
Abstract
We used 344 mitochondrial control region (717 bp) sequences from the finless porpoise (genus Neophocaena) from the northwestern Pacific to investigate the extent and manner in which past climatic oscillations may have shaped patterns of genetic diversity for this marine mammal. Both SplitsTree and Analysis of Molecular Variance (AMOVA) revealed the presence of a deep divergence among N. phocaenoides in subtropical waters compared with N. asiaeorientalis in temperate waters. Results from Migrate-n indicated that migration increased along the continent during the early Pleistocene period. Migration increased, although to a lesser extent than that during the Pleistocene, along the marginal shelf in the Yellow/Bohai Sea during the Last Glacial Maximum (LGM) due to a shortening coastline. Our results suggest that the current patterns of genetic diversity of Neophocaena vary at a hierarchy on a temporal and spatial scale and phylogeographic history should be taken into account when examining species population structure and taxonomy.
Similar content being viewed by others

Introduction
Understanding the relative roles of climate change and geography in driving patterns of genetic diversity is of great scientific importance given the increasing loss of biodiversity. Rapid speciation events and dispersal patterns shaped by paleogeological and climatic oscillations have been well documented around the world1 because these oscillations have led to significant changes in the geomorphological structure and size of habitats. The northwestern Pacific is one of the world's largest subduction zones, which was formed in the Cenozoic period and isolated from the Pacific during the Pleistocene glacial cycles with division into three independent refugia (Fig. 1): the South China Sea refugium (from the exposed Taiwan Strait to Borneo Island), the East China Sea refugium (surrounded by Taiwan, northern China, Korea and southern Japan) and the Sea of Japan refugium2.

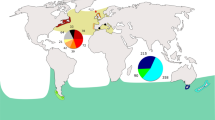
Sampling sites and SplitsTree of Neophocaena based on control regions.
Four haplogroups/subgroups are marked with different colors according to their clustering pattern (light blue: N. phocaenoides from PRE; dark blue: N. phocaenoides from Taiwan Strait; yellow: N. a. asiaeorientalis from the Yangtze River; and purple: N. a. sunameri from the Yellow/Bohai Sea and East China Sea). Five shared haplotypes are indicated in black. Seven populations are represented on the map with colors corresponding to the SplitsTree results. The numbers in the brackets indicate the sampling size of each locale. Dashed lines indicate 50, 100 and 200 m isobaths. The arrows indicate the direction of the Guangdong coastal current (dashed lines) and the South China Sea warm current (solid lines) in the summer. The box on the top right indicates the global distribution of Neophocaena. The map was generated using ArcGIS 9.2. The coastline and hydrologic data are available on the web at http://www.naturalearthdata.com/downloads/ (Public Domain; date accessed: Jun 2011) and http://www.mapcruzin.com/download-shapefile/asia-waterways-shape.zip (Open Database License 1.0; date accessed: Jun 2011).
Climate change and tectonic shifts greatly contributed to the genetic diversity and structure of marine organisms in the northwestern Pacific in two major aspects. First, the connection of modern islands (Ryukyu Archipelago, Taiwan, the Philippines and Sumatra) interrupted the gene flow among marine refugia, ultimately leading to great genetic divergence for many marine fauna and flora3. Moreover, the continental shelf in the Bohai Gulf, the Beibu Gulf in China and the Java Sea in southeast Asia became exposed as the sea level dropped to approximately 120–140 meters below the current level4. This sea level change resulted in a severe genetic bottleneck associated with habitat loss5 and increased gene flow among populations that once were geographically distant. While a phylogenetic partition corresponding to the three refugia has been well documented for many marine species6,7,8, a lack of genetic structure has been generally observed within the separate refugia, particularly in the East China Sea, which is much smaller in size than the South China Sea2.
Unlike many other marine organisms, cetaceans are characterized by complex life histories. They are long-lived, have a long generation time and display an incredible amount of variation in their behavioral ecology. Their complex life history likely influences the extent to which paleoclimatic oscillations affect patterns of genetic diversity in cetaceans; therefore, they merit full investigation9. The dispersal and migration patterns of marine organisms, such as fish10,11, crustaceans12 and algae13, are greatly influenced by changing currents, which homogenize genetic diversity between refugia because these organisms can travel thousands of kilometers in spawning form. Nevertheless, patterns of genetic diversity may be complex for species with unique life histories. For instance, the dispersal and migration pattern of seahorses (Hippocampus) are constrained by their reproductive strategy (i.e., internal brooding and the release of fully developed young) and behavioral ecology (i.e., site fidelity as adults)14. Lourie et al.15 identified population fragmentation for shallow-water seahorse species (H. barbouri and H. kuda) due to the Pleistocene separation of marine basins and expansion and isolation by distance for deep-water species (H. spinosissimus and H. trimaculatus), the latter of which were less affected by a change in coastline.
