
Abstract
High pressure structure, stability, metallization and superconductivity of PbH4(H2)2, a H2-containing compound combining one of the heaviest elements with the lightest element, are investigated by the first-principles calculations. The metallic character is found over the whole studied pressure range, although PbH4(H2)2 is metastable and easily decompose at low pressure. The decomposition pressure point of 133 GPa is predicted above which PbH4(H2)2 is stable both thermodynamically and dynamically with the C2/m symmetry. Interestedly, all hydrogen atoms pairwise couple into H2 quasi-molecules and remain this style up to 400 GPa in the C2/m structure. At high-pressure, PbH4(H2)2 tends to form the Pb-H2 alloy. The superconductivity of Tc firstly rising and then falling is observed in the C2/m PbH4(H2)2. The maximum of Tc is about 107 K at 230 GPa. The softening of intermediate-frequency phonon induced by more inserted H2 molecules is the main origin of the high Tc. The results obtained represent a significant step toward the understanding of the high pressure behavior of metallic hydrogen and hydrogen-rich materials, which is helpful for obtaining the higher Tc.
Similar content being viewed by others

Introduction
In recent decades, many scientists have devote to searching for the high-temperature superconducting materials. For the lightest element, hydrogen (H), Ashcroft applied the BCS theory to propose that the metallic hydrogen will be a room-temperature superconductor under high pressure1. This suggestion has motivated considerable experimental and theoretical activities. However, solid hydrogen remains insulating character at extremely high pressure, at least up to 342 GPa2. Due to the extremely high and experiment unreachable pressure, as a alternative, Ashcroft proposed that the hydrogen-rich alloys shall transform into metal under relatively lower pressure due to the chemical precompressions from the comparable weight elements3. Thus, hydrogen-rich group-IV hydrides have been extensively explored, such as CH4, SiH4, GeH4, SnH4 and PbH4. All of them show up interesting new structures and novel properties under pressure. CH4 is still an insulator up to the pressure of 520 GPa4. Although Eremets et al. experimentally reported the metallization and superconductivity of SiH4 above 60 GPa5, for the controversial result it might be understood as superconductivity of amorphous silicon, silicon hydrides, or platinum hydrides6,7. And theoretical prediction indicates that the stable SiH4 can behave as metal and exhibit superconductivity above 220 GPa with the superconducting transition temperature (Tc) of about 20 K (The Coulomb parameter  , the below is same.)8. GeH4 has lower metallization pressure than silane9,10 and the highest Tc reaches to 73 K at 220 GPa11. Furthermore, the metallization pressure of SnH4 decreases, the highest Tc is close to 83 K at 120 GPa12.
, the below is same.)8. GeH4 has lower metallization pressure than silane9,10 and the highest Tc reaches to 73 K at 220 GPa11. Furthermore, the metallization pressure of SnH4 decreases, the highest Tc is close to 83 K at 120 GPa12.
It is clearly that the metallization pressure of group-IV hydrides decreases with increase of atomic number of heavy element, which is obviously less than that of solid H2. Unfortunately, the Tc of group-IV hydrides is also greatly decreased. By analyzing the crystal feature, we find that the quasi-molecular H2 units exist in the high-pressure structures of GeH4 and SnH4. And these H2 units have been found to contribute significantly to the superconductivity. Then, whether can the Tc be improved by intercalating H2 into group-IV hydrides? H2-containing compounds of CH4-H2 have been fabricated up to 30 GPa, such as CH4(H2)2, (CH4)2H2, CH4(H2)4, CH4H213. But both metallization and superconductivity are still lack. For the SiH4-H2 system, the crystal structure, phase diagram and metallization under pressure of SiH4(H2)2 were extensively explored14,15,16,17,18,19,20,21,22. The Tc of SiH4(H2)2 is as high as 107 K at 250 GPa23, which is visibly higher than that of SiH4. Following the experimental observation24, we have also theoretically investigated the structural, phase transition, metallization and superconductivity of GeH4(H2)2 under pressure25,26. The predicted Tc of GeH4(H2)2 is close to 100 K at 250 GPa, higher than that of GeH4. These results inevitably encourage us further to seek for high-temperature superconductors and study the superconductivity in these H2-containing compounds. However, it is necessary to decrease the work pressure of superconducting. For examples, the decomposition pressures are as high as 248 GPa for SiH4(H2)2 and 220 GPa for GeH4(H2)2, respectively, above which they are stable superconducting materials.
