
Abstract
P-glycoprotein (Pgp) is an ATP-binding cassette (ABC) transporter that alternates between inward- and outward-facing conformations to capture and force substrates out of cells like a peristaltic pump. The high degree of similarity in outward-facing structures across evolution of ABC transporters allowed construction of a high-confidence outward-facing Pgp atomic model based on crystal structures of outward-facing Sav1866 and inward-facing Pgp. The model adhered to previous experimentally determined secondary- and tertiary- configurations during all-atom molecular dynamics simulations in the presence or absence of MgATP. Three long lasting (>100 ns) meta-stable states were apparent in the presence of MgATP revealing new insights into alternating access. The two ATP-binding pockets are highly asymmetric resulting in differential control of overall structural dynamics and allosteric regulation of the drug-binding pocket. Equilibrated Pgp has a considerably different electrostatic profile compared to Sav1866 that implicates significant kinetic and thermodynamic differences in transport mechanisms.
Similar content being viewed by others


Introduction
P-glycoprotein (Pgp/ABCB1) is an ATP-binding cassette (ABC) transporter1 and perhaps the most promiscuous drug pump known in nature. The function of Pgp has been likened to a hydrophobic "vacuum cleaner"2 in which it intercepts small molecules within the inner leaflet of the lipid bilayer and forces them to the outer leaflet or even directly out of cells. Pgp provides a critical function in protecting cells from herbicides and other xenotoxins3,4, but in the case of chemotherapies, Pgp can be a major cause of multidrug resistance4. Pgp recognizes an extraordinarily large number of diverse molecules. To date, thousands of chemically different and therapeutically important substrates have been identified with molecular weights ranging from 360 dalton (aldosterone), ~1.2 kD (valinomycin and cyclosporin A) and even the 4 kD amyloid peptide5,6,7,8,9,10,11. The original x-ray structure of mouse Pgp revealed inward-facing conformations with drugs bound12 and the atomic models recently have been improved in protein geometry and Ramachandran favorability13. As with all ABC membrane transporters, Pgp has two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs), which contain the ABC signature motif. The NBDs provide the power stroke of the pump by utilizing the energy from ATP-binding and hydrolysis to catalyze the translocation of substrates through the TMDs and across the lipid bilayer. The complete transport cycle of Pgp is known to include large conformational changes that couple the drug-binding sites in the TMDs14,15 to the dimerization of the NBDs upon binding nucleotides16,17.
In addition to rigorous biochemical characterization of the transport cycle, crystal structures of bacterial exporters and Pgp provided snapshots of the extremes of the catalytic cycle: higher affinity substrate binding (inward-facing conformation) versus nucleotide-bound (outward-facing conformation) having a lower affinity for substrates. These endpoints of drug transporter conformation dynamics embody the essence of the "alternating access" mechanism originally proposed nearly 50 years ago18. Pgp crystallized in an inward-facing conformation in which the two NBDs are disengaged, i.e. the interface is broken, allowing an opening of the TMDs to the lipid bilayer for drug entry into the internal drug-binding cavity12,13. Two slightly different inward-facing ATP-free Pgp conformations were apparent in the asymmetric unit of the original crystal structures, revealing structural degrees of freedom in the absence of an NBD interface. Related efflux ABC transporters captured in outward-facing conformations demonstrate much stronger structural constraint in the form of NBD dimerization in the presence of bound nucleotides. It is a foregone conclusion that Pgp will adopt a well-defined and constrained outward-facing conformation in the presence of nucleotide, but such a structure has not yet been described in atomistic terms.
Details of how substrates trigger conformational change from inward-facing to outward-facing, how ATP-binding results in reduced affinity for substrates and how transport is "peristaltic" - i.e. drugs do not slip backwards and evade the efflux mechanism - are difficult to discern from static crystal structures alone. Furthermore, structures of both inward- and outward-facing conformations for the same drug pump are not yet available, precluding computational molecular dynamics (MD) studies of the full transport cycle. In order to better understand these mechanisms in atomic detail, we have produced a high-confidence homology model of mouse Pgp in an outward-facing conformation. We used a modeling approach based on the crystal structure of outward-facing Sav1866 (PDB: 2ONJ19) bound to ANP and our recently refined inward-facing crystal structure of mouse Pgp (PDB: 4M1M13). Our approach avoids the risk of incorrect secondary structure assignment associated with building models solely from primary sequence phylogeny and secondary structure prediction. Rather, we used the corrected inward-facing Pgp crystal structure to guide the placement of secondary structure boundaries on a mutated Sav1866 template. The tertiary inter-domain packing and helix registry of the outward-facing Pgp initial model was validated by previous cysteine-scanning and disulfide crosslinking experiments20,21,22,23,24,25,26,27,28,29.
Four groups have described and characterized homology models of the outward-facing conformation of human Pgp using Sav1866 as the starting template for model building30,31,32,33. The focus of all four modeling efforts was primarily to analyze the outward-facing conformation or the effect of conformational change on the drug-binding pocket. Little emphasis was placed on the allosteric effect of ATP on Pgp conformational dynamics. During our model building, particular attention was paid to achieving a starting model with two Mg-ATP molecules bound to the canonical nucleotide binding sites. We used this high-confidence initial Pgp model in extensive all-atom MD simulations to probe inter-domain interactions, roles of ATP and to ascertain critical residues functioning in structural dynamics of outward-facing Pgp. Asymmetric dynamic features between two "halves" of mouse Pgp were governed by the ATP-binding sites. Three distinct meta-stable conformations of Pgp-MgATP were identified. Correlations between ATP-binding at the NBDs and conformational changes in TMDs were apparent. The simulations also shed light on allosteric effects of nucleotide binding and a “halting” mechanism in which Pgp adopts a non-hydrolyzable ATP-binding conformation to conserve ATP in the absence of drug. Electrostatic potential analysis suggests different resting states between Pgp and Sav1866, offering a plausible explanation for different thermodynamic equilibria in trapping conformations for crystallization.