The finless porpoise (genus Neophocaena) is a cetacean taxon found in warm coastal waters ranging from the Persian Gulf to the northwestern Pacific. Despite the wide distribution of finless porpoises, only two species (N. phocaenoides and N. asiaeorientalis) are currently recognized, primarily based on morphological data (dorsal ridge, groove and tubercled patch)16. The first species is a narrow-ridged form, N. asiaeorientalis, which is exclusively found in temperate waters. This species is further divided into two subspecies: N. a. sunameri (the range of which includes Korea, Japan, the East China Sea and the Yellow and Bohai Sea in China) and N. a. asiaeorientalis (found in the freshwater environment of the Yangtze River). The genetic structures among the N. asiaeorientalis populations17,18,19,20,21,22 suggest limited dispersal. The second species, N. phocaenoides, which is a wide-ridged form, widely ranges in tropical marine waters from the South China Sea (including Taiwan Strait) to the Persian Gulf. Unfortunately, little is known about this species, particularly those inhabiting the rim of Indian Ocean. Although some early studies suggested a lack of genetic structure within the South China Sea17,23, which appears contrast with the congeneric finding of N. asiaeorientalis in warm waters17,18,19,20,21,22, these studies relied on small sample sizes (6 < n < 18); thus, the results should be treated with caution. A sympatric area was documented in the Taiwan Strait for N. phocaenoides and N. asiaeorientalis, but it has been proposed that these two species have been breeding in isolation since the Last Glacial Maximum (LGM, approximately 18 ka)23.
Because the finless porpoise is found along a continuous distribution and in shallow coastal waters < 50 meters24, this cetacean is likely affected by paleoclimatic changes, making it an ideal candidate organism for investigating the impact of glacial oscillations on the genetic structure and diversity of marine mammals and assessing whether the subduction zone leads to differing patterns of genetic diversity across its latitude. We traced the divergence and demographic history of seven geographic populations (n = 344), including N. phocaenoides and the two sub-species under N. asiaeorientalis, in the northwestern Pacific (including the Yangtze River) by testing two divergence hypotheses. First, we tested the recent divergence hypothesis (RDH, Fig. 2a) proposed by Yang et al.25, which suggests that N. a. sunameri in the Yellow Sea is the most ancestral species and colonized the Yangtze River approximately 22 ka with subsequent colonization of the South China Sea approximately 2 ka. Based on the RDH, samples were categorized into three populations corresponding to their (sub)species definition17,20,25. Second, we evaluated the stepwise divergence hypothesis (SDH, Fig. 2b–c) as proposed in this study. According to the SDH, migration along the northwestern Pacific increased during the early Pleistocene period (~1.5 Mya, see details in Results). Since then, divergence began to develop within N. phocaenoides in the northern South China Sea most likely as a result of isolation by distance, while their connection to N. asiaeorientalis in the East China Sea was further interrupted during the LGM (approximately 18 ka)23. However, decreasing sea levels simultaneously forced the coastal populations of N. asiaeorientalis into a refugium in the Okinawa Trough, which resulted in the homogenization of these previously divergent populations during LGM. These populations diverged again approximately 5 ka as sea levels rapidly increased26. Divergence within the Yangtze River occurred as a result of recent human disturbance approximately 50 years ago27 (dozens of generations; details in the Results and Discussion sections).

The number of migrations of the genus Neophocaena along the western Pacific over time.
The migration rates are indicated by the parameter M multiplied by 1,000. The time scale is shown in mutational units by generation. A box indicates a time interval covering the strengthening migration during 0.00815–0.00995 along the marine coast, which is separately translated into absolute time using substitution rates of 0.6%, 0.98% and 1.2% per million years at the bottom.
Results
Data delineation
The final dataset consisted of 344 sequences from most of the known range of Neophocaena in China (Fig. 1), including 75 sequences from the Pearl River Estuary (PRE in the northern South China Sea, GenBank accession numbers: KC408426-KC408436), 53 sequences from the Taiwan Strait (NSS = 16, SSS = 33, GenBank accession numbers: AF289280-AF289352), 154 sequences from the Yellow/Bohai Sea (NYS = 142, SYS = 18, GenBank accession numbers: HQ108421-HQ108437) and 62 sequences from the Yangtze River (Yangtze River, population WYR = 50, population EYR = 12, GenBank accession number: AF481866-AF481874). The sequences were 717 bp in length with 28 substitutions and one deletion.
Population structure and expansion
SplitsTree analysis delineated two haplotype groups exhibiting clear divergence (Fig. 1). The haplotype from the Pearl River Estuary (PRE) population clustered exclusively into H1 with the exception of GD03 and H2 comprised all of the samples from the Taiwan Strait (H2a), the East China Sea (including the Yellow/Bohai Sea, H2c) and the Yangtze River (H2b). The positions of the four lineages in the network were in accord with their geographic locations. However, surprisingly, H2a, which mostly consisted of N. phocaenoides from the Taiwan Strait, was connected to N. asiaeorientalis by multiple reticulations with a relatively long distance to N. phocaenoides from the PRE. Reticulations in SplitsTree analysis are generally treated as sign of historical gene flow (hundreds of generations); thus, the divergence within N. phocaenoides in the northern South China Sea (PRE and Taiwan Strait) should predate that between N. phocaenoides and N. asiaeorientalis across the Taiwan Strait. AMOVA revealed a significantly higher Fct value (0.23896, P = 0.00978) when the samples were grouped according to the distribution of H1, H2a, H2b and H2c compared with grouping according to (sub)species definition (H1 + H2a, H2b and H2c, 0.00650, P = 0.65689). AMOVA identified the optimal geographic subdivision for the sequence data by maximizing the genetic variance among groups for the overall variance, which suggested that the divergence in our samples was largely due to the divergence within N. phocaenoides.