As mentioned above, the combination the lightest H with one of the heaviest Pb seems to be a good way to improve the Tc and decrease the work pressure. Chemically, PbH4 still remains the most elusive of group-IV tetrahydrides. The pioneering theoretical work of Desclaux and Pyykkö predicted the structure and stability of PbH427,28. The theoretically predicted tetrahedral structure of an isolated molecule, with an equilibrium Pb-H distance of approximately 1.73 Å, was eventually confirmed by experiments29,30. But, Krivtsun et al.30 observed that the PbH4 molecules were kinetically unstable and readily decompose to Pb atomic layer and H2 in approximately 10 s. Recently, Zaleski-Ejgierd et al. theoretically investigated the structure and the stability of PbH4 under high pressure31. They found that PbH4 is stable thermodynamically above 132 GPa, in forms of Imma (132–296 GPa) and Ibam (>296 GPa) space groups. And PbH4 even keeps the metallic character covering the whole range of pressure31. However, the superconductivity is indeterminate, since the dynamic stable phase of PbH4 has been not discovered from experimental and theoretical aspects yet. By intercalating H2 units into PbH4 molecular crystal, e.g. PbH4(H2)2, how about the structure, stability and superconductivity? It is just the purpose of our study. In this work, we found out the stable phase of PbH4(H2)2 thermodynamically and dynamically and investigated its desired superconductivity. The decomposition pressure of 133 GPa is much lower than the metallization pressure of solid hydrogen, which is easily reached in experiments by diamond-anvil techniques. And the H2-H2 coupling under high pressure figures out the different superconducting mechanism.
Results
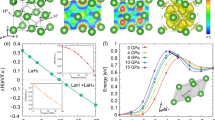
Covering the wide pressure range of 0–400 GPa, variable-cell structure prediction simulations with 1 to 4 PbH4(H2)2 formula units per cell (f.u./cell) were performed. We have calculated the enthalpies of searched structures of PbH4(H2)2 to examine the thermodynamical stability induced by pressure. For several competitive structures of PbH4(H2)2, the enthalpies (relative to the P-1 structure) as function of pressure are shown in Fig. 1. It is found that Pnnm phase is the most stablest below 40 GPa with the lowest enthalpy value. Starting from 40 GPa up to 135 GPa, PbH4(H2)2 transfers into P-1 phase. Upon further compression, the C2/m becomes to the most stablest phase above 135 GPa. As a result, there are two structural phase transitions existing in the range of 0–400 GPa. Three low-enthalpy structures were obtained, orthorhombic Pnnm (4 f.u./cell), triclinic P-1 (2 f.u./cell) and monoclinic C2/m (2 f.u./cell), respectively, as shown in Supplementary Fig. S1 online. The lattice parameters of these three structures at different pressures are also listed in Table S1 of the supplementary information online. From the crystal configurations at different pressures, PbH4 tetrahedral molecule does not exist in PbH4(H2)2 and all of hydrogen atoms construct the H2 quasi-molecules separating from Pb atoms.

Calculated enthalpies per PbH4(H2)2 unit as the function of pressure.
Enthalpy difference versus pressure for competitive structures of PbH4(H2)2, referenced to the P-1 phase. The decomposition enthalpies into PbH4 + 2H2, PbH3 + 5/2H2, PbH2 + 3H2, PbH + 7/2H2 and Pb + 4H2 were also plotted. The inset exhibits the change of enthalpies induced by ZPE correction, which indicates that the decomposition pressure of the C2/m structure decreases as 133 GPa.
However, it was reported that the hydrogen-rich materials is easily decomposed10,11,15,16,17,22,23,24,25,31,32. Hence, we must check the stability by mean of estimating the decomposition enthalpy. For PbH4(H2)2, there are five possible decomposition paths as PbH4(H2)2 → Pb + 4H2, 2PbH4(H2)2 → 2PbH + 7H2, PbH4(H2)2 → PbH2 + 3H2, 2PbH4(H2)2 → 2PbH3 + 5H2 and PbH4(H2)2 → PbH4 + 2 H2, respectively. For three system of PbH3, PbH2 and PbH, we searched their structures at different pressures. Structural parameters at different pressure regions are presented in Supplementary (Tables S2, S3 and S4) online. With help of the reported structures of Pmnm, P6/mmm, Imma and Ibam for PbH431, fcc, hcp and  for Pb33, P63m, C/2c and Cmca for H234 corresponding stable pressures, the decomposition enthalpies were calculated and plotted in Fig. 1. PbH4(H2)2 is unstable and decomposes into Pb + 4H2 blow 120 GPa and PbH4 + 2H2 in the pressure range of 120–160 GPa. Namely, both Pnnm and P-1 phases are metastable. PbH4(H2)2 is only stabilized above the pressure of 160 GPa, displaying the symmetry of C2/m.