Results and Discussion
Secondary and tertiary structure validity
Protein secondary structure generally remains unchanged during conformational cycling of enzymes under physiological condition34 to avoid unfolding or misfolding during catalytic cycling. ABC transporters are expected to have a highly stable secondary structure throughout conformational dynamics due to biophysical constraints of the bilayer and from what is already known about tertiary structure. Approximately 30% of Pgp is embedded in the membrane where the low dielectric field will tend to maintain alpha helices. Another ~40% of Pgp is represented as nucleotide binding domains (NBDs), which are the most conserved domains in the ABC superfamily. Multiple crystal structures of ABC transporters show that the NBD secondary- and tertiary- structures do not significantly vary between inward- and outward- facing conformations. Furthermore, Pgp secondary structure does not change during its catalytic cycle based on experimental observations35,36,37. Unbiased all-atom MD simulations of the mouse Pgp homology model based on full equilibration and sufficient sampling size (Text S1 and Figure S1) showed completely stable secondary structure components across the entirety of six independent simulations including consistent folded helix and beta sheet components (Figure S2). The average secondary structure components based on the last 50 ns of trajectories (Figure S3) reveal that the total secondary structure components of Pgp are also conserved between orthologs and between any given conformation regardless of outward-facing (Sav1866 and homology model), inward-facing (mouse Pgp crystal structures) or intermediates (MD simulation end points). In our simulations, ~95% of the secondary structure remained essentially unchanged throughout conformational dynamics. The slight fluctuation was due to dynamic features of the protein in the connecting regions between two distinct secondary structure components due to fluctuating φ and ψ angles. These local regions of Pgp fluctuated between two or three dominant secondary structures in a highly dynamic manner (Figure S4).
Inter-TM packing and helix registry of outward-facing Pgp initial model and equilibrated models was validated by previous disulfide crosslinking experiments based on double cysteine mutation (5–7 Å Cu-phenanthroline spacer). The following pairs were previously observed to form disulfide bonds and are mapped to atomic structures in Figure 1: L328-L971 (L332-L975 in human Pgp) between TM6 and TM1220; V129-G935 (V133-G939 in human Pgp) and C133-A931 (C137-R935 in human Pgp) between TM2 and TM1121; N292-G770 (N296-G774 in human Pgp) and G296-F766 (G300-F770 in human Pgp) between TM5 and TM822; pairs between TM3 and TM9 as D173-N816 (D177-N820 in human Pgp)23 and L171-N816 (L175 and N820 in human Pgp)29. Most residue pairs maintain direct contact between inward-facing and outward-facing conformations except for the three conformation-sensitive pairs L328-L971, D173-N816 and L171-N816. The Cα distance of residue pair L328-L971 is 12.4 Å apart in the inward-facing crystal structure (PDB: 4M1M; Figure 1A) but is 19.8 Å in the outward-facing conformation due to the opening of the drug pocket to the outer leaflet (Figure 1B). The formation of a disulfide bond would lock the protein in the inward-facing conformation, which explains the inhibition of verapamil-stimulated ATPase activity20. The two Cα of D173-N816 are 19.9 Å apart in the inward-facing crystal structure (Figure 1A) but are much closer in the outward-facing model with a Cα distance of ~8.0 Å. The side chains are in direct contact in the outward-facing model preventing the two Cα from closer approach, but would favor disulfide bond formation when mutated to Cys-Cys and crosslinked with Cu-phenanthroline as previously described23 (Figure 1B). The equilibrated structure of Pgp-MgATP showed little change in the pairwise distances except for L328-L971 due to partial closing of ECL1-ECL3.

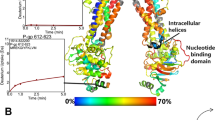
Residue pairs observed in cross-linking experiments20,21,22,23.
Inward-facing crystal structure (PDB: 4M1M-A) (A), initial structure of outward-facing Pgp (B) and equilibrated outward-facing Pgp with MgATP (C) are shown. Pgp is shown as cartoon representation with half1 in pink and half2 in ice blue. The highlighted residues are drawn as spheres in various colors: L328-L971 (L332-L975 in human Pgp) in red, V129-G935 (V133-G939 in human Pgp) and C133-A931 (C137-R935 in human Pgp) in gray, N292-G770 (N296-G774 in human Pgp) and G296-F766 (G300-F770 in human Pgp) in green and D173-N816 (D177-N820 in human Pgp) in blue. MgATP is drawn in spheres colored by elements as H in white, N in blue, C in cyan, O in red and P in tan. In the satellite panels of (C), Cα of highlighted residues are drawn as spheres and side chains are drawn as licorice colored by elements. Cα distances are labeled in black dash. The distances in A and B are single measurements and the distances in C are averages of the last 50 ns equilibrate trajectories over triplicate simulations.
Other crosslinks, including various pairs between TM1-TM1124, maintain similar positions between the inward-facing and outward-facing conformation and slightly favor the outward-facing conformation. Several cross-linked pairs at TM4/TM5-TM12 and at TM6-TM10/TM1125 are also compatible with the inward-facing crystal structures but are prevented in the outward-facing conformation, explaining inhibition of verapamil-stimulated ATPase activity when crosslinked. In addition, various pairwise positions in the drug-binding cavity between TM6, TM11 and TM12 are also compatible with both inward-facing and outward-facing structure26. These structural features reveal the registry and rotation of the helices in the outward-facing structures are in strong agreement with existing crosslinking data. We conclude from crosslinking experiments and the structural comparison between inward-facing and outward-facing conformations that conformational changes during drug transport are governed primarily by shifts in inter-domain distances between TM6-TM12 and TM3-TM9 whereas other TMs maintain relatively fixed inter-domain packing.
Asymmetric features of mouse Pgp structure and dynamics
In contrast to the symmetric Sav1866 template, mouse Pgp has evolved with a high degree of asymmetry between its two "halves", which share only 59.4% sequence similarity. More specifically, 77.3% similarity lies between two NBDs and only 43.5% similarity between two halves of TMDs (Figure 2). The differences in the primary sequence result in the asymmetry of chemical properties between pseudo symmetric residues and patches. In general, charged residues (K, R, E, D) are highly hydrophilic with the largest entropy profiles whereas hydrophobic residues prefer to pack together and stabilize each other in the aqueous phase. Therefore, the analysis of different chemical properties based on the residue types of each region can help explain their different dynamics (Figure 2). Specifically, TM1 and TM2 (Figure 2 A) are 5-residues longer and with more charged residues than their pseudo symmetric domains TM6 and TM7 (Figure 2 A′). The intracellular end of TM3 (Figure 2B) has more charged residues than TM9 (Figure 2 B′). These features render larger mobility of half1 than half2 at TMDs. Even though the ATP-binding residues are conserved between site 1 and site 2 (Figure 3), their surrounding residues are substantially different. WA1 and WB1 are surrounded by more charged residues (Figure 2 D and E) than WA2 and WB2 (Figure 2 D′and E′). Whereas LSGGQ2 is surrouded by more charged residues (Figure 2 C and F′) than that of LSGGQ1 (Figure 2 C′and F). In sum, our observations reveal a more hydrophilic site 1 (WA1 + WB1 + LSGGQ2) than site 2 (WA2 + WB2 + LSGGQ1) and indicate different thermodynamics and kinetics between the two sites, consistent with multiple findings in the literature37,38,39. A previous MD study40 was performed using the inward-facing conformation with ATP bound at each of the Walker A motifs but with both LSGGQ motifs completely dissociated. Even though the study was done at the near-opposite conformation and different ATP binding environment compared to our study, asymmetric structural dynamics between two NBDs was also observed. The asymmetry between the two halves of Pgp must therefore play a major role in the overall function and mechanism of the transporter.