Because the Fst suggested no differentiation between the NSS and SSS20 populations, we combined these two populations into an SS population for the purpose of demographic analyses. A negative Fu's F was detected in all tested populations except the PRE population. However, demographic expansion was only statistically supported in two populations in which a large scale of habitat loss was expected during the LGM (Table 1): the Yellow/Bohai Sea (SYS) and the East China Sea (NYS) populations. This value was close to significant for the SS population (P = 0.077), which was also expected to experience massive habitat loss during lower sea levels. Consistent with Fu's F, a unimodal distribution of mismatch was observed in SYS and NYS populations (Fig. S1). In the NYS population, we found no support for the spatial expansion model, but the non-significance of the raggedness value supported the sudden demographic expansion model (Table 1). However, the contribution of these two expansion models could not be distinguished in the SYS population because of the much smaller sample size (n = 19). Using a mutation rate of 0.94 × 10−8, the latest expansions dated back to 0.47, 0.48 and 0.59 Mya for the SYS, NYS and SS populations, respectively (Table 1). The effective population size (Ne) before expansion was estimated to be approximately 10% of the current Ne for the NYS and SYS populations and 15% for SS according to a Bayesian Skyline Plot (BSP) (Fig. S2).
Historical migration
Migrate-n uses the parameter M (M = m/u, where m is the migration rate and u is the mutation rate of the loci) to describe the mutation-scaled migration rate between populations. The Bayesian inference indicated a high level of gene exchange around the LGM (6–38 ka based on three different mutation rates of 0.6%, 0.98% and 1.2% per million years) between the populations from the Yellow Sea (NYS and SYS) and the lower reach of the Yangtze River (EYR, Fig. 2). The gene flow was symmetrical between the SYS and EYR populations but moved in a southern direction between the SYS and NYS populations (Fig. 2b); the gene flow between the SYS and NYS populations could be associated with the displacement of N. a. sunameri from the present-day Yellow/Bohai Sea during marine regression.
Surprisingly, a more significant migration was detected prior to the LGM, when the value of M was an order of magnitude higher than at any other time. We dated this period using a wide range of substitution rates (0.6%, 0.98% and 1.2% per million years), which all fall into the range of early Pleistocene (2.04–2.49 Mya, 1.25–1.52 Mya and 1.02–1.24 Mya, respectively, Fig. 2). The migration was found to have occurred twice in the northern direction (from the PRE population to the SSS population) and once in the southern direction (from the NSS population to the SSS population) in the South China Sea during the early Pleistocene period (Fig. 2a). In contrast, migration was mostly symmetrical in the Yellow/Bohai and East China Sea with the exception of that between SYS and NYS (Fig. 2b). Gene flow between the East China Sea and the Yangtze River was also evident but less than half of that determined between coastal populations. In addition, the migration fluctuated less in the Yangtze River (Fig. 2c) than in any other region, indicating a relatively stable environment in the inland river.
Tests of divergence history
Based on a rejection algorithm, two divergence hypotheses were tested by evaluating the susceptibility of empirical data using coalescent simulation. Under the SDH, θ was estimated to be 0.0463, 0.00543, 0.099, 0.00183, 0.0027 and 0.00163 for SS, NYS, SYS, EYR, PRE and WYR, respectively. Using μ = 0.94 × 10−8, we calculated corresponding point estimates for Ne as 24628, 28883, 52660, 9734, 14362 and 8670, respectively. Bottleneck and follow-up expansion were expected in the Yellow/Bohai Sea (NYS/SYS) and Taiwan Strait (NSS plus SSS), which experienced vast habitat loss during the marine regression. However, the time of expansion derived from the τ value predated the divergence history. To preclude a possible error caused by methodological sensitivity and resolution, we further tested the hypothesis with and without the demographic change during the LGM. In the former, the Ne values remained consistent after the early Pleistocene period. In the latter, the three populations that exhibited a bottleneck signal (NYS, SYS and SS) were assumed to have experienced an 85 to 90% decline during the LGM (Fig. S2). Under the RDH, the point estimates for θ and Ne were 0.0071, 0.0031 and 0.0096 and 37766, 16489 and 50904, respectively, for N. phocaenoides, N. a. asiaeorientalis and N. a. sunameri. To ensure that the model testing was robust for a wide range of substitution rates in mtDNA, the simulation was rerun using 25% and 75% quantiles of the posterior distribution of θ (Table S1).
The value of s between genes and populations was estimated to be 33 according to the SDH and 15 according to the RDH. Figure 2 shows the distribution of s for the 100 simulated genealogies using the point estimate of Ne derived from the mode value of θ. The observed s significantly deviated from the values simulated based on the RDH and SDH with constant demographic history across the range of Ne (Fig. 3a–b). When the demographic decline in the Yellow/Bohai Sea, East China Sea and Taiwan Strait during the LGM was taken into consideration even though the SDH was rejected when using the upper bound of θ, we could not reject the SDH with the lower bound and mode value of θ, which suggested a more reasonable Ne (Fig. 3c and Table S1).

Models used to test the divergence history (left) and s values for 100 simulated coalescent genealogies (right).
(a) Recent divergence hypothesis; (b) Stepwise divergence hypothesis with constant demographic history; and (c) Stepwise divergence hypothesis with demographic decline (thin lines) in the Yellow/Bohai Sea and the Taiwan Strait during the LGM. The bars show the expected distribution of the s value using the mode of Ne with the 95% CI indicated by dashed lines. The red lines show the s values for the actual sequences under each hypothesis.
Discussion
Previous studies have demonstrated that N. asiaeorientalis populations are significantly structured in the northwestern Pacific17,18,19,20,21,22,25; Wang et al. proposed that the patterns of genetic divergence between N. phocaenoides and N. asiaeorientalis may have been caused by a geographic obstacle formed during the LGM23. In contrast with previous studies that suggested no genetic structure among N. phocaenoides18,23, our analyses, which included a much greater sample size, revealed the presence of extensive genetic divergence for Neophocaena within the South China Sea (between H1 and H2a in Fig. 1). Other than the glacial land bridge in the Taiwan Strait, there was no apparent geographic barrier inside the northern South China Sea, suggesting that such divergence might have been caused by either unknown paleoclimatic factors or isolation by distance. Moreover, the divergence proposed by the SplitsTree network at the species and subspecies level was relatively shallower than the divergence observed within the South China Sea (Fig. 1). Taken together, these results indicate that the divergence stratification of Neophocaena along the Chinese coast (including the Yangtze River) was most likely the product of multiple factors on different temporal and spatial scales, which are presented in further details below.
