
Abstract
New polynitrides containing metastable forms of nitrogen are actively investigated as potential high-energy-density materials. Using a structure search method based on the CALYPSO methodology, we investigated the stable stoichiometries and structures of cesium polynitrides at high pressures. Along with the CsN3, we identified five new stoichiometric compounds (Cs3N, Cs2N, CsN, CsN2 and CsN5) with interesting structures that may be experimentally synthesizable at modest pressures (i.e., less than 50 GPa). Nitrogen species in the predicted structures have various structural forms ranging from single atom (N) to highly endothermic molecules (N2, N3, N4, N5, N6) and chains (N∞). Polymeric chains of nitrogen were found in the high-pressure C2/c phase of CsN2. This structure contains a substantially high content of single N-N bonds that exceeds the previously known nitrogen chains in pure forms and also exhibit metastability at ambient conditions. We also identified a very interesting CsN crystal that contains novel N44− anion. To our best knowledge, this is the first time a charged N4 species being reported. Results of the present study suggest that it is possible to obtain energetic polynitrogens in main-group nitrides under high pressure.
Similar content being viewed by others
Introduction
The pursuit for new and efficient energy source has always been a focus of scientific research. Nitrogen, which is abundant in nature, may be sought as a high-energy-density material (HEDM). This is possible if one can transform the diatomic N2 molecules into single- or double-bonded polynitrogen, utilizing the large energy difference between the single and double/triple bonds1,2,3,4,5,6,7,8,9. Solely single-bonded polynitrogen, for example, has an estimated energy capacity of 4.6 eV/mol, about three times that of the most powerful energetic materials known today10. The realization of polynitrogens has been actively experimented in nitrides, due to the fact that the charged nitrogen species in such materials often show improved kinetic stability over pure polynitrogens. Before 1999, the azide anion (N3−), which exists in metal azides (i.e., NaN3, Pb(N3)2), was the only known polynitrogen ion. Producing new polynitrogens involves substantial experimental difficulties due to the metastability of the products. However, in 1999, Christe et al. reported a successful synthesis of marginally stable N5+ AsF6− crystal, which contains the second polynitrogen ion, the N5+ cation11. Subsequently, thermally more stable N5+ SbF6− and N5+ Sb2F11− have been synthesized12. Around the same time, the N5− anion13, neutral N4 molecule14 have also been isolated experimentally. These successes led to an increasing interest in polynitrogen compounds and to the search of other stable polynitrongen forms in recent years.
Theoretical studies have often been employed to guide experiments and to identify unknown polynitrogens. In the past, many polynitrogen forms have been predicted to be metastable, from neutral (N3 to N60) to charged molecules (N42+, N5−, N6+, N6−, N62+); several of which have already been realized in experiments15,16,17,18. Prior to the synthesis of N5+ AsF6−, for example, quantum mechanical calculations19 already predicted that the N5+ could be detected experimentally. Previous theoretical studies of polynitrogens have primarily focused on single molecules, while the experimental realizations often proceeded in solid state, seeking for suitable matrices. Prediction of solid-state polynitrogen materials had been formidable due to the complexity of the potential-energy surface. Such a task only became possible recently attributing to newly developed heuristic algorithms and the high-performance computation capability. In recent studies20,21, a novel LiN5 crystal consisting of stable N5− anions has been predicted, which represents one of the first predictions of the polynitrogen compounds beyond ordinary stoichiometries. The LiN5 crystal is energetically stable only at high pressures, but exhibit mechanical stability at ambient conditions which provides the possibility for its recovery. Moving forward, in the present study we investigated the closed-related Cs-N system. Since Cs has the lowest ionization potential in alkali metals, it could lose the valence electron more easily than Li. Electrons acquired by polynitrogen anions in the Cs-N crystals reduce the electrons sharing between nitrogen atoms which ultimately change the bonding order. Thus, one expects that mixing reactive Cs with nitrogen will result in more diverse structures and higher energy densities of the products.