Comparison of the pseudo-symmetric halves of mouse-Pgp.
Structures of the four parts (two TMDs and two NBDs) of the mouse-Pgp are separately demonstrated in parallel for comparison. The structures are drawn in cartoon: helices as tubes, beta-sheets as arrows, turn and coil as wires. The residues are colored by residue type: non-polar residues in white, polar-residues in green, basic residues in blue and acidic residues in red. Psuedosysmetric regions with distinctive residue type composition are in squares for Half1 (A-F) and Half2 (A′-F′).

Pgp-MgATP Contact Map.
H-bonds are shown as black lines, aromatic interaction is as orange lines and coordination bonds are as aqua blue lines. Amino acid numbering corresponds to mouse Pgp.
The root-means-square fluctuation (RMSF) (Figure 4) reveals asymmetric dynamics of apo-Pgp in equilibrated states (Figure 4, blue line). The results suggested that MgATP-binding reduced flexibility and introduced more symmetry to the protein dynamics (Figure 4, red line). In general, apo-Pgp has larger RMSF than Pgp-MgATP across all regions. Certain locations are particularly noteworthy: ECL1 (Figure 4, peak A) has the largest RMSF of the entire structure for both apo-Pgp (3.5 Å) and Pgp-MgATP (2.8 Å). ECL4 (Figure 4, peak A′) has the biggest RMSF of the TMs in half2. In apo-Pgp, ATP-binding site 1 (Figure 4 peak B, peak C, peak D, peak E′ and peak F) is more flexible than site 2 (Figure 4 peak B′, peak C′, peak D′, peak E and peak F′). Specifically, MgATP-binding reduces the flexibility of all the ATP-binding signature motifs at NBDs: Walker A (WA1: G423NSGCGK429; WA2: G1067SSGCGK1072), Walker B (WB1:I547LLLDE552; WB2:I1192LLLDE1197), LSGGQ (LSGGQ 1: L527SGGQ531; LSGGQ2: L1172SGGQ1176), A-loop (A-loop1: Y397PSR400; A-loop2: Y1040PTR1043). In addition, ATP-binding also reduced the flexibility of TMDs at various levels, which reveals an allosteric effect of nucleotide binding to the TMDs.

Root-mean-square fluctuation (RMSF) per residue at equilibrium.
The RMSFs are measured using the Cα of each residue from trajectories of the last 50 ns simulations and are averaged over triplicate simulations for both apo-Pgp in blue and Pgp-MgATP in red. 2D cartoon representatives of the secondary structures are in pink. Domains are shown in the same sequence order as that of the RMSF (helix as spring, coil and turn as straight line, beta sheet as arrow). The Figures of the two pseudo-symmetric halves (half1: residue 40-626, half2: residue 688-1271) are displayed in parallel for comparison. Labeled regions are region A (ECL1), region A′ (ECL4), region B (IH1), region B′ (IH3), region C (A-loop1), region C′ (A-loop2), region D (WA1), region D′ (WA2), region E (LSGGQ1), region E′ (LSGGQ2), region F (WB1), region F′ (WB2). In apo-Pgp, ATP-binding site 1(B + C + D + E′ + F) is more flexible than site 2 (B′ + C′ + D′ + E + F′).
Three distinctive meta-stable states of ATP-binding
While MD is a powerful technique for probing all-atom contributions to protein function, only nanosecond to microsecond timeframes are currently feasible for a system as large as Pgp (~209,000 atoms). On the order of millisecond to second timescales would be required to observe a full catalytic cycle for an active transporter. In addition, MD is largely classical in mechanics as opposed to quantum and therefore hydrolysis of chemical bonds, such as those in ATP and the resulting release of energy, is not currently feasible to simulate by MD. Taken together, there are limitations in analyzing Pgp function by MD. However, MD simulations can access information in a small window of the catalytic cycle and provide value biochemical insights. In this work, we started with a scenario in which dual ATP-bound outward-facing Pgp was simulated by MD and we observed the sequential events when ATP was removed from the system. ATP-binding represents one of the most conserved properties of all ABC transporters. An ATP-interaction map, based on the MD simulations is shown in Figure 3. All the binding residues are conserved between site 1 and site 2. The ATP-Mg2+ binds to mouse Pgp as in Figure S5. The H-bonds and coordination bonds started in the range between 1.63–2.20 Å in the initial structure (2.5 Å cutoff was used as the definition). All bonding interactions between ATP-Mg2+ and Walker A/Walker B stayed within the cutoff with remarkable stability but the interactions between ATP and LSGGQ motifs resulted in distinct meta-states.
A previous study33 used steered MD (SMD) to probe the effect of conformational change on drug-binding. In this case, the two ATP binding sites were equilibrated separately and the ATP site in NBD1 included an engaged E556 residue (mouse-Pgp E552) whereas ATP binding at NBD2 had a disengaged E1201 residue (mouse Pgp E1197). A skewed NBD orientation was observed in both cases in their study. We note that SMD requires placing restraints on the protein and the application of bias forces in order to cause a relatively fast conformational change within a short timeframe. We describe the first unrestrained (unbiased) MD study of outward-facing Pgp in which both Mg-ATP were placed at canonical binding sites and equilibrated simultaneously. We found that both E552-Mg2+ and E1197-Mg2+ were engaged with nucleotide throughout the entire duration of all simulations. The interactions are very strong, since both glutamates were associated with Mg2+ as bidentate clamps. Taken together, double-occlusion of two ATPs help stabilize an NBD sandwich configuration (proper NBD alignment) prior to a hydrolysis event.