Reconstruction of the historical migration suggests that the gene flow between the Yellow/Bohai Sea (NYS and SYS populations) and the lower reach of the Yangtze River (EYR population) increased during the LGM (approximately 18 ka). This increase corresponds to coastline development during this time period. Since the LGM, a series of climatic and geographic events associated with a drop in sea level, have occurred28 and exposed the entire Yellow/Bohai Seabed to air by extending the coastline seaward by more than 1,200 km to the north28,29. As a result, N. asiaeorientalis, which is found in the contemporary Yellow/Bohai Sea, lost its entire habitat, bringing the NYS and the SYS populations much closer geographically (approximately 150 km vs. 700 km as determined by measuring the central point of the sampling area). This shift in geography may have resulted in the increased gene flow we identified between the NYS and SYS populations during the early Holocene period (Fig. 2). The river mouth of the Yangtze River invaded eastward by approximately 300 km, which may have caused N. a. sunameri near the river mouth to move into the freshwater environment as the sea receded. The evidence of migration between the EYR and SYS populations might indicate hybridization between N. a. sunameri from the Yellow/Bohai Sea and N. a. asiaeorientalis from the lower reach of the Yangtze River, but this hypothesis remains to be tested. In addition, the river mouth of the Yangtze River splits into at least six river mouths with the northern one occurring around Chejudo Island30. This split created a corridor between N. a. sunameri from the NYS population and N. a. asiaeorientalis from the lower reach of the Yangtze River (EYR population). In conclusion, migration among pairwise populations of EYR, NYS and SYS likely simultaneously increased during the period of low sea levels, a possibility that is well supported by the comparable levels of historical migration rates among the NYS-EYR and SYS-EYR populations (Fig. 2) and the degree of interpopulation differentiation18. In addition, we detected reticulated evolution within the H2c (including both NYS and SYS) populations, which was most likely due to historical gene flow when the marginal shelf in Bohai/Yellow Sea was still exposed approximately 5 ka.
Apart from the geomorphological changes in the river mouth, the Yangtze River drainage was maintained throughout the glacial and interglacial periods of the Pleistocene period31. This relatively stable environment resulted in N. a. asiaeorientalis being less vulnerable during the interglacial period. We therefore expected that patterns of isolation by distance would dominate the divergence that has occurred in the Yangtze River. Supporting this scenario, Yang et al.25 found that the five subpopulations of N. a. asiaeorientalis in the Yangtze River followed an isolation-by-distance pattern. Although they failed to demonstrate this pattern when other populations from the Yellow/Bohai Sea and Japanese waters were included, patterns of isolation by distance might have been obscured by other geographic factors, such as the sea level fluctuation in the East China Sea, isolation of the East China Sea and possible vicariance within Japanese waters. It is therefore likely that the present population structure found within the Yangtze River is the result of recent population degradation27 and/or habitat fragmentation caused by rapid development in recent decades32.
Despite historical (hundreds of generations) migration events between N. a. sunameri and N. a. asiaeorientalis around the Yangtze River mouth, Chen et al.17 have argued against ongoing gene flow because they observed the presence of a significant population structure based on mtDNA and nuclear markers. However, the level of genetic differentiation between N. a. sunameri from the Yellow/Bohai Sea and N. a. asiaeorientalis from the Yangtze River (Fst = 0.070)17 was much lower than that generally found among other populations (Fst: 0.166–0.601)18. We therefore propose that the divergence between N. a. sunpameri and N. a. asiaeorientalis could have occurred recently (dozens of generations) due to a recent estuarine development.
During the Pleistocene glaciation, Borneo Island was connected to the main continent with a narrow connection at the Boshi Strait, isolating the South China Sea from the Pacific29. Thus N. phocaenoides from the Taiwan Strait and the South China Sea were expected to have originated from the same refugium33. Surprisingly, we observed that N. phocaenoides from the PRE and the conspecific Taiwan Strait population exhibited the greatest amounts of genetic divergence (Fig. 1). The extent of genetic divergence between these populations was higher than the genetic divergence between the two N. asiaeorientalis subspecies. Our results therefore strongly reject the hypothesis that N. phocaenoides formed a single population in the South China Sea17,23. Significant genetic structure has also been documented for other species found in the South China Sea. For example, a mangrove species (Ceriops tagal) also exhibits significant genetic divergence between Borneo Island and the west Malay Peninsula34. An isolated lineage of mullet (Mugil cephalus) was also found exclusively along the coast of the Philippines with a hybridization lineage found in the northern South China Sea2. Similar genetic patterns were also reported for snail (Echinolittorina eidua)35, sea horse (Hippocampus kuda)36 and wrasse (Thallasoma hardwicki) species37, patterns largely caused by the deep basin in the central region of the South China Sea, which acts as a geographic barrier. In contrast to the populations in these studies, the N. phocaenoides populations (PRE and NSS/SSS) in our study are located on the same side of the South China Sea without any apparent geographic obstacles, yet they exhibit significant genetic divergence. During the Pleistocene glaciations, the northern coastline of the South China Sea moved seaward as a whole, resulting in a relatively stable morphological structure and a constant geographic distance between the PRE and NSS/SSS populations. Thus, N. phocaenoides from the northern region of the South China Sea was less vulnerable to the geomorphological development associated with sea level fluctuations. Instead, a strong signal for strengthening migration was detected much earlier than the LGM. To overcome the possible bias caused by the uncertainty of the substitution rate, we used a range of CR substitution rates reported in other cetacean species, which resulted in a long-term interval during the early Pleistocene period. Many reasons might be responsible for the increasing migration during this phase, including climate transition38,39 and tectonic change40. However, the discussion will remain open without a precise dating of divergence.