The ambient-pressure chemistry for the Cs-N system has been well studied. The CsN3 is the only energetically stable phase, formed by the Cs+ cation and double-bonded N3− anion22. Same N3− anions also exist in other alkali metal azides (LiN3, NaN3, KN3). The N3− has considerable metastability at ambient conditions which results in many applications for example as a propellant in automobile airbags. The decomposition of the N3− in CsN3 is highly exothermic and also environmental friendly. Previous theoretical studies23 suggested that the energy density of CsN3 can be further enhanced at high pressure, where the nitrogen atoms form extended structures. Herein, we present a thorough theoretical investigation of the Cs-N system with other possible stoichiometries at ambient and high pressures, ranging from the Cs3N to the CsN5. By changing the density of the electron donors (Cs) in the system, one is able to manipulate the bond strength of the polynitrogen anions such that the single N−N bonds can be stabilized at much lower pressures than that required for pure nitrogen. Six novel stoichiometric Cs-N compounds with fascinating structures were predicted which may be synthesized at high pressure and recovered at ambient conditions. This study revealed the possibility of the formation of several new polynitrogen forms in solids, including tetrazadiene (N4), pentazole (N5), hexazine (N6) and extended chains (N∞). The bonding nature in these polynitrogens is of great important to nitrogen chemistry and to the understanding of metal-nitrogen interactions.
Results and Discussion
Figure 1 shows the enthalpies of formation, ∆Hf, for the energetically most favorable Cs1-xNx structures obtained in the structure search calculated at four pressures. The ∆Hf of each Cs-N structure was calculated relative to the enthalpy of cesium and nitrogen solids, using a fractional representation Cs1-xNx (0 < x < 1), ∆Hf(Cs1-xNx) = H(Cs1-xNx) − (1−x)H(Cs solid)—xH(N solid). The energetically most favorable structures known for solid cesium (bcc, fcc, C2221, tI4, oC16 and dhcp phases) and solid nitrogen (α-, γ-, ε- and cubic gauche phases) were used as reference structures in their corresponding stable pressure ranges. In Fig. 1, the energetically stable phases at each pressure are shown by solid squares, which are connected by the convex hull (solid lines), whereas the unstable or metastable phases are shown by open squares. At ambient pressure, the only stable stoichiometry revealed in the structure search is the experimentally known CsN324. At high pressures, five new stoichiometries, Cs3N, Cs2N, CsN, CsN2 and CsN5, previously unknown for the Cs-N system, were predicted to become energetically stable in different pressure ranges. Detailed pressure-composition phase diagram for the Cs-N system is presented in Fig. 2, noting here the predicted structure changes in CsN3, CsN2 and CsN at high pressure. Specifically, the predicted pressure ranges of stability are, 18 to 64 GPa for Cs3N, 16 to 100 GPa and above for Cs2N, 7.5 to 100 GPa and above for CsN, 4 to 100 GPa and above for CsN2, 0 to 26 GPa and 81 to 100 GPa and above for CsN3 and 14 to 100 GPa and above for CsN5. It may strike as a surprise that energetically new stoichiometries, other than the commonly known CsN3 are calculated to be stable at high pressure. On the face of it, one sees the enigma of these stoichiometries violating the ‘octet rule’ for main-group elements. However, a closer look into the crystal structures reveals that all predicted stoichiometries are suited well with different bonding patterns of the polynitrogens and can be explained by traditional electron count. The calculated phonon dispersion relations also confirmed the mechanical and dynamical stability of these structures (Supplementary Figs 1–7). The crystal structures of the predicted Cs-N phases are presented in Fig. 3a–j, whereas the crystallographic parameters are provided in Supplementary Table 1. In what follow these structures will be analyzed and their relevance to energy storage applications will be discussed.

Relative enthalpies of formation of Cs-N phases with respect to elemental cesium and nitrogen solids.
The convex hulls connecting stable phases (solid squares) are shown by solid lines. Unstable/metastable phases are shown by open squares.

Predicted pressure-composition phase diagram of Cs-N crystal phases.

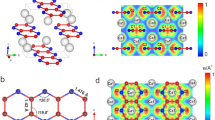
Structures of predicted stable Cs-N crystals.
