
Abstract
No definitive therapy exists to treat human metastatic tumors. We reported previously that down-regulation of Lin-7C is essential for metastasis of human squamous cell carcinomas (hSCCs). In this study, we investigated the chemical restoration of Lin-7C expression and demonstrated its effectiveness for suppressing the metastatic potential in human cancer cells. Ingenuity Pathway Analysis (IPA) identified candidate chemical agents, i.e., apomorphine, caffeine, risperidone, quetiapine and mirtazapine. Among them, mirtazapine, an antagonist of HTR2C, an upstream molecule of Lin-7C, caused substantial up-regulation of the Lin-7C/β-catenin pathway in a metastatic hSCC cell line and human melanoma-derived cell line in vitro and up-regulation did not contribute to cellular proliferation. Moreover, the antimetastatic effect of mirtazapine in these metastatic cell lines in vivo also was evident in multiple organs of immunodeficient mice with no marked side effects. The current data offer novel information for further study of antimetastatic activity in association with enhanced Lin-7C/β-catenin pathway activation with mirtazapine.
Similar content being viewed by others

Introduction
Despite accumulating evidence in clinical investigations, a certain proportion of patients who undergo chemotherapy to treat malignant tumors develop metastasis with a possible poor prognosis. Cancer cells may acquire the ability to metastasize in response to multiple molecular events. The metastatic process itself could partly represent cell-cell interactions; therefore, identification of their specific molecular markers for cancer metastasis is critical. Of them, the loss of cell-cell adhesion via the cadherin-catenin complex in squamous cell carcinoma (SCC) cells results from this irreversible modification. Aberrant expression of β-catenin (CTNNB1), an essential component of cadherin-based adherent junctions, is among the most important molecular event that contributes to metastasis1. In melanoma, while accumulation of nuclear translocation of β-catenin promotes oncogenic activity2, its down-regulation is associated with metastasis in vivo3. In addition, high phosphorylation of β-catenin in nuclei with overexpression of Dickkpf-1 is correlated with tumoral invasiveness and lymph node metastasis in human SCC (hSCC)4. Moreover, we previously reported that Lin-7C (also known as VELI3 or MALS-3) is required to suppress the metastatic potential of hSCC cells through β-catenin signaling5. β-catenin, whose function is cellular adhesion through the Wnt signaling pathway6, is down-regulated in the metastatic lesions of hSCC7 with decreased expression of E-cadherin, which is associated with specific stages of tumoral differentiation8. This evidence suggested the hypothesis that Lin-7C expression in metastatic cancer cells is part of the β-catenin signaling pathway that regulates β-catenin and that metastatic suppression can be achieved when a specific chemical agent for Lin-7C is used.
In the current study, we describe the novel efficacy of mirtazapine, which shows that the drug suppresses metastasis by activation of the Lin-7C/β-catenin pathway in vitro and in vivo.
Results
IPA
IPA clearly showed that 5-hydroxytryptamine receptor 2C (HTR2C) can be linked to Lin-7C (Figure 1). Furthermore, five pharmaceutical reagents, i.e., apomorphine, caffeine, risperidone, quetiapine and mirtazapine, were characterized as ligands for HTR2C (Figure 1), suggesting that they may affect Lin-7C expression.

IPA of Lin-7C-related genes and their interactive nodes in hSCC cells relative to human normal oral keratinocytes cells.
Each line and arrow between proteins represent known functional and physical interactions, with lines indicating direct relationships, i.e., the two molecules have physical contact. (Rx: HTR2C agonist and antagonists).
Selection of reagents affecting Lin-7C
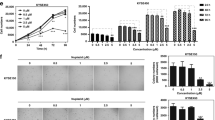
Of the reagents identified by IPA, apomorphine, caffeine and risperidone showed no marked increased sensitivity for Lin-7C mRNA expression in metastatic cell lines, SAS-H1, compared with the mirtazapine- and quetiapine-treated cells (Figure 2a, c–f). The optimal concentrations of mirtazapine and quetiapine for high Lin-7C mRNA levels were 10 μM and 100 μM, respectively. In addition, mirtazapine-treated SAS-H1 cells had a significantly elevated level (about a 4-fold increase) of Lin-7C mRNA compared with quetiapine-treated SAS-H1 cells (Figure 2a, f). Therefore, we adopted mirtazapine for further analyses. SAS cells showed no apparent effect of mirtazapine (Figure 2b).

qRT-PCR analysis of Lin-7C gene expression in hSCC cell lines.
While mirtazapine affects up-regulation of the Lin-7C expression in the SAS-H1 cells (a), other candidate HTR2C ligands show no expected effect for Lin-7C expression in the SAS and SAS-H1 cell lines (b–f). The results are expressed as the means ± standard error of the mean of values from three assays. The control cells were treated by 0.05% DMSO. DMSO = dimethyl sulfoxide.
Lin-7C status in hSCCs and effects of mirtazapine on Lin-7C and CTNNB1 expression
The expression levels of the three genes of interest (Lin-7C, calcium/calmodulin-dependent serine protein kinase [CASK] and CTNNB1) were unchanged (or the expression levels of Lin-7C and CASK were lower) in the SAS cells with or without mirtazapine (Figure 3a). In contrast, all three genes were highly expressed by SAS-H1 with mirtazapine compared to their counterparts (Figure 3b, c). In the mirtazapine-treated G-361 cells, while CASK expression was unexpectedly unchanged, significant up-regulation of Lin-7C and CTNNB1 was detected (Figure 3c).