The unimodal mismatch distribution and significantly negative Fu F value strongly support the historical expansion of N. a. sunameri in the Yellow/Bohai Sea, while the multimodal mismatch distribution and large positive Fu F value suggest a constant demographic history for N. phocaenoides in the northern region of the South China Sea (Fig. S1 and Table 1). This discordance between different areas may be explained by the changes the coastline endured during the glaciation period, which may have resulted in multiple bottleneck events. During the deglaciation phase, the Yellow/Bohai Sea remained exposed before the rapid transgression that occurred at approximately 4–5 ka when the sea level was 50–60 meters lower than the present level26. We therefore expected a subsequent expansion in the Yellow/Bohai Sea. Unfortunately, the expansion was estimated to have occurred much earlier than the time of the rapid transgression using either the mismatch distribution or the Bayesian skyline method. This result could simply be due to the sensitivity of the methods and the use of a single locus and might be improved by using a more appropriate genetic marker (e.g., a microsatellite) and the recently developed likelihood-based method41, which detects recent demographic changes more robustly.
We reconstructed an evolutionary and biogeographic history of the genus Neophocaena in the northwestern Pacific and determined that patterns of genetic diversity coincide with past climatic events. First, we determined that the gene flow in the northwestern Pacific was promoted during the early Pleistocene period. Second, we not only confirmed that the presence of genetic divergence between the South China Sea and East China Sea correlated with an interruption in gene flow during the LGM23, we, more importantly, detected two additional and contrasting levels of genetic divergence in separate refugia during the LGM. We found significant population structure within N. phocaenoides in the South China Sea and argued that such patterns of isolation by distance may have been driven by a warm and relatively stable environment and have persisted until the present (approximately 1.5 Mya). However, even though the LGM interrupted the gene flow between the South China Sea and East China Sea, it otherwise increased the gene flow within the East China Sea refugium. The patterns of isolation by distance have reassumed a major role in these areas due to the extension of the coastline resulting in the latest expansion and contemporary divergence within the Yellow/Bohai Sea.
To conclude, the biogeographical history proposed here improves our understanding of the stratification of the divergence of the genus Neophocaena in the northwestern Pacific. Moreover, we provide new insight into the divergence within the South China Sea. Many regional populations of the genus Neophocaena are now near extinction27,42 and based on our results, we believe there is an urgent need to incorporate the phylogeographic history of this genus into a review of its taxonomy, particularly for less-studied populations in tropical areas.
Methods
Sample collection
We combined a total of 344 control region sequences collected from previous studies18,19,22,25,43. All sequences were aligned using the Clustal program44. The population and taxonomic identification definitions were obtained from source studies (Fig. 1), including N. phocaenoides from the South China Sea and Taiwan Strait (including NSS, SSS and PRE populations); N. a. asiaeorientalis exclusively found in the Yangtze River (EYR and WYR populations) and N. a. sunameri from the NYS and SYS populations.
Divergence
Tree splits were reconstructed using the method by Huson and Bryant45 to assess the extent of the compatibility of the data. Splits were calculated with NeighborNet distance transformation46 and equal angle splits transformation47 using the SplitsTree v.4.12.845 program. Splits were compatible only if they co-existed on the same tree.
To test the hypothesis that the current genetic structure of Neophocaena was determined by the refugium structure along the west Pacific, hierarchical analysis of molecular variance (AMOVA) was performed using 1,000 permutations48. AMOVA identified the optimal geographical subdivision for the sequence data by maximizing the genetic variance among groups for the overall variance. For this purpose, we conducted two separate analyses. First, we subdivided the populations into three groups according to their current taxonomic classification: (1) N. phocaenoides (PRE, NSS and SSS populations); (2) N. a. asiaeorientalis (WYR and EYR populations); and (3) N. a. sunameri (NYS and SYS populations). Second, we further divided the N. phocaenoides into two separate groups (PRE and NSS plus SSS) following the reticulated network relationship of CR (details in the Results and Fig. 1).
Demography
Population expansion was evaluated by calculating Fu's F and its significance was assessed via 1,000 simulations49 comparing the number of observed haplotypes with the expected value in a random sample. The mismatch distribution, which describes expansion by generating a unimodal distribution of variations between paired sequences in expansive populations and a multimodal distribution in constant populations, was determined50. When a unimodal distribution was detected, a sum of squared deviation (SSD) and raggedness index were calculated to describe the fitness of the observed and expected mismatch distributions. The significance of Fu's F, SSD and raggedness were corrected for multiple tests using the false discovery rate (FDR) method51. The date of expansion was determined using the online tool http://www.uni-graz.at/zoowww/mismatchcalc/mmc1.php (date accessed: Dec, 2013) developed by Schenekar and Weiss52. The upper and lower bounds of the mtDNA substitution rate were set at 0.6 to 1.2%53 with a point estimate using 0.94%54,55. τ is the crest of the mismatch distribution, which was estimated using Arlequin v.3.1156. Lee et al.57 studied the reproduction of N. asiaeorientalis by examining 111 ovaries and testis and suggested that the species would reach sexual maturity above 5 years old. Maximum ages were generally reported to be between 23 and 2558. Given that the fecundity remains high for much of the adulthood of this long-lived species, the generation time should be close to the average age for reproductively mature females. Thus, absolute years were translated into generations using an average generation of 15 years. To quantify the demographic decline and thus provide a framework for testing the biological history of species, we reconstructed a demographic history for each population using the Bayesian Skyline Model59 implemented in BEAST 1.5.360. We used five piecewise intervals for Ne with uniform prior distribution for each interval and a gamma distribution for a transition:transversion ratio (K). Each Bayesian skyline was run in 10 million steps three times. Results from all three runs were combined to construct the BSP. The convergence of analysis was judged according to the Effective Sampling Size (>200).