(a) Cmcm structure of Cs3N. (b) C2/m structure of Cs2N. (c) Low-pressure C2/m structure of CsN. (d) High-pressure P-1 structure of CsN. (e) Low-pressure C2/m structure of CsN2. (f) High-pressure C2/c structure of CsN2. (g,h) Low-pressure C2/m and P21/m structures of CsN3. (i) High-pressure C2/m structure of CsN3. (j) Cmc21 structure of CsN5. Nitrogen and cesium atoms are represented by blue and green spheres, respectively.
Cs-N phases analogous to known metal nitrides
In the Cs-rich end, the Cs3N crystal adopts an orthorhombic Cmcm structure (Fig. 3a) between 18 and 64 GPa. Chemical structure of Cs3N is very similar to that of lithium nitride Li3N22. In the Cs3N, nitrogen atoms form N3− anions with fully occupied subshells. The N-N separations in the Cmcm structure are large, i.e., 3.16 Å at 20 GPa, which limits its capability of forming polynitrogens. It does, however, have an extremely strong nitrogen base, which, if can be recovered at ambient conditions, may be sufficient to deprotonate hydrogen leading to possible applications. The CsN crystal adopts a monoclinic C2/m structure between 7.5 and 44 GPa (Fig. 3c) in which the nitrogen atoms form double-bonded N22− anion (Fig. 4b). The N-N bondlength in the C2/m structure is 1.21 Å (at 20 GPa), which is a typical value for a double bond. The calculated Mayer bond order (MBO)25 for the N22− anion in the CsN is 2.20, also consistent with double bonding (Table 1). Same N22− anion has been known to exist in alkaline earth diazenides26,27,28,29. A related N24− anion, or deprotonated hydrazine (N2H4), was found in the C2/m structure of Cs2N (Fig. 3b). The C2/m structure of Cs2N is structurally similar to the C2/m phase of CsN, with one additional Cs atom at the 4i Wyckoff position. The two nitrogen atoms in the N24− are nearly single-bonded, as shown by the MBO value of 1.34. Calculated N-N bond length in Cs2N is 1.34 Å (at 20 GPa), similar to the value in the cubic gauche phase of nitrogen. The N24− have been synthesized in noble metal pernitrides30,31,32; many of the products exhibit remarkable properties (e.g., superconductivity, low compressibility) and, therefore, are often considered as technologically important materials.

Forms of polynitrogen discovered in the Cs-N crystals.
(a) Isolated N3– in the Cs3N. (b) Diazene N22− and hydrazine N24− in CsN and Cs2N. (c) Azide N3− in CsN3. (d) Tetrazadiene N44− in CsN. (e) Pentazole N5− in CsN5. (f) Hexazine N62− in CsN3. (g) Spiral nitrogen chains in CsN2.
At ambient pressure, the CsN3 crystal has the I4/mcm structure with azide N3− anion (Fig. 4c). The double-bond feature of the N3− is shown nicely by the calculated MBO value of 2.10 (Table 1). At high pressure, the I4/mcm structure is predicted to transform to a C2/m structure (Fig. 3g) at 7 GPa and then to a P21/m structure (Fig. 3h) at 16 GPa (Fig. 2). These results are in good agreement with previous studies23,33. Another C2/m structure is predicted to have lower enthalpy than the P21/m structure above 49 GPa (Fig. 3i). Energetically, however, the CsN3 becomes less stable already at 26 GPa with respect to the decomposition of CsN2 and CsN5, but regains the stability at above 81 GPa. Interestingly, if we treat the N6 as a unity located at its center of mass, the C2/m structure of CsN3 and the C2/m phase of Cs2N would have the same nitrogen sublattice and Cs positions. In the C2/m structure, the nitrogen atoms form cyclic N62− anions34,35,36,37. A neutral cyclic N6 molecule is the nitrogen analogue of benzene which has a planar D6 h symmetry (Fig. 4f). In the CsN3, the charged N6 has a planar distortion for non-aromatic π system which results in four non-equivalent N-N bonds (Fig. 5a). The bonding pattern of the N62− is illustrated by the electron localization function (ELF) map38 drawn on the plane (Fig. 5b). The regions with the maximum ELF values on the plane are identified as six σ-bonds and six lone pairs. Above and below the plane, the π electrons have cyclic delocalization counterbalanced by the positively charged nitrogen cores. Such arrangement of charges induces an electric quadrupole along the 6-fold axis of the N62−, which may interact favorably with the Cs+ located on the axis. Similar cation-π interactions should also present in the high-pressure phases of LiN3 and KN334,36,37. The calculated MBO values for the N-N bonds in the N62− anion are between 1.17 and 1.33 (Fig. 5a), slightly stronger than single bonds (1.0).