In order to better demonstrate the conformational effects in NBDs upon MgATP binding, Cα distance between the pairs K429-S1173 (site 1) and K1072-S528 (site 2) were monitored over time (Figure 5). In apo-Pgp, both ATP-binding site 1 and site 2 (Figure 5A and C) demonstrated constant fluctuations between these residue pairs. However, site 1 showed a larger flexibility than that of site 2. The residue distance at site 1 also increased up to 20 Å whereas the residue distance at site 2 remained below 12 Å. A larger separation of NBDs at site 1 compared to site 2 was a consistently observed feature. On the other hand, MgATP-binding markedly stabilized the residue pairs at both site 1 (Figure 5B) and site 2 (Figure 5D) with nearly no fluctuation observed. Three distinct meta-stable states were revealed at the binding regions (Figure 5 E-G).

Effect of ATP on conformation dynamics and stability.
The MgATP, K429/K1072 (2xWalker A) and S1173/S528 (2xLSGGQ) are drawn in licorice with H in white, N in blue, C in cyan, O in red, P in gold and Mg2+ in pink. The Cα of the ATP binding residues are drawn as green spheres, pairwise distances are drawn in dashed lines (E-G). H-bonds <2.5 Å are in solid black lines and H-bonds >2.5 Å are in dashed lines (E1, E2, F1, F2, G1 and G2). Orange arrows point to the directions of conformational change (F1, F2 and G1). The protein is shown in cartoon with half1 in pink and half2 in ice blue. (A) site 1 residue pair K429(WA)-S1173(LSGGQ) and (C) site 2 residue pair K1072(WA)-S528(LSGGQ) from the three apo-Pgp simulations. The same residue pairs in the three simulations of Pgp-MgATP are shown in (B) (site 1) and (D) (site 2). Conformation 1, “double-tight occluded” (E) (equilibrated structure from run3) consists of ATP in a tight sandwich at site 1 (E1) and site 2 (E2) with the K-S distance of ~9.4 Å. Conformation 2, “double-loose occluded” (F) (equilibrated structure from run2) is at loose sandwich between Walker A and LSGGQ with the K-S distance of ~11.1 Å due to the shift of ATP(Oγ)-S528/S1173(HG) to ATP(Oα)-S528/S1173(HG) at both site (F1, F2). This conformation has a slightly tilted Walker A to the intracellular direction and tilted LSGGQ to the membrane direction for both sites. Conformation 3, “loose-tight occluded” (G) (equilibrated structure from run1) adopts detached LSGGQ on site 1 (G1) with a K-S distance of 12.2 Å and the ATP locked site 2 (G2) with a K-S distance of ~9.4 Å.
Previous work has characterized alternating access mechanisms of ATP-binding and hydrolysis of Pgp37,38,41. Our MD simulations support these observations but also provide atomistic details of several different proposed meta-stable occluded nucleotide-bound states of Pgp. The simulations of Pgp-MgATP may represent catalytic states previously characterized38,41 in which, in the absence of substrate, the complex adopts either a high affinity "double-tight occluded" resting state (Figure 5 E-E2) or a low affinity "double-loose occluded" resting state (Figure 5 F-F2) with both nucleotides bound. These kinetic studies also showed that drug-binding facilitates the ATP-binding pocket shift from either resting state to a "loose-tight occluded" transition state in which only one ATP is tightly bound (Figure 5 G-G2). In "double-loose occluded", both LSGGQ motifs in each ATP-binding site shift the binding from the Oγ of ATP to the Oα that are shared by both ATP and ADP. A previous biochemical experiment shows that the mutation of S528A and S1173A together completely disabled the ATPase activity of mouse-Pgp39. The binding of ATP(Oγ)-S528/S1173(HG) is required for ATP hydrolysis but this bond is not formed in "double-loose occluded" state. The non-hydrolyzable "double-loose occluded" state may implicate an ATP conserving or “halting” mechanism which could prevent ATP hydrolysis in the absence of drug.
The so-called "basal" ATPase activity of Pgp is routinely observed in the absence of Pgp substrates. However, any ATPase activity of Pgp requires the presence of phospholipids42, which are present in the detergent "mixed" micelles surrounding purified Pgp preparations or when Pgp is reconstituted into liposomes. The lipids, in principle, would thus have access to the hydrophobic cavity of Pgp as with other substrates. Indeed, a previous study43 has shown that lipids, especially short chain phosphatidyl cholines, are transported by Pgp. It is highly likely therefore, that "basal" ATPase activity is a normal consequence of occasional lipid flippase activity of Pgp occurring in detergent micelles, liposomes or the cell membrane. In this case, lipid and drug substrate could cause similar structural dynamic effects on Pgp if they gain full access to the substrate-binding pocket. We therefore propose that partial intrusion of lipids as seen in our simulations prompt less favorable NBD alignment compared to proper binding of a substrates. This model could be therefore described as a "primed" state, which allows Pgp longer time to sample higher-affinity inward-facing conformations for completely enveloping substrates prior to collapse and transport.
The "loose-tight occluded" conformation (Figure 5 G-G2) is in agreement with an asymmetric state identified in a kinetic study38 in which one low affinity (Kd = 0.74 mM) site and one “occluded” high affinity (Kd = 6 μM) site appeared after the ejection of the drug. After drug ejection, the hydrolyzed site 1 is proposed to avoid adopting a conformation that will result in another ATP hydrolysis event before the protein resets to an inward-facing conformation. Inward-facing conformations are also more accessible to ATP exchange to prepare for another round of the drug transport cycle. The "loose-tight occluded" conformation is also in agreement with the previous kinetic studies37 in which site 2 demonstrated increased stability and decreased accessibility after site 1 is hydrolyzed during drug ejection. The equilibrated conformation of apo-Pgp from our simulation also suggested that hydrolysis of site 2 is crucial to the transformation of Pgp from the outward-facing conformation to the inward-facing conformation. We extended one apo-Pgp simulation to 400 ns, but no opening was observed at site 2, which suggested that extra thermal energy beyond background levels and static repulsion between ADP and Pi generated from ATP hydrolysis16 is likely required for site 2 to disengage and allow dissociation of NBDs.