Migration rate through time
To determine the immigration and emigration rate through time, M (the mutational-scaled effective immigration rate) was estimated using the coalescent-based program Migrate-n under a Bayesian inference and sequence model61. A random tree was used as a starting genealogy and the initial theta was estimated from the Fst calculations. The parameters of the first run were used as starting values for the next run until a convergent result was obtained. Adaptive heating was set to 1.0, 1.2, 1.5 and 1,000. The run was performed with 2,000,000 genealogies and 25% of which were discarded as burn-in. Three long chains were run for each set of analyses and multiple long chains were combined for final estimates. The stepping stone model was followed as the animals were expected to migrate along the coastline (Fig. S3). The parameters for between the EYR and NYS populations were also calculated because one study proposed that the Yangtze River mouth extended to the Cheju Island during the glaciation30, thus potentially providing a corridor for migration between the lower reach of the Yangtze River and the northern East China Sea. The time units were translated into absolute years using the mutation rate and generation interval as suggested above.
Test of phylogeographical hypotheses
A coalescent method was adopted to test the phylogeographical hypotheses by constraining the genealogies into a hypothetical evolutionary model and assessing the model's fit by comparing the observed genetic pattern with the range of simulated patterns62. The CR ML tree was first constructed using the Mega 5.063 program. The observed value for s of Slatkin and Maddison, which is the minimum number of sorting events required to explain the evolutionary model, was calculated using the Mesquite ver. 2.74 program (available at http://mesquiteproject.org, date accessed: Oct, 2012). For either RDH or SDH, 100 coalescent CR genealogies were constructed. The s of the simulated genealogies was calculated and plotted against their frequency and the 95% confidence interval for the simulated s was automatically adopted by the program. The hypothetical population history was rejected if the observed s fell outside the 95% confidence interval of the simulated s. In this study, simulations were conducted using a single locus. Thus, special attention should be paid to the error associated with Ne. To overcome this limitation, the upper and lower bounds of the 50% CI of θ from the results of Migrate-n were followed to ensure that the most reasonable θ was included as described by Maddison and Knowles64. The Ne was calculated using the equation θ = 2Neμ.
References
Rawlings, L. H. & Donnellan, S. C. Phylogeographic analysis of the green python, Morelia viridis, reveals cryptic diversity. Mol. Phylogenet. Evol. 27, 36–44 (2003).
Liu, J. X., Gao, T. X., Wu, S. F. & Zhang, Y. P. Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol. Ecol. 16, 275–288 (2007).
Rohfritsch, A. & Borsa, P. Genetic structure of Indian scad mackerel Decapterus russelli: Pleistocene vicariance and secondary contact in the Central Indo-West Pacific Seas. Heredity 95, 315–326 (2005).
Lambeck, K., Esat, T. & Potter, E. K. Links between climate and sea levels for the past three million years. Nature 419, 199–206 (2002).
Liu, J. X. et al. Late Pleistocene divergence and subsequent population expansion of two closely related fish species, Japanese anchovy (Engraulis japonicus) and Australian anchovy (Engraulis australis). Mol. Phylogenet. Evol. 40, 712–723 (2006).
Shen, K. N., Jamandre, B. W., Hsu, C. C., Tzeng, W. N. & Durand, J. D. Plio-Pleistocene sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus. BMC Evol. Biol. 11, 83 (2011).
Collins, J. S. H., Lee, C. & Noad, J. Miocene and Pleistocene crabs (Crustacea, Decapoda) from Sabah and Sarawak. J. Syst. Palaeontol. 1, 187–226 (2003).
Williams, S. T. & Duda Jr, T. F. Did Tectonic Activity Stimulate Oligo–Miocene Speciation in the Indo-west Pacific? Evolution 62, 1618–1634 (2008).
Harlin-Cognato, A., Markowitz, T., Wursig, B. & Honeycutt, R. Multi-locus phylogeography of the dusky dolphin (Lagenorhynchus obscurus): passive dispersal via the west-wind drift or response to prey species and climate change? BMC Evol. Biol. 7, 131 (2007).
Jamandre, B. W., Durand, J. D. & Tzeng, W. N. Phylogeography of the flathead mullet Mugil cephalus in the north-west Pacific as inferred from the mtDNA control region. J. Fish Biol. 75, 393–407 (2009).
Wang, M. et al. Genetic diversity in the mtDNA control region and population structure in the Sardinella zunasi Bleeker. Afr. J. Biotechnol. 7, 24 (2010).
Tzeng, T. D. Population Structure of the Sword Prawn (Parapenaeopsis hardwickii) (Decapoda: Penaeidae) in the East China Sea and Waters Adjacent to Taiwan Inferred from the Mitochondrial Control Region. Zool. Stud. 46, 561–568 (2007).