(a) The planer N62− anion isolated from the C2/m structure of CsN3 at 100 GPa. (b) The ELF map shown the (1 1 –2) cross-section of the C2/m structure of CsN3 at 100 GPa. (c) The ELF isosurface (value = 0.82) illustrated on a plane containing N44− in the P-1 structure of CsN at 50 GPa. (d) The ELF map shown on the (2 –2 –1) cross-section of the P-1 structure of CsN at 50 GPa. Numbers next to the N-N bond represent the bondlength (black, in Å) and MBO value (red), respectively.
The CsN5 crystal forms a Cmc21 structure (Fig. 3j) that is stable above 14 GPa and metastable at ambient pressure (Fig. 1), as the fact that phonon calculations carried out at ambient pressure in Fig. 6a reveal no imaginary modes. Its chemical structure is similar to that of the LiN5 we previously studied20. The nitrogen atoms in CsN5 form a planar N5− anion of the D5 h symmetry (Fig. 4e). Within the plane the N atoms are bonded by five σ-bonds. Above and below the plane the six π electrons form an aromatic set of π bonds that stabilizes the ring. Calculated N−N distances in the N5− are of the same length, i.e., 1.33 Å at ambient pressure, which is an intermediate between the single (1.45 Å) and double bonds (1.20 Å). Calculated MBO value of 1.43 also confirms this bonding nature (Table 1).

Phonon dispersion curves for the Cmc21 structure of CsN5 (a) and C2/c structure of CsN2 (b) and at ambient pressure.
CsN phase with energetic N44− anions
At pressures higher than 44 GPa, we identified a very interesting CsN crystal (Fig. 3d) that contains novel N44− anion (Fig. 4d). To our best knowledge, this is the first time a charged N4 species being reported in literature. The neutral N4 molecule has been successfully isolated in the gas phase a decade ago14. Dissociation of the N4 molecule was found to be highly exothermic, releasing ~ 800 kJ energy per mole. Such high energy content, if realized in a controlled manner, would place N4 among modern high-energy materials such as TATB, RDX and HMX39. On the other hand, the lifetime of the N4 molecule is only around 1 microsecond, which limits its practical usages. Compared with the neutral counterpart, the charged N44− as predicted in the CsN crystal is substantially stabilized by strong cation-anion interactions, which is expected to have an extended lifetime. Furthermore, due to the additional electrons acquired from Cs, the N44− anion has a lower degree of electrons-sharing compared with the neutral counterpart and therefore has a higher energy content.
The predicted N44− anion in CsN has an open-chain structure (Fig. 4d). Its Lewis structure has two terminal single-bonds (σ) and one internal double-bond (σ and π). The average bonding strength of the N44− is weaker than that of the neutral N440. The bonding pattern of the N44− is illustrated by the ELF isosurface and cross section (Fig. 5c,d). The maximum of the ELF in N44− appears as lobes outside the nitrogen atoms, which are identified as the lone pairs. Between the nitrogen atoms the smaller high ELF regions correspond to three σ bonds. The π electrons are delocalized and distributed among nitrogen atoms, which is not directly visualized by the ELF but can be inferred from the similar bond lengths and strengths of the three N-N bonds (Fig. 5c). Clearly, all three N-N bonds in the N44− (1.10, 1.10 and 1.26) are slightly stronger than the single bonds (1.0) but much weaker than the double bonds (2.0). As well, the N-N bond in the middle of the molecule (1.26) appears to be slightly stronger than the two terminal counterparts (1.10), showing some resemblance to the Lewis structure. In bulk, crystalline CsN is a semiconductor with a very small, indirect band gap of 0.25 eV (Supporting Information).