Conformational change and allosteric regulation of ATP-binding
The conformational changes were monitored using residue pairs from pseudo-symmetric counterparts (Figure 6). Six different averaged pair distances at the “extracellular gate” in the six simulations demonstrated large randomness in this region (Figure 6A). Therefore, large flexibility and numerous meta-stable states with similar energy are implicated. In addition, it was also shown that the three residue pairs are strongly inter-correlated in distance patterns, which suggests that the opening/closing of the extracellular gate occurs simultaneously for all TMs. When compared to the initial structure, the A79-T736 pair distance markedly decreased for all simulations whereas the other pairs at the extracellular gate fluctuated within a small dynamic range from the initial distance (Figure 6 A, G and H). The simulations demonstrate that the dynamic structure of the extracellular gate oscillates between open and closed forms rather than a relatively fixed conformation.

Conformational change illustrated with landmark residues.
Landmark residue distances at each regions of the protein are measured using the average of the last 50 ns trajectories from each simulation. The protein is shown as cartoon representation with half1 in pink and half2 in ice blue. The Cα of the landmark residues are drawn as spheres in green (D-L). P atoms of lipid bi-layer are drawn in semi-transparent sphere in gold. MgATP is drawn in licorice with H in white, N in blue, C in cyan, O in red, P in gold and Mg2+ in pink. Orange arrows point to the directions of conformational change. The conformational changes are monitored using residue pair from two pseudo-symmetric counterparts: Extracellular gate (A) uses A79-T736 (ECL1-ECL3), G207-G850(TM3-TM9), G325-E968(ECL2-ECL4), drug binding pocket (B) uses higher end F71(TM1)-F728(TM7) pair and lower end Q343 (TM6)-Q986 (TM12) pair, ATP-binding sites (C) uses K429-S1173 (site 1) and K1072-S528 (site 2). The predominant conformation observed from all three runs of apo-Pgp (E, G, I and K) and predominant conformation observed from 30 ns-100 ns of run1, 30-50 ns of run2 and 30-200 ns of run3 of Pgp-MgATP (F, H, J and L) are different in various aspects.
In our simulations, the apo-Pgp adopts an “A” shape at the drug-binding pocket open towards intracellular direction (Figure 6I) whereas Pgp-MgATP adopts a “V” shape open towards extracellular direction (Figure 6J). The result indicates that MgATP-binding potentiates the outward-facing conformation by promoting the opening of TM1-TM7 at the higher end of drug-binding pocket and the closing of TM6-TM12 at the low drug-binding pocket. Even though these simulations did not include substrates, the ejection of drug would require a “V” shape of the drug-binding pocket and not a top blocked “A” shape as observed in the inward-facing conformation. In addition, a stronger correlation was observed in conformational change between site 1 (K429-S1173) and TMDs (Q343-Q986) than that of site 2 (K1072-S528) (Figure 6B and C). This suggests the possibility that the activity of site 1 is the dominant “thermal engine” while TM6 is the “transmission” for the conformational change of the drug-binding pocket.
In one study27, the crosslinking of the upper region of TMDs was affected by ATP binding at the NBDs. Specifically, ATP binding inhibited the crosslink of pairs of human Pgp between TM6 and TM12 at L339C-V982C (mouse L334-V978) and L332C-L975C (mouse L328-L971) but promoted the crosslink of F343C-V982C (mouse F339-V978). In our equilibrated trajectory, the Cα distance between F339-V978 (10.41 Å) is ~5 Å shorter than that of L334-V978 (15.5 Å) and L328-L971 (16.4 Å) pairs and therefore F339-V978 is more favored of disulfide formation than L334-V978 and L328-L971. The effect of ATP is to introduce and maintain an outward-facing conformation with a “V” shape at drug binding pocket that inhibits or promotes these crosslinks. Even though our simulations can not describe the inward-facing to outward-facing transition triggered by ATP binding, the difference of these three residue pairs are clearly maintained in the simulations in the presence of ATP and is in agreement with the experimental observation. The same group conducted crosslinking experiments between the WalkerA-LSGGQ motifs and observed increased ATP hydrolysis28. Since ATP hydrolysis requires a precise conformation of the WalkerA-LSGGQ sandwich, the crosslinking between the WalkerA-LSGGQ was suggested to essentially reduce the degrees of freedom for the NBDs enhancing alignment and hydrolysis. Our simulations showed a twisted (improper) NBD alignment in the absence of ATP binding (Figure 6K), which could indicate that a crosslink between the two NBDs assists the correction of a misalignment, favoring enhanced ATP binding. We did not simulate crosslinking per se, but our structure showed direct side-chain contact between the crosslinked pair LSGGQ (P517C) and Walker A (I1050C) (mouse P513-I1046) showing agreement between our homology model and the biochemical data.
In addition to ligand effects (drug-binding and ATP-binding/hydrolysis), water is a major driving force of protein conformational change by hydrophobic effect and hydrostatic pressure on both sides of the membrane. Solvent accessible surface area (SASA) is an indirect measurement for conformational change. For both models, SASA of TMDs decreases while the SASA of NBDs increases, which indicates that the closure of TMDs is coupled with the opening of NBDs. The results suggest water efflux from the TMDs and water influx into the NBDs due to the change of conformation in these areas (Figure 7). Such water flux is directly quantifiable using the change of SASA in percentage compared to that of initial structure. The results indicated that the efflux of water from the TMDs and the influx of water into the NBDs are strongly correlated processes. It also showed that the MgATP binding provided the counterforce in this process to maintain a different hydration distribution favoring the outward facing conformation as opposed to that of apo-Pgp, which favors the inward-facing conformation.

Solvent Accessible Surface Area (SASA).
The SASA are measured using the last 50 ns trajectories and averaged over the triplicate simulations for both apo-Pgp and Pgp-MgATP models. The changes of SASA are calculated against the initial structure. The apo-Pgp showed larger total SASA increase (3.0%) than Pgp-MgATP (0.6%). The apo-Pgp demonstrated larger decreases (−7.8% vs. −3.6%) in TMDs and larger increases in NBDs (11.3% vs. 4.3%) compared Pgp-MgATP.