Cheang, C. C., Chu, K. H. & Ang, P. O., Jr Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Mol. Ecol. 19, 2933–2948 (2010).
Foster, S. J. & Vincent, A. C. J. The life history and ecology of seahorses, Hippocampus spp.: implications for conservation and management. J. Fish Biol. 65, 1–61 (2004).
Lourie, S. A., Green, D. M. & Vincent, A. C. J. Dispersal, habitat differences and comparative phylogeography of Southeast Asian seahorses (Syngnathidae: Hippocampus). Mol. Ecol. 14, 1073–1094 (2005).
Jefferson, T. A. & Wang, J. Y. Revision of the taxonomy of finless porpoises (genus Neophocaena): The existence of two species. J. Mar. Anim. Ecol. 4, 3–16 (2011).
Chen, L., Bruford, M., Xu, S., Zhou, K. & Yang, G. Microsatellite variation and significant population genetic structure of endangered finless porpoises (Neophocaena phocaenoides) in Chinese coastal waters and the Yangtze River. Mar. Biol. 157, 1453–1462 (2010).
Li, X. et al. Analysis of global and local population stratification of finless porpoises (Neophocaena phocaenoides) in Chinese waters. Mar. Biol. 158, 1791–1804 (2011).
Yang, G. et al. Population Genetic Structure of Finless Porpoises, Neophocaena phocaenoides, in Chinese Waters, Inferred from Mitochondrial Control Region Sequences. Mar. Mamm. Sci. 18, 336–347 (2002).
Yang, G. & Zhou, K. Y. Genetic variation of three populatioins of finless porpoise (Neophocaena phocaenoides) in Chinese waters. Acta Zool. Sin. 43, 411–419 (1997).
Yoshida, H., Yoshioka, M., Shirakihara, M. & Chow, S. Population structure of finless porpoises (Neophocaena phocaenoides) in coastal waters of Japan based on mitochondrial DNA sequences. J. Mammal. 82, 123–130 (2001).
Zheng, J. S., Xia, J. H., He, S. P. & Wang, D. Population genetic structure of the Yangtze finless porpoise (Neophocaena phocaenoides asiaeorientalis): implications for management and conservation. Biochem. Genet. 43, 307–320 (2005).
Wang, J. Y., Frasier, T. R., Yang, S. C. & White, B. N. Detecting recent speciation events: the case of the finless porpoise (genus Neophocaena). Heredity (Edinb) 101, 145–155 (2008).
Amano, M., Nakahara, F., Hayano, A. & Shirakihara, K. Abundance estimate of finless porpoises off the Pacific coast of eastern Japan based on aerial surveys. Mamm. Study 28, 103–110 (2003).
Yang, G., Guo, L. I., Bruford, M. W., Wei, F. & Zhou, K. Mitochondrial phylogeography and population history of finless porpoises in Sino-Japanese waters. Biol. J. Linn. Soc. 95, 193–204 (2008).
Lee, Y. G., Choi, J. M. & Oertel, G. F. Postglacial Sea-Level Change of the Korean Southern Sea Shelf. J. Coast. Res. 24, 118–132 (2008).
Mei, Z. et al. Accelerating population decline of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis). Biol. Conserv. 153, 192–200 (2012).
Xu, X. & Oda, M. Surface-water evolution of the eastern East China Sea during the last 36,000 years. Mar. Geol. 156, 285–304 (1999).
Wang, P. Response of Western Pacific marginal seas to glacial cycles: paleoceanographic and sedimentological features. Mar. Geol. 156, 5–39 (1999).
Xiao, S. et al. The History of the Yangtze River Entering Sea since the Last Glacial Maximum: a Review and Look Forward. J. Coast. Res. 20, 599–604 (2004).
Cao, G. et al. Characteristics and Runoff Volume of the Yangtze River's Paleovalley at Nanjing Reach in the Last Glacial Maximum. Acta Geogr. Sin. 64, 331–338 (2009).
Yang, S. L. et al. Delta response to decline in sediment supply from the Yangtze River: evidence of the recent four decades and expectations for the next half-century. Est. Coast. Shelf Sci. 57, 689–699 (2003).
Lin, W., Zhou, R., Porter, L., Chen, J. & Wu, Y. Evolution of Sousa chinensis: A scenario based on mitochondrial DNA study. Mol. Phylogenet. Evol. 57, 907–911 (2010).
Liao, P. C., Havanond, S. & Huang, S. Phylogeography of Ceriops tagal (Rhizophoraceae) in Southeast Asia: the land barrier of the Malay Peninsula has caused population differentiation between the Indian Ocean and South China Sea. Conserv. Genet. 8, 89–98 (2007).
Reid, D. G. et al. Comparative phylogeography and species boundaries in Echinolittorina snails in the central Indo-West Pacific. J. Biogeogr. 33, 990–1006 (2006).
Lourie, S. A., Green, D. M. & Vincent, A. C. J. Dispersal, habitat differences and comparative phylogeography of Southeast Asian seahorses (Syngnathidae: Hippocampus). Mol. Ecol. 14, 1073–1094 (2005).
Chen, C. et al. Population Structure and Genetic Variability of Six Bar Wrasse (Thallasoma hardwicki) in Northern South China Sea Revealed by Mitochondrial Control Region Sequences. Mar. Biotechnol. 6, 312–326 (2004).
Li, Q. et al. Paleoceanography of the mid-Pleistocene South China Sea. Quat. Sci. Rev. 27, 1217–1233 (2008).