for Pb33, P63m, C/2c and Cmca for H234 corresponding stable pressures, the decomposition enthalpies were calculated and plotted in Fig. 1. PbH4(H2)2 is unstable and decomposes into Pb + 4H2 blow 120 GPa and PbH4 + 2H2 in the pressure range of 120–160 GPa. Namely, both Pnnm and P-1 phases are metastable. PbH4(H2)2 is only stabilized above the pressure of 160 GPa, displaying the symmetry of C2/m.
Besides, it has well-known that quantum effects related to hydrogen atoms are very important. The hydrogen zero-point energy (ZPE) has significantly revised the structural stability as in the cases of solid hydrogen34 and hydrogen-rich materials9,11. To judge the effect on stability, we also calculated the ZPEs of PbH4(H2)2, PbH4 and H2 in the range of 100–200 GPa using the quasiharmonic approximation35. As the insert shown in Fig.1, the ZPE effect does not change the order of the phase transitions but lowers the decomposition pressure of the C2/m structure into ~133 GPa. This decomposition pressure of PbH4(H2)2 is obviously lower than 248 GPa of SiH4(H2)223 and 220 GPa of GeH4(H2)225, which indicates that PbH4(H2)2 will exist in the wider pressure range. For this stability, the subsequent crystal structural, electronic, phonon and electron-phonon coupling (EPC) calculations are focused on the C2/m structure above 133 GPa and typical results are presented at 200 GPa.
For C2/m structure, Pb atoms occupy the crystallographic 2a sites and four non-equivalent H atoms sit on the 4i sites under high pressure. All of H atoms pairwise coupling into two types of quasi-molecules as shown in Fig. 2a. The nearest distance between Pb and H atom is about 2 Å. In this dense structure, we can not find any plumbane molecules existing, but H2 quasi-molecules distribute around Pb atoms and are ordering (Fig. 2). This kind of ordered arrangements of H2 units is clearer at high pressure, while H2 units tend to be inordering at low pressure18,25. A visible character of Pb and H2 in layers is observed from (001)-plane (Fig. 2b) or (010)-plane (Fig. 2c). Noticeably, the layered feature is also a common phenomenon in some hydrogen-rich systems. With the increase of pressure, all of the lattice constants of C2/m structure in a, b and c directions decrease. However, the H-H bond lengths in H2 quasi-molecules marked as d1H−H (formed by H1 and H2 sites shown in Fig. 2a) and d2H−H (formed by H3 and H4 sites shown in Fig. 2a) firstly increase then decrease as shown in Fig. 3a. There are three kinds of intermolecular distances among H2 molecules in the C2/m structure, all of them are monotonously decreased with the pressurizing, as shown in Fig. 3b. Reviewing the high-pressure structural character, we find that part hydrogen atoms form H2 units with the other hydrogen atoms strongly bonding with Si in Ccca phase of SiH4(H2)223, while all of hydrogen atoms pairwise coupling into H2 quasi-molecules with the nearest distance of ~1.7 Å between Ge and H in P21/c phase of GeH4(H2)225. As a comparison, with the help of analysis of atomic distances, the intermolecular and intramolecular couplings of H2 gradually strengthen, while the interaction between H and the heavy atom evidently weakens from SiH4(H2)2 to GeH4(H2)2 and then to PbH4(H2)2.

High-pressure crystal structure of PbH4(H2)2.
(a) C2/m structure at 200 GPa. Large and small spheres represent Pb and H atoms, respectively. H1-H4 mark four non-equivalent H atoms on the crystallographic sites. (b,c) show the C2/m structure normal to the (001) and (010) plane, respectively.

The H-H bond lengths in H2 unit and the H2-H2 intermolecular distances.
For C2/m structure, two types of H-H bond lengths in H2 (a) and three kinds of distances among H2 molecules (b) change with pressure (133–350 GPa).