Residue type and electrostatic potential analyses between Pgp and Sav1866
The Sav1866 crystal structure was used as the template for the initial structure of the outward-facing conformation of mouse Pgp. However, the homology modeling process was not complete until MD simulation produced thermodynamically favored conformations. This was due to the differences in the electrostatic and hydrophobic properties between Sav1866 and mouse Pgp. The partial closure of TMs was observed during equilibrium for all simulations for mouse-Pgp regardless of ATP-binding. Our starting structure based on Sav1866 is more likely to represent a high-energy short-lived transition state that is driven by ATP hydrolysis at the moment of drug ejection. Residue type analysis (Figure 8) and electrostatic potential (Figure S6) analysis on both mouse Pgp and Sav1866 explains why each protein favored inward-facing- and outward-facing- conformations, respectively, for crystallography. The analysis of residue type differences (Figure 8) reveal that only polar and non-polar residues are included in the drug-binding pocket (Figure 8A and C) of mouse Pgp whereas 9 pairs (18 total) of extra charged residues exist in the same region of Sav1866 (Figure 8B and D). The conterparts of the 18 charged residues in mouse Pgp are 4 aromatic residues, 5 hydrophobic residues and 9 neutral polar residues. The hydrophobicity of mouse Pgp in the drug-binding pocket is substantially larger than that of Sav1866. In addition, the position of the charged residues in Sav1866 would tend toward repulsion thereby promoting the outward-facing comformation with an open substrate pocket towards extracellular space (Figure 8D). The comparison in electrostatic potential shows an overall neutral region in mouse Pgp but highly charged region in Sav1866 at TMDs. In contrast, the interfaces between two NBDs in mouse Pgp (Figure S6 B1 and B2) are more charged than that of Sav1866 (Figure S6 B), which indicates more hydrophilic NBDs in mouse Pgp than Sav1866. Since the hydrophobic effect has a stronger attraction (~10 kcal/mol) than electrostatic interaction (~5 kcal/mol), mouse Pgp favors closed TMDs and open NBDs, whereas Sav1866 favors the exact opposite configurations. These differences imply that mouse Pgp and Sav1866 employ rather different transport mechanisms, particularly in regard to the role of water.

Residue type comparison between mouse P-pg and Sav1866 at TMDs.
Both proteins are drawn in cartoon: helices as tubes, beta-sheets as arrows, turn and coil as wires. The residues are colored by residue type: non-polar residues in white, polar-residues in green, basic residues in blue and acidic residues in red. There are only polar and non-polar residues in the drug-binding pocket (A) of mouse Pgp whereas there are 9 pairs (18 total) charged residues in the same region of Sav1866 (B). The counterpart residues in the mouse P-pg are shown in text blocks: mouse P-pg half1 in red, half2 in blue, Sav1866 both halves in green. These residues are drawn in spheres from top view (C) for mouse P-pg and (D) for Sav1866.
Methods
Structure construction of outward-facing conformation of Pgp
Rigorous atomic modeling of three-dimensional (3D) protein structure requires correct primary amino acid sequence alignment and accurate 3D structural predictions for the gap regions in the alignment. In our case, an additional challenge included a template with fewer amino acid residues compared to the homology sequence (a Sav1866 dimer consists of 1156 residues whereas mouse Pgp contains 1282 residues). Therefore, Pgp has an additional ~10% of 3D structure information that must be gleaned from sources other than the alignment. An automatic homology modeling method (e.g. MODELLER44) predicted unreasonable secondary structure for the gaps in the alignment. However, since the inward-facing conformation of mouse Pgp has been resolved, 3D structure corresponding to these gaps can be extended to the outward-facing conformation homology model prior to equilibration. In addition to combining both sequence and structural alignments, a rational homology modeling process should also carefully examine any effects of protein function manifested in differences between the template and the output structures. We used a rational and systematic approach of homology modeling to produce a plausible starting structure of the outward-facing conformation of mouse Pgp.
a. Partial modeling based on sequence alignment on outward-facing Sav1866 crystal structure
Primary amino acid sequence alignments were generated using the EMBOSS Needle pairwise alignment tool at the EMBL (https://www.ebi.ac.uk/Tools/psa/emboss_needle) using the PAM500 similarity matrix, a gap opening penalty of 50 and a gap extension penalty of 10 (Figure S7). To eliminate skewed alignments resulting from the Pgp linker region, a mouse Pgp monomer (1–1276) with linker omitted (residues 626–692) was aligned to two tandem copies of Sav1866 monomer with two C-terminal residues (577 and 578) removed from the first copy (i.e. 1–576 and 1–578). The resulting alignment revealed an extended Pgp N-terminus (1–47) that has no counterpart in Sav1866. These residues, including four C-terminal mouse Pgp residues (1273–1276) were trimmed and the alignment was regenerated using the same parameters (Figure S7A). The final alignment revealed only four single and one double amino acid insertion for mouse Pgp and only one double insertion for Sav1866. Secondary structure obtained from both the Sav1866 crystal structure (PDB: 2ONJ) and mouse Pgp crystal structure (PDB: 4M1M) was overlaid above/below the respective amino acid sequence in the alignment (Figure S7A). The procedure revealed an impressive positional conservation of secondary structure between the bacterial- and mammalian- transporters, despite the very different protein conformations represented in the crystal structures. Exceptions in the form of secondary structure mismatches include mouse Pgp first transmembrane domain (TM1) and first extracellular loop (ECL1; residues 1–106), mouse Pgp ECL4 (residues 692 to 747) and Sav1866 TM1/ECL1 (residues 1–59). The final alignment (transporter core), using the same weighting parameters and omitting the secondary structure mismatches, resulted in a good overall amino acid sequence identity of 30%, a high degree of amino acid similarity of 63% and unchanged positions of the insertions/deletions.
