesis. Chem. Soc. Rev. 38, 3010–3021 (2009).
Holland, H. L. C–H activation. Curr. Opin. Chem. Biol. 3, 22–27 (1999).
Krest, C. M. et al. Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem. 288, 17074–17081 (2013).
Yosca, T. H. et al. Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450. Science 342, 825–829 (2013).
Hubbard, B. K. & Walsh, C. T. Vancomycin assembly: Nature’s way. Angew. Chem., Int. Ed. 42, 730–765 (2003).
Bugg, T. D. H. Introduction to Enzyme and Coenzyme Chemistry. 2nd ed. Blackwell Publishing Ltd.: Oxford, U.K,., Chapter 6 (2004).
Silverman, R. B. The Organic Chemistry of Enzyme-Catalyzed Reactions. 2nd ed.; Academic Press: San Diego, Chapters 3–6 (2002).
Denisov, I. G., Makris, T. M., Sligar, S. G. & Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277 (2005).
Grobe, N. et al. Mammalian cytochrome P450 enzymes catalyze the phenol-coupling step in endogenous morphine biosynthesis. J. Biol. Chem. 284, 24425–24431 (2009).
Novo, E. & Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogen. Tissue Rep. 1, 5 (2008).
Brady, J. D., Sadler, I. H. & Fry, S. C. Pulcherosine, an oxidatively coupled trimer of tyrosine in plant cell walls: Its role in cross-link formation. Phytochemistry 47, 349–353 (1998).
Fujimoto, D., Horiuchi, K. & Hirama, M. Isotrityrosine, a new crosslinking amino-acid isolated from ascaris cuticle collagen. Biochem. Biophys. Res. Commun. 99, 637–643 (1981).
Rouau, X. et al. Dehydrotrimer of ferulic acid from maize bran. Phytochemistry 63, 899–903 (2003).
Ban, F., Lundqvist, M. J., Boyd, R. J. & Eriksson, L. A. Theoretical studies of the cross-linking mechanisms between cytosine and tyrosine. J. Am. Chem. Soc. 124, 2753–2761 (2002).
Mitrasinovic, P. M. Cross-linking between thymine and indolyl radical: Possible mechanisms for cross-linking of DNA and tryptophan-containing peptides. Bioconjugate Chem. 16, 588–597 (2005).
Williamson, N. R., Fineran, P. C., Leeper, F. J. & Salmond, G. P. C. The biosynthesis and regulation of bacterial prodiginines. Nature Rev. Microbiol. 4, 887–899 (2006).
Fürstner, A. Chemistry and biology of roseophilin and the prodigiosin alkaloids: A survey of the last 2500 years. Angew. Chem., Int. Ed. 42, 3582–3603 (2003).
Withall, D. M., Haynes, S. W. & Challis, G. L. Stereochemistry and Mechanism of Undecylprodigiosin Oxidative Carbocyclization to Streptorubin B by the Rieske Oxygenase RedG. J. Am. Chem. Soc. 137, 7889–7897 (2015).
Cerdeño, A. M., Bibb, M. J. & Challis, G. L. Analysis of the prodiginine biosynthesis gene cluster of Streptomyces coelicolor A3(2): new mechanisms for chain initiation and termination in modular multienzymes. Chem. Biol. 8, 817–829 (2001).
Haynes, S. W., Sydor, P. K., Corre, C., Song, L. & Challis, G. L. Stereochemical elucidation of Streptorubin B. J. Am. Chem. Soc. 133, 1793–1798 (2011).
Justicia, J. et al. Bioinspired terpene synthesis: a radical approach. Chem. Soc. Rev. 40, 3525–3537 (2011).
Kimball, D. B. & Haley, M. M. Triazenes: a versatile tool in organic synthesis. Angew. Chem. Int. Ed. 41, 3338–3351 (2002).
Patrick, T. B., Willaredt, R. P. & DeGonia, D. J. Synthesis of biaryls from aryltriazenes. J. Org. Chem. 50, 2232–2235 (1985).
Gross, M. L., Blank, D. H. & Welch, W. M. The triazene moiety as a protecting group for aromatic amines. J. Org. Chem. 58, 2104–2109 (1993).
Braese, S. The virtue of the multifunctional triazene linkers in the efficient solidphase synthesis of heterocycle libraries. Acc. Chem. Res. 37, 805–816 (2004).
Nicolaou, K. C. et al. New synthetic technology for the synthesis of aryl ethers: construction of C-O-D and D-O-E ring model systems of vancomycin. J. Am. Chem. Soc. 119, 3421–3422 (1997).
Voica, A.-F., Mendoza, A., Gutekunst, W. R., Fraga, J. O. & Baran, P. S. Guided desaturation of unactivated aliphatics. Nat. Chem. 4, 629–635 (2012).
Han, G., McIntosh, M. C. & Weinreb, S. M. A convenient synthetic method for amide oxidation. Tetrahedron Lett. 35, 5813–5816 (1994).
Pines, S. H., Purick, R. M., Reamer, R. A. & Gal, G. New aspects of intramolecular hydrogen transfer in some ortho-substituted aryl radicals. J. Org. Chem. 43, 1337–1342 (1978).
Maegawa, T. et al. Efficient and selective deuteration of phenylalanine derivatives catalyzed by Pd/C. Synlett 845–847 (2005).
Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (Grant Nos. 21172128, 21372139 and 21221062), the Ministry of Science and Technology of China (Grant No. 2012CB722605) and Shenzhen Municipal Government (Grant No. SZSITIC CXB201104210014A).
Author information
Authors and Affiliations
Contributions
H.T. and H.F. conceived and design this subject, H.T. and H.Y. conducted the experimental work, H.T., C.Z. and H.F. analysed the results, H.T. and H.F. co-wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tian, H., Yang, H., Zhu, C. et al. Transition metal-free intramolecular regioselective couplings of aliphatic and aromatic C-H bonds. Sci Rep 6, 19931 (2016). https://doi.org/10.1038/srep19931
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19931
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