At 200 GPa, the lattice parameters of C2/m structure are a = 7.184 Å, b = 2.807 Å and c = 2.973 Å, as well as the angle β = 68.1° (see Supplementary Table S1 online). The d1H−H and d2H−H are 0.78 Å and 0.82 Å, respectively. The intermolecular distance of H2-H2 is less than that between Pb and H atoms. With the lattice parameters, calculated electronic structures show that PbH4(H2)2 is metallic at 200 GPa. For SiH4(H2)2 and GeH4(H2)2 reported previously, they remain the characteristics of insulator under low pressure. The insulator-to-metal transition occurs at 92 GPa in SiH4(H2)2 and at 48 GPa in GeH4(H2)2, respectively. However, we didn’t find the transition point of PbH4(H2)2. It seems to be metal even in ambient pressure, which consist with PbH431. So the low pressure metallization does not come from the intercalation of H2 molecules. Comparing with Si and Ge, Pb has larger ionic radius which results in more strong itinerant property of valent electrons. Figure 4 shows the projected density of state (PDOS) at several selected pressures. According to the electronic PDOS at Fermi level we can draw a conclusion that at low pressure in Pnnm structure the Pb-p electrons make the most contribution to density of state and exhibit properties of a nearly free-electron metal (Fig. 4a,b). As the pressure increases, the strengthening of H2-H2 interaction leads to the overlap of H-s wave functions. The contribution of H-s electrons to Fermi surface increases. PDOS tends to be uniform distribution and the bandwidth further broadens from 100 GPa to 300 GPa (Fig. 4c–f). It indicates that with the increase of pressure PbH4(H2)2 mainly like to be Pb-H2 alloy. The Pb interlayer interaction is connected by these H2 molecules. To gain more insight into the bonding nature of PbH4(H2)2, the electron location function (ELF) of C2/m phase at 200 GPa was calculated. ELF shown in Fig. 5 displays the electronic location around Pb and H atoms as well as the nearly free-electron-like distribution among Pb atoms. However, the high ELF values between Pb and H atoms (Fig. 5a) and of intermolecular H2 (Fig. 5b) indicate that the electrons become delocalized, suggesting a feature of nearly free-electron metal.

Electronic PDOS at different pressures.
Calculated PDOS of PbH4(H2)2 at different pressures of 5 GPa (a) and 20 GPa (b) for P-1 phase, 100 GPa for Pnnm phase (c), 160 GPa (d), 200 GPa (e) and 300 GPa (f) for C2/m phase. The lines at zero indicate the Fermi level.

ELF of PbH4(H2)2.
Calculated ELF isosurface of PbH4(H2)2 for C2/m at 200 GPa with the ELF value of 0.75. (a,b) highlight the sections on (001) and (010) planes, respectively.
The phonon dispersion curves for C2/m structure at 200 GPa (Fig. 6) and other selected pressure point (see Supplementary Fig. S2 online) were calculated to explore the lattice dynamics of PbH4(H2)2. The absence of any imaginary frequencies implies the dynamical stability of C2/m phase under high pressure. The whole phonon spectrum can be divided into three parts. By combining with the phonon density of states (PhDOS) projected on atoms shown in Fig. 7a, in the case of 200 GPa, we find that the low-frequency vibration below 215 cm−1 mainly come from the vibrations Pb atoms. The intermolecular strong phonon coupling among H2 molecules appear in the intermediate-frequency range of 295–1876 cm−1. After a large gap, in high frequency area above 2695 cm−1, the H-H vibration in H2 formed by H3 and H4 sites mainly contributes in the range of 2695–2898 cm−1, while the vibration in H2 formed by H1 and H2 sites around 3220 to 3380 cm−1. Comparing these three systems of Si-, Ge- and Pb-based, we find a strong phonon coupling between silicon and hydrogen in SiH4(H2)223, very weak phonon coupling between metal and hydrogen in GeH4(H2)225 as well as PbH4(H2)2. The H-H vibration in H2 molecule is the strongest in PbH4(H2)2. From the Eliashberg phonon spectral function α2F(ω) and the integrated EPC parameter λ(ω) shown in Fig. 7b, the intermediate-frequency (295–1876 cm−1) vibrational modes of H2 molecules contribute 81.5% of total λ. This percentage is larger than 66% in Si-based and 75% in Ge-based case. This result highlights the significant role played by H2 molecules on the electron-phonon interaction.

Phonon spectrum.
Calculated phonon spectrum of C2/m structure at 200 GPa.

Phonon properties and Eliashberg spectral function.