CsN2 phase with linear-polymeric nitrogen
The CsN2 crystal is predicted to become energetically stable near 4 GPa. Between 4 and 40 GPa, CsN2 adopts the C2/m structure in which the nitrogen atoms still keep the diatomic form (Fig. 3e). Above 40 GPa, CsN2 transforms to the C2/c structure (Fig. 3f). In the C2/c structure the nitrogen atoms form one-dimensional helical chains that extend infinitely in the crystal, which is the only polymeric form of nitrogen discovered in the present study (Fig. 4g). One-dimensional chains of similar geometry have been predicted to exist in pure oxygen under high pressure5. According to the Zintl-Klemm rule, the negatively charged nitrogen can behave like oxygen under certain conditions. The repeating unit of the chain in the C2/c structure contains eight nitrogen atoms (Fig. 7a). In its Lewis structure, an extending N84− unit should have 6 single bonds and 2 double bonds, forming a 3:1 ratio. This assignment is confirmed by the calculated MBO values for the four independent N-N bonds in the chain, e.g., 1.03, 1.17, 1.17 and 1.78, respectively (Fig. 7a). The calculated bond distances are 1.43, 1.39, 1.39 and 1.32 Å, respectively, also correspond with a 3:1 ratio, although the differences are less distinct than those in the bond orders. The predicted C2/c phase of CsN2 is an insulator, as illustrated by the calculated band structure and electronic density-of-states (DOS) (Fig. 7c,d). The calculated band gap is indirect, with the magnitude of ~1.3 eV. The valence states occupied by nitrogen are separated from each other and grouped into subsets, which, in the order of increasing energy, are characteristic of 2sσg/2sσu*, 2pσg/2pπu/2pπg* states. Notable contributions of nitrogen to the bands above and below the band gap are clearly seen in Fig. 7c.

(a) The ELF isosurface (value = 0.93) illustrated on a plane containing N84− in the C2/c structure of CsN2 at ambient pressure. Numbers next to the N-N bond represent the bondlength (black, in Å) and MBO value (red), respectively. (b) The ELF map shown on the (1 1 –1) cross-section of the C2/c structure of CsN2 at ambient pressure. (c,d) Electronic band structure and projected DOS of the C2/c structure calculated at ambient pressure.
Significantly, phonon calculations carried out at ambient pressure in Fig. 6b reveal no imaginary modes for the C2/c phase of the CsN2, suggesting that it is both mechanically and dynamically stable and might be quench recoverable. Furthermore, the minimum pressure required for the synthesis of this phase, ~40 GPa, is well within the current capability of high-pressure techniques. Nitrogen chains with such high content of single N-N bonds are not only promising for energy storage applications, but also of great impact in the rational design of polynitrogens. Polymeric nitrogen phases that are consisting of one-dimensional chains have been extensively studied for decades2,3. For pure nitrogen, however, electron count requires all such chains to have alternating single and double bonds which fixes the ratio of single bonds to double bonds to 1:1. The present study suggests that one may be able to increase the content of the single bonds simply by mixing the chains with electropositive metals. The negatively charged nitrogen chains are also stabilized by strong cation-anion interactions41 and therefore are expected to be more stable than their neutral counterparts.
Methods
The search for energetically stable crystalline structures was made on various stoichiometry of CsNx (x = 1/3, 1/2, 1, 2, 3, 4, 5 and 6) using simulation cells containing up to four formula units. Structure searches for all stoichiometries were carried out at four pressures, e.g., 0, 20, 50 and 100 GPa, using the particle swarm optimization methodology as implemented in the CALYPSO code42,43. In recent studies, it was shown that the approach was successful on the prediction of high pressure structures on both elemental and binary compounds, such as dense oxygen44, Bi2Te345 and Xe-Fe complex46. Total energy calculations, geometrical optimizations and electronic structure calculations were performed within the framework of density functional theory (DFT) using the VASP (Vienna Ab Initio simulation package) program47. Exchange and correlation of the electrons were treated by the generalized gradient approximation with the Perdew-Burke-Ernzerhof functional48. The projector-augmented wave (PAW) method49 was employed with the Cs and N potentials adopted from the VASP potential library. The Cs and N potentials have 5s25p66s1 and 2s22p3 as valence states, respectively and use an energy cutoff of 600 eV for the planewaves. A dense k-point grid50 with the spacing of 2π × 0.03 Å−1 was used to sample the Brillouin zone which was shown to yield excellent convergence for total energies (within 1 meV/atom).