b. Model finalization using structural alignment on inward-facing Pgp crystal structure
Six gaps consisted of Pgp residues (Phe374, Pro398, Tyr1017, Pro1044, Ser1137 and Tyr1138) that were not represented in Sav1866, but all reside in coil loops that could easily accommodate the addition of each in their respective tertiary structure. The remaining two gaps were located in Pgp intracellular helix 1 (IH1) which was modeled to preserve the positioning of the conserved double aromatic motif (IH1: Trp158 and Phe159 and IH3: Trp799 and Phe800) present in the pseudo-symmetric counterpart IH3 whereas Asp801 and Asp802 was not modeled. The alignment was used as input to a PyMOL script that mutated the Sav1866 core (with TM1 and ECL1 removed) one residue at a time to Pgp side chains, taking advantage of optimal choice of side chain rotamers within PyMOL to minimize clashes. The homology structure has 0.78 Å α-carbon RMSD from the template structure of Sav1866 (Figure S7B). The missing residues and fragments were then truncated into the homology structure using fragments of the inward-facing conformation (PDB: 4M1M-A). Structure fragments of residue 30 to 107 (TM1), residue 681 to 747 (TM7) were added without disturbing the integrity of the connecting helices. In order to achieve maximum local structural alignment between the outward-facing homology model and the inward-facing crystal structure, loops corresponding to missing residues were replaced using the inward-facing crystal structure as the structural template. Specifically, fragment 368 to 385 in crystal structure (PDB: 4M1M) was aligned to the region with missing residue 374 and the same procedure was applied to the following: fragment 390 to 410 for missing residue 398, fragment 1009 to 1032 for missing residue 1017, fragment 793 to 808 for missing residues 801, 802, fragment 1034 to 1054 for missing residue 1044 and fragment 1129 to 1139 for missing residue 1137, 1138 (Figure S8). The rationale was to use the inward-facing crystal structures to model these gaps by transferring the corresponding secondary structure to the outward-facing homology model (Figure S9). In contrast, using automatic homology modeling program MODELLER generated incorrect structure folding in these regions (Figure S10).
c. Structure optimization and validation
The structure was then processed using Phenix to optimize protein geometry and Ramachandran favorability. The final protein geometry was evaluated using the MolProbity server, which reported 3.27% poor rotamers, 0.68% Ramachandran outliers, 95.4% Ramachandran favored, 0% Cβ deviations (>0.25 Å), 0% bad backbone bonds and 0% bad backbone angles. In order to further examine the validity of the homology model, a morph calculation was performed for the transition between inward-facing (crystal structure) and outward-facing (homology model) conformations (Figure S11). The results revealed no incompatible helix and beta-sheet during the transitions. The positioning of TMs is also validated by several previous disulfide crosslinking experiments on Pgp20,21,22,23,24,25,26,27,28,29 (Figure 1).
d. Modeling of MgATP binding
Mg2+ coordinated ATP4− (MgATP2−) was chosen as the ligand for the NBDs of mouse Pgp. The homology template Sav1866 crystal structure has ANP in the ATP-binding sites but lacks Mg2+ that is important for ATP hydrolysis. Therefore, MgACP as modeled in the crystal structure of the human mitochondrial solute transporter ABCB10 in the inward-facing conformation, (PDB code 4AYT)45, was used as template for the positioning of ATP in mouse Pgp because of 100% conservation of ATP binding residues between the two proteins. The β carbon in ACP was replaced with β oxygen to generate initial coordinates of MgATP2−.
All-atom molecular dynamics simulations
All MD simulations utilized CHARMM22 with the CMAP correction of all-atom protein force field46 and CHARMM36 all-atom lipid force field47. The force field of ATP4− was implemented by CHARMM36 general force field48. Protein were placed in 120 × 120 (Å) POPC bi-layer and dissolved in a water box of the padding distance maintains >12 Å from every direction in equilibrium. Free ions with the concentration of 0.15 M NaCl and 0.002 M of MgCl2 were added to mimic physiological ionic condition and to neutralize the total system charge. All atomistic MD simulations were performed using NAMD2.9. The details of lipid-bilayer modeling and system control of 15 ns multi-step pre-equilibration and 200 ns production phase MD simulations are described in the Text S2. We will make the coordinate files produced in this study available upon request.
References
Linton, K. J. & Higgins, C. F. Structure and function of ABC transporters: the ATP switch provides flexible control. Pflugers Archiv: Eur J Phy 453, 555–567, 10.1007/s00424-006-0126-x (2007).
Raviv, Y., Pollard, H. B., Bruggemann, E. P., Pastan, I. & Gottesman, M. M. Photosensitized labeling of a functional multidrug transporter in living drug-resistant tumor cells. J Bio Chem 265, 3975–3980 (1990).
Rogan, A. M., Hamilton, T. C., Young, R. C., Klecker, R. W., Jr & Ozols, R. F. Reversal of adriamycin resistance by verapamil in human ovarian cancer. Science 224, 3 (1984).
Shen, D. W. et al. Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification. Science 232, 643 (1986).
Gottesman, M. M. & Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem 62, 385–427 (1993).
Zamora, J. M., Pearce, H. L. & Beck, W. T. Physical-chemical properties shared by compounds that modulate multidrug resistance in human leukemic cells. Mol Pharmacol 33, 454–462 (1988).
Seelig, A. A general pattern for substrate recognition by P-glycoprotein. Eur J Biochem 251, 252–261 (1998).
Schinkel, A. H., Wagenaar, E., Mol, C. A. & van Deemter, L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest 97, 2517–2524 (1996).
Ueda, K. et al. Human P-glycoprotein transports cortisol, aldosterone and dexamethasone, but not progesterone. J Bio Chem 267, 24248–24252 (1992).
Kuhnke, D. et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer's amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol 17, 347–353 (2007).
Shukla, S., Wu, C.-P. & Ambudkar, S. V. Development of inhibitors of ATP-binding cassette drug transporters: present status and challenges. Expert Opin Drug Metab Toxicol 4, 205–223 (2008).
Aller, S. G. et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722, 10.1126/science.1168750 (2009).
Li, J., Jaimes, K. F. & Aller, S. G. Refined structures of mouse P-glycoprotein. Protein Sci 23, 34–46, 10.1002/pro.2387 (2014).
Pascaud, C., Garrigos, M. & Orlowski, S. Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J 333, 351–358 (1998).
Martin, C. et al. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol 58, 624–632 (2000).
Smith, P. C. et al. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol Cell 10, 139–149 (2002).
Zaitseva, J. et al. A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J 25, 3432–3443 (2006).
Jardetzky, O. Simple allosteric model for membrane pumps. Nature 211, 969–970 (1966).
Dawson, R. J. & Locher, K. P. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS letters 581, 935–938, 10.1016/j.febslet.2007.01.073 (2007).