Calculated phonon density of states (PhDOS) (a) and the Eliashberg phonon spectral function α2F(ω) and electron-phonon integral λ(ω) (b) for the C2/m structure at 200 GPa.
At 200 GPa, the calculated total EPC constant λ is 1.296 for C2/m PbH4(H2)2. From Si to Ge and then to Pb case, the λ gradually decreases from 1.625 to 1.43 and then to 1.296, which implies a weak coupling between metal and hydrogen. However, the phonon frequency logarithmic average ωlog rises gradually, from 871 K in SiH4(H2)2 to 1051 K in PbH4(H2)2. This means more higher Debye temperature in PbH4(H2)2. Based on the obtained α2F(ω) and λ(ω), we now can analyze the superconductivity using the modified McMillan equation by Allen and Dynes36,

With the typical choice of the Coulomb pseudopotential  3, a remarkable large Tc of 103 K was obtained for C2/m phase of PbH4(H2)2, which is comparable with those of copper oxide superconductors.
3, a remarkable large Tc of 103 K was obtained for C2/m phase of PbH4(H2)2, which is comparable with those of copper oxide superconductors.
To figure out the pressure effect on superconductivity in PbH4(H2)2, in addition, the Tc values at several typical pressure points were calculated and shown in Supplementary Fig. S3 online. An interesting phenomenon exhibits the superconductivity firstly strengthening before weakening. The Tc has a maximum between 140 and 350 GPa, ~107 K for  . Seen from the distances of among H2 molecules shown in Fig. 3b, the monotonously decreasing makes a hint of “hardening” of intermediate-frequency phonon with the increase of pressure. The phonon spectra shown in Supplementary Fig. S2 online confirm this point. To analyze this phenomenon of Tc variations, we have further calculated the Eliashberg phonon spectral function and the EPC strength at different pressures, the results are presented in Supplementary Fig. S4 online. With the increase of pressure, the calculated EPCs are 1.280, 1.296, 1.379 and 1.341 for 180 GPa, 200 GPa, 250 GPa and 300 GPa, respectively, which shows a tendency of first increase and then decrease similar to Tc. In the Tc rising zone, the contribution of Pb-H coupling to the EPC strength is decreased from 14.3% at 180 GPa to 12% at 200 GPa and the phonon vibration of H-H in H2 units also weakens the EPC (The contribution is from 7.3% to 6.5% corresponding pressures.). However, the contribution of H2-H2 coupling to the EPC is strengthening from 78.4% at 180 GPa to 81.5% at 200 GPa. So the initial rising of Tc results from the contribution increasing of H2-H2 for the EPC. As shown in Fig. S4 online, from 75.8% at 250 GPa to 73.5% at 300 GPa, the decrease of contribution of H2-H2 for the EPC leads to the fall of Tc. The result further reveals the significance of H2-H2 coupling to superconductivity in PbH4(H2)2.
. Seen from the distances of among H2 molecules shown in Fig. 3b, the monotonously decreasing makes a hint of “hardening” of intermediate-frequency phonon with the increase of pressure. The phonon spectra shown in Supplementary Fig. S2 online confirm this point. To analyze this phenomenon of Tc variations, we have further calculated the Eliashberg phonon spectral function and the EPC strength at different pressures, the results are presented in Supplementary Fig. S4 online. With the increase of pressure, the calculated EPCs are 1.280, 1.296, 1.379 and 1.341 for 180 GPa, 200 GPa, 250 GPa and 300 GPa, respectively, which shows a tendency of first increase and then decrease similar to Tc. In the Tc rising zone, the contribution of Pb-H coupling to the EPC strength is decreased from 14.3% at 180 GPa to 12% at 200 GPa and the phonon vibration of H-H in H2 units also weakens the EPC (The contribution is from 7.3% to 6.5% corresponding pressures.). However, the contribution of H2-H2 coupling to the EPC is strengthening from 78.4% at 180 GPa to 81.5% at 200 GPa. So the initial rising of Tc results from the contribution increasing of H2-H2 for the EPC. As shown in Fig. S4 online, from 75.8% at 250 GPa to 73.5% at 300 GPa, the decrease of contribution of H2-H2 for the EPC leads to the fall of Tc. The result further reveals the significance of H2-H2 coupling to superconductivity in PbH4(H2)2.