Conclusion
New compounds containing polymeric forms of nitrogen (polynitrogens) have been actively investigated as potential high-energy-density materials over the past decades. The energy contents of the polynitrogens result from the large differences in bond dissociation energies between single, double and triple N-N bonds. This fact makes the polynitrogen compounds metastable and highly endothermic. In the present study, swarm-intelligence structure searches were carried out to predict stable stoichiometries and structures of cesium polynitrides at high pressures. The goal is to discover metastable Cs1-xNx structures containing polynitrogens that are energetically stable at high pressures. Along with the known CsN3, we found five more stoichiometries, namely, Cs3N, Cs2N, CsN, CsN2 and CsN5, to also become stable provided suitable pressure conditions. In the predicted crystals, the Cs atoms behave as electron donors whose concentration strongly influences the N-N bonding. This enables the nitrogen atoms to adopt versatile forms in molecules and extended structures, ranging from small clusters made of a few atoms (N2, N3, N4, N5, N6) to one-dimensional infinite chains (N∞). In particular, the N44− anion and the N∞ chain as found in the high-pressure phases of CsN and CsN2 contain an exceptionally high content of the single N-N bonds, which, if realized in controlled manner, may find applications as high energy carriers. At pressures above 40 GPa, the CsN2 structure with the N∞ chains is energetically stable, suggesting that it may be prepared by high-pressure synthesis. Moreover, this structure is mechanically stable at ambient conditions which may make an ambient-pressure recovery possible. The present study provides new insights to the understanding of polynitrogens and encourages experimental exploration of these promising materials in the future.
Additional Information
How to cite this article: Peng, F. et al. Exotic stable cesium polynitrides at high pressure. Sci. Rep. 5, 16902; doi: 10.1038/srep16902 (2015).
References
Mailhiot, C., Yang, L. H. & McMahan, A. K. Polymeric nitrogen. Phys. Rev. B 46, 14419 (1992).
Alemany, M. & Martins, J. L. Density-functional study of nonmolecular phases of nitrogen: metastable phase at low pressure. Phys. Rev. B 68, 024110 (2003).
Mattson, W., Sanchez-Portal, D., Chiesa, S. & Martin, R. Prediction of New Phases of Nitrogen at High Pressure from First-Principles Simulations. Phys. Rev. Lett. 93, 125501 (2004).
Zahariev, F., Hu, A., Hooper, J., Zhang, F. & Woo, T. Layered single-bonded nonmolecular phase of nitrogen from first-principles simulation. Phys. Rev. B 72, 214108 (2005).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 124, 244704 (2006).
Yao, Y., John, S. T. & Tanaka, K. Metastable high-pressure single-bonded phases of nitrogen predicted via genetic algorithm. Phys. Rev. B 77, 052103 (2008).
Ma, Y., Oganov, A., Li, Z., Xie, Y. & Kotakoski, J. Novel High Pressure Structures of Polymeric Nitrogen. Phys. Rev. Lett. 102, 065501 (2009).
Wang, X. et al. Cagelike Diamondoid Nitrogen at High Pressures. Phys. Rev. Lett. 109, 175502 (2012).
Hirshberg, B., Gerber, R. B. & Krylov, A. I. Calculations predict a stable molecular crystal of N8. Nat. Chem. 6, 52–56 (2013).
Uddin, J., Barone, V. & Scuseria, G. E. Energy storage capacity of polymeric nitrogen. Molecular Physics 104, 745–749 (2006).
Christe, K. O., Wilson, W. W., Sheehy, J. A. & Boatz, J. A. N5+: A Novel Homoleptic Polynitrogen Ion as a High Energy Density Material. Angew. Chem. Int. Ed. 38, 2004 (1999).