Lisiecki, L. E. & Raymo, M. E. Plio-Pleistocene climate evolution: treads and transitions in glacial cycle dynamics. Quat. Sci. Rev. 26, 56–59 (2007).
Duan, Y. New Tectonic Movement and the Formation and Evolution of the Bohai Sea and their Effects on the Current Geological Environment. Chin. J. Geol. Hazard. Control. 9, 106–113 (1998).
Girod, C., Vitalis, R., Leblois, R. & Fréville, H. Inferring Population Decline and Expansion From Microsatellite Data: A Simulation-Based Evaluation of the Msvar Method. Genetics 188, 165–179 (2011).
Preen, A. Distribution, abundance and conservation status of dugongs and dolphins in the southern and western Arabian Gulf. Biol. Conserv. 118, 205–218 (2004).
Xia, J., Zheng, J. & Wang, D. Ex situ conservation status of an endangered Yangtze finless porpoise population (Neophocaena phocaenoides asiaeorientalis) as measured from microsatellites and mtDNA diversity. ICES Journal of Marine Science: J. Cons. Int. Explor. Mer. 62, 1711–1716 (2005).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Huson, D. H. & Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 23, 254–267 (2006).
Bryant, D. & Moulton, V. Neighbor-Net: An Agglomerative Method for the Construction of Phylogenetic Networks. Mol. Biol. Evol. 21, 255–265 (2004).
Dress, A. W. M. & Huson, D. H. Constructing Splits Graphs. IEEE/ACM Trans. Comput. Biol. Bioinform. 1, 109–115 (2004).
Excoffier, L., Smouse, P. & Quattro, J. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992).
Fu, Y. Statistical tests of neutrality of mutaions against population growth, hitchhiking and background selection. Genetics 147, 915–925 (1997).
Harpending, H. C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 66, 591–600 (1994).
Storey, J. D. A direct approach to false discovery rates. J. R. Stat. Soc. Series. B. Stat. Methodol. 64, 479–498 (2002).
Schenekar, T. & Weiss, S. High rate of calculation errors in mismatch distribution analysis results in numerous false inferences of biological importance. Heredity 107, 511–512 (2011).
Dalebout, M. L. et al. Worldwide structure of mtDNA diversity among Cuvier's beaked whales (Ziphius cavirostris): implications for threatened populations. Mol. Ecol. 14, 3353–3371 (2005).
Baker, C. S. et al. Abundant mitochondrial DNA variation and worldwide population structure in humpback whales. Proc. Natl. Acad. Sci. USA 90, 8239–8243 (1993).
McMillan, W. O. & Bermingham, E. The phylogeographic pattern of mitochondrial DNA variation in the Dall's porpoise Phocoenoides dalli. Mol. Ecol. 5, 47–61 (1996).
Excoffier, L., Laval, G. & Schneider, S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform. Online 1, 47–50 (2005).
Lee, Y. R. et al. Age and reproduction of the finless porpoises, Neophocaena asiaeorientalis, in the Yellow Sea, Korea. Anim. Cells. Syst. 17, 366–373 (2013).
Jefferson, T. A., Robertson, K. M. & Wang, J. Y. Growth and Reproduction of the Finless Propoise in Southern China. Raffles. B. Zool. 10, 105–113 (2002).
Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 22, 1185–1192 (2005).
Drummond, A. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Peter, B. & Michal, P. Unified Framework to Evaluate Panmixia and Migration Direction Among Multiple Sampling Locations. Genetics 185, 313–326 (2010).
Slatkin, M. & Maddison, W. P. A cladistic measure of gene flow inferred from phylogenies of alleles. Genetics 123, 603–613 (1989).
Tamura, K. et al. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance and Maximum Parsimony Methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Maddison, W. P. & Knowles, L. L. Inferring Phylogeny Despite Incomplete Lineage Sorting. Syst. Biol. 55, 21–30 (2006).
Acknowledgements
This research was supported by the Ocean Park Conservation Foundation, Hong Kong, the Australia and New Zealand Bank, the National Natural Science Foundation of China (40976082 and 41276147) and the National Key Technology R and D Program (grant no. 2011BAG07B05-3). We would like to thank OPCFHK and AFCD for sample collection and those who contributed samples or helped in sample collection, including Mr. Xi Chen and Mr. Yingku Wang.
Author information
Authors and Affiliations
Contributions
Y.W. managed the research project. Y.W. and W.L. conceived the ideas and W.L. performed most of the initial research. D.G. and J.X. contributed the data from the PRE. W.L. collected other data from the literature and W.L. conducted the analyses and drafted the manuscript, which was approved by C.H.F., L.K., Y.W. and J.X.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Info
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Lin, W., Frère, C., Karczmarski, L. et al. Phylogeography of the finless porpoise (genus Neophocaena): testing the stepwise divergence hypothesis in the northwestern Pacific. Sci Rep 4, 6572 (2014). https://doi.org/10.1038/srep06572
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06572
This article is cited by
-
Mitochondrial genomics reveals the evolutionary history of the porpoises (Phocoenidae) across the speciation continuum
Scientific Reports (2020)
-
Genetic footprint of population fragmentation and contemporary collapse in a freshwater cetacean
Scientific Reports (2017)
-
Cryptic diversity in the Japanese mantis shrimp Oratosquilla oratoria (Crustacea: Squillidae): Allopatric diversification, secondary contact and hybridization
Scientific Reports (2017)
-
Credibility of statistical downscaling under nonstationary climate
Climate Dynamics (2016)
-
Analysis of the diet of finless porpoise (Neophocaena asiaeorientalis sunameri) based on prey morphological characters and DNA barcoding
Conservation Genetics Resources (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