Discussion
Thus far, the stability of PbH4(H2)2 has been identified in the pressure range of 0–400 GPa. At low pressure it is metastable and possibly decomposes into Pb + H2 or PbH4 + H2. Above 133 GPa, it is stable not only thermodynamically but also dynamically. This high-pressure stable phase of C2/m exhibits the expected superconductivity of Tc ~ 107 K at 230 GPa, which is obviously higher than those of conventional group-IV hydrides such as silane, germane and stannane. Noticeably, the coupling between group-IV element and hydrogen reduces with the increase of atomic number. Namely, the contribution of group-IV element to total EPC decreases in hydrides from 33% in SiH4(H2)223 to 25% in GeH4(H2)225 and then to 12% in PbH4(H2)2. On the contrary, the coupling among H2 molecules strengthens as mentioned above. Particularly, we want to point out that the Tc (~100 K) is comparable for SiH4(H2)2, GeH4(H2)2 and PbH4(H2)2 at the same Coulomb pseudopotential, though the superconducting mechanism is incompletely same. The intercalating H2 molecules into group-IV hydrides really improves the Tc. From the phonon contribution to EPC, we find that the intermediate-frequency phonon is dominated. Comparing with corresponding SiH48, GeH411 and SnH412, it is clear that the intercalation of H2 molecules results in the softening of intermediate-frequency phonon. As increasing the content of hydrogen in group-IV elements, it results in enhancing the EPC strength that is dominated by the coupling of the H2 molecular in the AH4(H2)2 (A = Si, Ge, Sn and Pb) crystals. This is just the origin of higher Tc in H2-containing compounds. Furthermore, we infer that the higher Tc may be obtained if more H2 are inserted in group-IV hydrides. Actually, more future works are needed to advance the Tc and understand the superconductivity.
As a comparison, the high-pressure structure of PbH4(H2)2 is visibly different from other hydrogen-rich compounds with high Tc, such as CaH637 and (H2S)2H232. In high-pressure structures of CaH6 and (H2S)2H2, the H2 quasi-molecules have been broken, with the strong bonds forming between metal and hydrogen atoms. Although the EPC is mainly contributed by hydrogen, the superconducting mechanism is different. It is the H-H coupling in CaH6 and (H2S)2H2, while the H2-H2 coupling in PbH4(H2)2. It is interested that the H2 quasi-molecule form keeps all along at thus high pressure up to 400 GPa. At the same time, Pb is one of the heaviest elements. The combination with the lightest H is one of the most important physical problems in high-pressure research. Pb metal makes the metallization pressure of hydrogen-rich compound decrease. Remarkably, the decomposition pressure point (133 GPa) of PbH4(H2)2 is the lowest among these H2-containing compounds of Si-, Ge- and Pb-based. This value is much lower than the metallization pressure of bulk molecular hydrogen, which indicates the feasibility to experimentally observe. Hence, Pb-based hydrides are the potential candidates as high-Tc superconductors. Our finding may hopefully stimulate the potential high-Tc superconductors research in H2-containing hydrides.
Methods
The search for crystalline structures of PbH4(H2)2 phases was performed using particle swarm optimization methodology as implemented in the CALYPSO program38,39. Structural optimizations, enthalpies and electronic structures were calculated using the Vienna ab initio simulation (VASP) program40,41 and projector-augmented plane wave (PAW) potentials employing the Perdew-Burke-Ernzerhof (PBE) functional42. The 1s1 and 6s26p2 electrons were included in the valence space for H and Pb atoms, respectively. For the plane-wave basis-set expansion, an energy cutoff of 800 eV was used. Dense k-point meshes were employed to sample the first Brillouin zone (BZ) and ensured that energies converged to within 1 meV/atom. All forces acting on atoms were converged 0.001 eV/Å or less and the total stress tensor was reduced to the order of 0.01 GPa. With the noteworthy mass ratio 207:1 between Pb and H, we have involved the spin-orbit effect in this calculation.
Based on the optimized structures from VASP, lattice dynamics and superconducting properties were calculated using density functional perturbation theory43 and the Troullier-Martins norm-conserving potentials44, as implemented in the QUANTUMESPRESSO code45. The cutoff energies of 60 and 400 Ry were used for wave functions and charge densities, respectively. 12 × 12 × 8 Monkhorst-Pack k-point grid with Gaussian smearing of 0.03 Ry was used for the phonon calculations at 3 × 3 × 2 q-point mesh and double k-point grid was used in the calculation of the electron-phonon interaction matrix element.