Vij, A. et al. Polynitrogen chemistry. Synthesis, characterization and crystal structure of surprisingly stable fluoroantimonate salts of N5. J. Am. Chem. Soc. 123, 6308–6313 (2001).
Vij, A., Pavlovich, J. G., Wilson, W. W., Vij, V. & Christe, K. O. Experimental Detection of the Pentaazacyclopentadienide (Pentazolate) Anion, cyclo‐N5−. Angewandte Chemie 114, 3177–3180 (2002).
Cacace, F. Experimental Detection of Tetranitrogen. Science 295, 480–481 (2002).
Wasilewski, J. Stationary points on the lowest doublet and quartet hypersurfaces of the N3 radical: A comparison of molecular orbital and density functional approaches. J. Chem. Phys. 105, 10969–10982 (1996).
Manaa, M. R. Toward new energy-rich molecular systems: from N 10 to N 60. Chem. Phys. Lett. 331, 262–268 (2000).
Olah, G. A., Surya Prakash, G. K. & Rasul, G. N62+ and N42+ Dications and Their N12 and N10 Azido Derivatives: DFT/GIAO-MP2 Theoretical Studies1. J. Am. Chem. Soc. 123, 3308–3310 (2001).
Nguyen, M. T. & Ha, T.-K. Decomposition mechanism of the polynitrogen N 5 and N 6 clusters and their ions. Chem. Phys. Lett. 335, 311–320 (2001).
Pyykkö, P. & Runeberg, N. Ab initio studies of bonding trends: Part 9. The dicyanamide-carbon suboxide-dicyanoether-cyanogen azide isoelectronic series A = B = C = D = E1. Journal of Molecular Structure: THEOCHEM 234, 279–290 (1991).
Peng, F., Yao, Y., Liu, H. & Ma, Y. Crystalline LiN 5Predicted from First-Principles as a Possible High-Energy Material. J. Phys. Chem. Lett. 6, 2363–2366 (2015).
Shen, Y. et al. Novel lithium-nitrogen compounds at ambient and high pressures. Sci. Rep. 5, 14204 (2015).
Pringle, G. E. & Noakes, D. E. The crystal structures of lithium, sodium and strontium azides. Acta Crystallogr B 24, 262–269 (1968).
Wang, X., Li, J., Zhu, H., Chen, L. & Lin, H. Polymerization of nitrogen in cesium azide under modest pressure. J. Chem. Phys. 141, 044717 (2014).
Sangster, J. Cs-N (Cesium-Nitrogen). Eur.J.Mineral. 25, 556–557 (2004).
Mayer, I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 97, 270–274 (1983).
Auffermann, G., Prots, Y. & Kniep, R. SrN and SrN2: Diazenides by Synthesis under High N2‐Pressure. Angew. Chem. Int. Ed. 40, 547–549 (2001).
Vajenine, G. V. et al. Preparation, crystal structure and properties of barium pernitride, BaN2. Inorg. Chem. 40, 4866–4870 (2001).
Schneider, S. B., Frankovsky, R. & Schnick, W. Synthesis of Alkaline Earth Diazenides MAEN2 (MAE = Ca, Sr, Ba) by Controlled Thermal Decomposition of Azides under High Pressure. Inorg. Chem. 51, 2366–2373 (2012).
Wang, H., Yao, Y., Si, Y., Wu, Z. & Vaitheeswaran, G. Hidden Thermodynamic Ground State of Calcium Diazenide. J. Phys. Chem. C 118, 650–656 (2013).
Gregoryanz, E. et al. Synthesis and characterization of a binary noble metal nitride. Nat. Mater. 3, 294–297 (2004).
Crowhurst, J. C. Synthesis and Characterization of the Nitrides of Platinum and Iridium. Science 311, 1275–1278 (2006).
Yu, R., Zhan, Q. & De Jonghe, L. C. Crystal structures of and displacive transitions in OsN2, IrN2, RuN2 and RhN2. Angewandte Chemie 119, 1154–1158 (2007).