Loo, T. W. & Clarke, D. M. Inhibition of oxidative cross-linking between engineered cysteine residues at positions 332 in predicted transmembrane segments (TM) 6 and 975 in predicted TM12 of human P-glycoprotein by drug substrates. J Bio Chem 271, 27482–27487 (1996).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. Val133 and Cys137 in transmembrane segment 2 are close to Arg935 and Gly939 in transmembrane segment 11 of human P-glycoprotein. J Bio Chem 279, 18232–18238, 10.1074/jbc.M400229200 (2004).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. Disulfide cross-linking analysis shows that transmembrane segments 5 and 8 of human P-glycoprotein are close together on the cytoplasmic side of the membrane. J Bio Chem 279, 7692–7697, 10.1074/jbc.M311825200 (2004).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem Biophy Res Commun 395, 436–440, 10.1016/j.bbrc.2010.04.057 (2010).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. ATP hydrolysis promotes interactions between the extracellular ends of transmembrane segments 1 and 11 of human multidrug resistance P-glycoprotein. Biochemistry 44, 10250–10258, 10.1021/bi050705j (2005).
Loo, T. W. & Clarke, D. M. The packing of the transmembrane segments of human multidrug resistance P-glycoprotein is revealed by disulfide cross-linking analysis. J Bio Chem 275, 5253–5256 (2000).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J Bio Chem 278, 13603–13606, 10.1074/jbc.C300073200 (2003).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. Nucleotide binding, ATP hydrolysis and mutation of the catalytic carboxylates of human P-glycoprotein cause distinct conformational changes in the transmembrane segments. Biochemistry 46, 9328–9336, 10.1021/bi700837y (2007).
Loo, T. W., Bartlett, M. C., Detty, M. R. & Clarke, D. M. The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J Bio Chem 287, 26806–26816, 10.1074/jbc.M112.376202 (2012).
Loo, T. W. & Clarke, D. M. Identification of the distance between the homologous halves of P-glycoprotein that triggers the high/low ATPase activity switch. J Bio Chem 289, 8484–8492, 10.1074/jbc.M114.552075 (2014).
Ravna, A. W., Sylte, I. & Sager, G. Molecular model of the outward facing state of the human P-glycoprotein (ABCB1) and comparison to a model of the human MRP5 (ABCC5). Theor Biol Med Model 4, 33, 10.1186/1742-4682-4-33 (2007).
Globisch, C., Pajeva, I. K. & Wiese, M. Identification of putative binding sites of P-glycoprotein based on its homology model. ChemMedChem 3, 280–295, 10.1002/cmdc.200700249 (2008).
Prajapati, R. & Sangamwar, A. T. Translocation mechanism of P-glycoprotein and conformational changes occurring at drug-binding site: Insights from multi-targeted molecular dynamics. Biochim Biophy Acta 1838, 2882–2898, 10.1016/j.bbamem.2014.07.018 (2014).
Wise, J. G. Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 51, 5125–5141, 10.1021/bi300299z (2012).
Fields, P. A. Review: Protein function at thermal extremes: balancing stability and flexibility. Comp Biochem Phys A 129, 417–431 (2001).
Sonveaux, N., Shapiro, A. B., Goormaghtigh, E., Ling, V. & Ruysschaert, J. M. Secondary and tertiary structure changes of reconstituted P-glycoprotein. A Fourier transform attenuated total reflection infrared spectroscopy analysis. J Bio Chem 271, 24617–24624 (1996).
Sharom, F. J., Liu, R., Romsicki, Y. & Lu, P. Insights into the structure and substrate interactions of the P-glycoprotein multidrug transporter from spectroscopic studies. Biochim Biophy Acta 1461, 327–345 (1999).
Vigano, C., Julien, M., Carrier, I., Gros, P. & Ruysschaert, J. M. Structural and functional asymmetry of the nucleotide-binding domains of P-glycoprotein investigated by attenuated total reflection Fourier transform infrared spectroscopy. J Bio Chem 277, 5008–5016, 10.1074/jbc.M107928200 (2002).
Siarheyeva, A., Liu, R. & Sharom, F. J. Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J Bio Chem 285, 7575–7586, 10.1074/jbc.M109.047290 (2010).
Tombline, G., Bartholomew, L., Gimi, K., Tyndall, G. A. & Senior, A. E. Synergy between conserved ABC signature Ser residues in P-glycoprotein catalysis. J Bio Chem 279, 5363–5373, 10.1074/jbc.M311964200 (2004).
O'Mara, M. L. & Mark, A. E. Structural characterization of two metastable ATP-bound states of P-glycoprotein. PloS one 9, e91916, 10.1371/journal.pone.0091916 (2014).
Qu, Q., Russell, P. L. & Sharom, F. J. Stoichiometry and affinity of nucleotide binding to P-glycoprotein during the catalytic cycle. Biochemistry 42, 1170–1177, 10.1021/bi026555j (2003).
Callaghan, R., Berridge, G., Ferry, D. R. & Higgins, C. F. The functional purification of P-glycoprotein is dependent on maintenance of a lipid-protein interface. Biochim Biophy Acta 1328, 109–124 (1997).
van Helvoort, A. et al. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell 87, 507–517 (1996).
Sali, A. & Overington, J. P. Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci 3, 1582–1596, 10.1002/pro.5560030923 (1994).
Shintre, C. A. et al. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. PNAS 110, 9710–9715, 10.1073/pnas.1217042110 (2013).
MacKerell, A. D. J. & Bashord, D. M. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102, 3586–3616, 10.1021/jp003919d (1998).
Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843, 10.1021/jp101759q (2010).
Vanommeslaeghe, K. et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comp Chem 31, 671–690, 10.1002/jcc.21367 (2010).
Acknowledgements
We acknowledge funding support from NIH grant DP2 OD008591 and XSEDE award TG-MCB140067.
Author information
Authors and Affiliations
Contributions
L.P. and S.G.A. wrote the main manuscript text and L.P. prepared all figures. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY INFO
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Pan, L., Aller, S. Equilibrated Atomic Models of Outward-Facing P-glycoprotein and Effect of ATP Binding on Structural Dynamics. Sci Rep 5, 7880 (2015). https://doi.org/10.1038/srep07880
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07880
This article is cited by
-
Different structures of berberine and five other protoberberine alkaloids that affect P-glycoprotein-mediated efflux capacity
Acta Pharmacologica Sinica (2019)
-
Endoplasmic reticulum-targeting doxorubicin: a new tool effective against doxorubicin-resistant osteosarcoma
Cellular and Molecular Life Sciences (2019)
-
Allosteric Role of Substrate Occupancy Toward the Alignment of P-glycoprotein Nucleotide Binding Domains
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