Additional Information
How to cite this article: Cheng, Y. et al. Pressure-induced superconductivity in H2-containing hydride PbH4(H2)2. Sci. Rep. 5, 16475; doi: 10.1038/srep16475 (2015).
References
Ashcroft, N. W. Metallic Hydrogen: A High-Temperature Superconductor? Phys. Rev. Lett. 21, 1748 (1968).
Narayana, C., Luo, H., Orloff, J. & Ruoff, A. L. Solid hydrogen at 342 GPa: no evidence for an alkali metal. Nature 393, 46–49 (1998).
Ashcroft, N. W. Hydrogen Dominant Metallic Alloys: High Temperature Superconductors? Phys. Rev. Lett. 92, 187002 (2004).
Martinez-Canales, M. & Bergara, A. No evidence of metallic methane at high pressure. High Pressure Res. 26, 369–375 (2006).
Eremets, M. I., Trojan, I. A., Medvedev, S. A., Tse, J. S. & Yao, Y. Superconductivity in Hydrogen Dominant Materials: Silane. Science 319, 1506–1509 (2008).
Zhou, X. F. et al. Superconducting high-pressure phase of platinum hydride from first principles. Phys. Rev. B 84, 054543 (2011).
Kim, D. Y., Scheicher, R. H., Pickard, C. J., Needs, R. J. & Ahuja, R. Predicted Formation of Superconducting Platinum-Hydride Crystals under Pressure in the Presence of Molecular Hydrogen. Phys. Rev. Lett. 107, 117002 (2011).
Martinez-Canales, M. et al. Novel Structures and Superconductivity of Silane under Pressure. Phys. Rev. Lett. 102, 087005 (2009).
Zhang, C. et al. Superconductivity in Hydrogen-rich Material: GeH4 . J. Supercond. Nov. Magn. 23, 717–719 (2010).
Zhang, C. et al. Structural transitions of solid germane under pressure. Europhys. Lett. 90, 66006 (2010).
Gao, G. et al. Superconducting High Pressure Phase of Germane. Phys. Rev. Lett. 101, 107002 (2008).
Tse, J. S., Yao, Y. & Tanaka, K. Novel Superconductivity in Metallic SnH4 under High Pressure. Phys. Rev. Lett. 98, 117004 (2007).
Somayazulu, M. S., Finger, L. W., Hemley, R. J. & Mao, H. K. High-Pressure Compounds in Methane-Hydrogen Mixtures. Science 271, 1400–1402 (1996).
Wang, S., Mao, H. K., Chen, X. J. & Mao, W. L. High pressure chemistry in the H2-SiH4 system. Proc. Natl. Acad. Sci. USA 106, 14763–14767 (2009).
Strobel, T. A., Somayazulu, M. & Hemley, R. J. Novel Pressure-Induced Interactions in Silane-Hydrogen. Phys. Rev. Lett. 103, 065701 (2009).
Yao, Y. & Klug, D. D. Silane plus molecular hydrogen as a possible pathway to metallic hydrogen. Proc. Natl. Acad. Sci. USA 107, 20893–20898 (2010).
Yim, W. L., Tse, J. S. & Iitaka, T. Pressure-Induced Intermolecular Interactions in Crystalline Silane-Hydrogen. Phys. Rev. Lett. 105, 215501 (2010).
Li, Y., Gao, G., Li, Q., Ma, Y. & Zou, G. Orientationally disordered H2 in the high-pressure van der Waals compound SiH4(H2)2 . Phys. Rev. B 82, 064104 (2010).
Chen, X. Q., Wang, S., Mao, W. L. & Fu, C. L. Pressure-induced behavior of the hydrogen-dominant compound SiH4(H2)2 from first-principles calculations. Phys. Rev. B 82, 104115 (2010).
Michel, K., Liu, Y. & Ozolins, V. Atomic structure and SiH4-H2 interactions of SiH4(H2)2 from first principles. Phys. Rev. B 82, 174103 (2010).
Shanavas, K. V., Poswal, H. K. & Sharma, S. M. First principles calculations on the effect of pressure on SiH4(H2)2 . Solid State Commun. 152, 873–877 (2012).
Wei, Y. K. et al. Pressure induced metallization of SiH4(H2)2 via first-principles calculations. Comp. Mater. Sci. 88, 116–123 (2014).
Li, Y. et al. Superconductivity at ~100 K in dense SiH4(H2)2 predicted by first principles. Proc. Natl. Acad. Sci. USA 107, 15708–15711 (2010).