Hou, D. et al. Series of phase transitions in cesium azide under high pressure studied by in situ x-ray diffraction. Phys. Rev. B 84, 064127 (2011).
Zhang, M., Yan, H., Wei, Q., Wang, H. & Wu, Z. Novel high-pressure phase with pseudo-benzene ‘N 6’ molecule of LiN 3. Europhys. Lett. 101, 26004 (2013).
Wang, X. et al. Polymerization of nitrogen in lithium azide. J. Chem. Phys. 139, 164710 (2013).
Prasad, D. L. V. K., Ashcroft, N. W. & Hoffmann, R. Evolving Structural Diversity and Metallicity in Compressed Lithium Azide. J. Phys. Chem. C 117, 20838–20846 (2013).
Zhang, J., Zeng, Z., Lin, H.-Q. & Li, Y.-L. Pressure-induced planar N6 rings in potassium azide. Sci. Rep. 4, (2014).
Becke, A. D. & Edgecombe, K. E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 92, 5397 (1990).
Evans, W. J. et al. Pressure-Induced Polymerization of Carbon Monoxide: Disproportionation and Synthesis of an Energetic Lactonic Polymer. Chem. Mater. 18, 2520–2531 (2006).
Korkin, A. A., Balkova, A., Bartlett, R. J., Boyd, R. J. & Rague Schleyer, von, P. The 28-electron tetraatomic molecules: N4, CN2O, BFN2, C2O2, B2F2, CBFO, C2FN and BNO2. Challenges for computational and experimental chemistry. The Journal of Physical Chemistry 100, 5702–5714 (1996).
Christe, K. O. Recent Advances in the Chemistry of N5+, N5− and High‐Oxygen Compounds. Propellants, Explosives, Pyrotechnics 32, 194–204 (2007).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 82, 094116 (2010).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 183, 2063–2070 (2012).
Zhu, L. et al. Spiral chain O4 form of dense oxygen. Proc. Natl. Acad. Sci. USA 109, 751–753 (2012).
Zhu, L. et al. Substitutional Alloy of Bi and Te at High Pressure. Phys. Rev. Lett. 106, 145501 (2011).
Lv, J., Wang, Y., Zhu, L. & Ma, Y. Predicted Novel High-Pressure Phases of Lithium. Phys. Rev. Lett. 106, 015503 (2011).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Perdew, burke and ernzerhof reply. Phys. Rev. Lett. 80, 891 (1998).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Acknowledgements
We thank the China Natural Science Foundation of China under 11304141, 11304140, 11304167, 11274136, 11104104, 11504007, 11025418 and 91022029, the 2012 Changjiang Scholars Program of China, Changjiang Scholar and Innovative Research Team in University (IRT1132), a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant and work at Carnegie was supported by EFree, an Energy Frontier Research Center funded by the DOE, Office of Science, Basic Energy Sciences under Award No. DE-SC-0001057 (salary support for H.L). The infrastructure and facilities used at Carnegie were supported by NNSA Grant No. DE-NA-0002006, CDAC. This work is also sponsored by the Program for Science and Technology Innovation Research Team in University of Henan Province Grant No.13IRTSTHN020. Part of the calculations was performed at the High Performance Computation Training and Research Facilities at the University of Saskatchewan.
Author information
Authors and Affiliations
Contributions
F.P. and Y.Y. conceived the idea. F.P., Y.H. and H.L. performed the calculations. F.P. and H.L. and Y.Y. wrote the manuscript with contribution from all.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Peng, F., Han, Y., Liu, H. et al. Exotic stable cesium polynitrides at high pressure. Sci Rep 5, 16902 (2015). https://doi.org/10.1038/srep16902
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16902
This article is cited by
-
Fe-N system at high pressure reveals a compound featuring polymeric nitrogen chains
Nature Communications (2018)
-
Novel triadius-like N4 specie of iron nitride compounds under high pressure
Scientific Reports (2018)
-
The polymerization of nitrogen in Li2N2 at high pressures
Scientific Reports (2018)
-
Diverse ruthenium nitrides stabilized under pressure: a theoretical prediction
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