Strobel, T. A., Chen, X. J., Somayazulu, M. & Hemley, R. J. Vibrational dynamics, intermolecular interactions and compound formation in GeH4-H2 under pressure. J. Chem. Phys. 133, 164512 (2010).
Zhong, G. et al. Structural, Electronic, Dynamical and Superconducting Properties in Dense GeH4(H2)2 . J. Phys. Chem. C 116, 5225–5234 (2012).
Zhong, G. et al. Superconductivity in GeH4(H2)2 above 220 GPa high-pressure. Physica B 410, 90–92 (2013).
Desclaux, J. P. & Pyykkö, P. Relativistic and non-relativistic Hartree-Fock one-centre expansion calculations for the series CH4 to PbH4 within the spherical approximation. Chem. Phys. Lett. 29, 534 (1974).
Pyykkö, P. & Desclaux, J. P. Dirac-Fock one-centre calculations show (114)H4 to resemble PbH4 . Nature 266, 336–337 (1977).
Wang, X. & Andrews, L. Infrared spectra of group 14 hydrides in solid hydrogen: experimental observation of PbH4, Pb2H2 and Pb2H4 . J. Am. Chem. Soc. 125, 6581–6587 (2003).
Krivtsun, V. M., Kuritsyn, Y. A. & Snegirev, E. P. Observation of IR absorption spectra of the unstable PbH4 molecule. Opt. Spectrosc. 86, 686–691 (1999).
Zaleski-Ejgierd, P., Hoffmann, R. & Ashcroft, N. W. High Pressure Stabilization and Emergent Forms of PbH4 . Phys. Rev. Lett. 107, 037002 (2011).
Duan, D. et al. Pressure-induced metallization of dense (H2S)2H2 with high-Tc superconductivity. Sci. Rep. 4, 6968 (2014).
Liu, A. Y., Garca, A., Cohen, M. L., Godwal, B. K. & Jeanloz, R. Theory of high-pressure phases of Pb. Phys. Rev. B 43, 1795 (1991).
Pickard, C. J. & Needs, R. J. Structure of phase III of solid hydrogen. Nat. Phys. 3, 473–476 (2007).
Ma, Y. & Tse, J. S. Ab initio determination of crystal lattice constants and thermal expansion for germanium isotopes. Solid State Commun. 143, 161–165 (2007).
Allen, P. B. & Dynes, R. C Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 12, 905 (1975).
Wang, H., Tse, J. S., Tanaka, K., Iitaka, T. & Ma, Y. Superconductive sodalite-like clathrate calcium hydride at high pressures. Proc. Natl. Acad. Sci. USA 109, 6463–6466 (2012).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 82, 094116 (2010).
Wang, Y., Lv, J., Zhu, L. & Ma, Y. CALYPSO: A Method for Crystal Structure Prediction. Comput. Phys. Commun. 183, 2063 (2012).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865 (1996).
Baroni, S., de Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Troullier, N. & Martins, J. L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 43, 1993 (1991).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Acknowledgements
The work was supported by the National Basic Research Program of China (973 Program) under Grant no. 2012CB933700, the Natural Science Foundation of China (Grant nos 11274335, 61274093, U1230202 and 91230203), the Shenzhen Basic Research Grant (Nos KQC201109050091A and JCYJ20120617151835515) and the Dong Guan foundation under Grant no. 2014509121212.
Author information
Authors and Affiliations
Contributions
G.H.Z. and C.L.Y. designed research. Y.C., C.Z. and T.T.W. performed research. Y.C., C.Z., T.T.W., C.L.Y., G.H.Z., X.J.C. and H.Q.L. analyzed the results. Y.C., C.L.Y. and G.H.Z. wrote the first draft of the paper and all authors contributed to revisions. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Cheng, Y., Zhang, C., Wang, T. et al. Pressure-induced superconductivity in H2-containing hydride PbH4(H2)2. Sci Rep 5, 16475 (2015). https://doi.org/10.1038/srep16475
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep16475
This article is cited by
-
Enhanced superconducting transition temperature via alloying In, Sn and Sb in PbH4 by using first-principles calculations
Journal of Materials Science (2023)
-
Structure, charge transfer, and superconductivity of M-doped phenanthrene (M = Al, Ga, and In): A comparative study of K-doped cases
Science China Physics, Mechanics & Astronomy (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