The effect of mirtazapine on Lin-7C, CASK and CTNNB1 mRNA expression levels.
Treatment with mirtazapine (10 μM) is associated with up-regulation of the genes examined in the SAS-H1 (b) and G-361 (c) cells but not in those in the SAS (a) cells. The results are expressed as the means ± standard error of the mean of values obtained in triplicate. Mir (−) = without mirtazapine; Mir (+) = with mirtazapine.
Mirtazapine affected the HTR2C protein expression in SAS-H1 and SAS cells, but did not have an impact on G361 cells (Figure 4b, Supplementary Figure S1). However, significant up-regulation of the HTR2C-downstream molecules (Lin-7C/CASK/CTNNB1) was detected only in the highly metastatic cancer cells (SAS-H1 and G-361) when they were treated with mirtazapine (10 μM) for 24 hours. In contrast, no Lin-7C up-regulation was detected in SAS cells (Figure 4b, Supplementary Figure 1).

Functional analyses of Lin-7C gene in hSCC cells (SAS, SAS-H1) and malignant melanoma-derived cells (G-361).
(a) qRT-PCR shows that Lin-7C mRNA expression levels in the SAS-H1and G-361 cells are significantly (P < 0.05, Mann-Whitney U test) lower than that in the SAS cells. (b) Western blot analyses of HTR2C, Lin-7C, CASK and CTNNB1 protein expression levels in SAS, SAS-H1 and G-361 cells with or without mirtazapine. Each blot is normalized to each of the β-actin protein levels. (The blots were cropped and the full-length blots were included in the supplementary information). Mir (−) = without mirtazapine; Mir (+) = with mirtazapine.
Prevention of metastatic potential by mirtazapine in vitro
To investigate the effects of mirtazapine on cellular proliferation, cellular growth was monitored for 7 days and no difference was found between the cells treated or not treated with mirtazapine in all cell lines examined (Figure 5). We further investigated whether mirtazapine prevented cellular invasiveness/migration. To investigate the effects of mirtazapine on invasive behavior, we performed an invasiveness assay with the SAS-H1 and G-361 cell lines. After incubation in the medium with mirtazapine, both cell lines showed significantly (P < 0.05) reduced invasiveness (Figure 6a, b). Furthermore, mirtazapine enhanced reduction of cellular migration in both cell lines (Figure 6c, d). These results suggested that mirtazapine can significantly prevent metastatic potential in vitro.

Comparison of cellular proliferation in the control group (without mirtazapine) and the mirtazapine-treated groups.
To determine the effect of mirtazapine on cellular proliferation, SAS (a), SAS-H1 (b) and G-361 (c) cells are seeded in 6-well plates. Cellular proliferation was measured throughout 7 days of treatment. The results are expressed as the means ± the standard error of the mean of values from three assays. Mir (−) = without mirtazapine; Mir (+) = with mirtazapine; h = hours.

The functional effect of mirtazapine for antimetastatic potential in human metastatic cancer cells.
Typical results of the invasiveness assays for SAS-H1 (a) and G-361 (b) cells are indicated. The number of mirtazapine-treated cells entering the pores decreases significantly (P < 0.05, Mann-Whitney U test) compared with the control cells. Original magnification, ×100; scale bars, 100 μm. (c), (d) Migration assay of mirtazapine-treated cells. The wound area in cultured mirtazapine-treated cells has significantly (P < 0.05, Mann-Whitney U test) decreased, whereas there is a gap in the control cells. Original magnification, ×100; scale bars, 100 μm. Mir (−) = without mirtazapine; Mir (+) = with mirtazapine.
Prevention of metastatic potential by mirtazapine in vivo
To determine the metastasis-suppressing effect of mirtazapine in vivo, we inoculated 2 × 106 SAS, SAS-H1 and G-361 cells into the tongues of nude mice and primary tumors developed in 4 weeks. The average body weight of the mice in the mirtazapine-treated group never was lower than that of the control group at any time (Supplementary Figure 2). To quantify the disseminated metastatic human tumoral cells, we designed a qRT-PCR method based on real-time amplification of Alu repeat sequences present within murine tissue. Although no visible metastatic lesions were observed in distant organs, the results of Alu PCR indicated that the mice inoculated with SAS-H1 and G-361 cells contained significantly (P < 0.05) more human genomic DNA in their submandibular gland, lung, liver and kidney compared with the tissue injected with SAS cells (Figure 7a–c). Therefore, mirtazapine has antimetastatic potential specific to the highly metastatic cancer cells that show lower expression of Lin-7C (Figure 4a).

Quantitative analysis of spontaneous metastasis using different human tumor cells.
qRT Alu PCR is used to compare spontaneous metastasis of SAS, SAS-H1 and G-361 cells with mirtazapine treatment. The degree of metastasis is presented as the relative amount of metastasis normalized against murine GAPDH and as the actual number of human cells/tongue. While the control cells in the SAS-H1 and G-361 cells spontaneously metastasize into the submandibular glands, lungs, liver and kidney, mirtazapine-treated cells in the SAS-H1 and G-361 cells are not seen in these organs, suggesting that mirtazapine inhibits micrometastasis. The asterisks indicate significant (P < 0.05, Mann-Whitney U test) differences between the control cells and mirtazapine-treated cells. NT = no mirtazapine treatment; N.C. = negative control without human genomic DNA.
Discussion
Lin-7C, also known as VELI-3 or MALS-3, is a PDZ protein that may mediate protein-protein interactions and be a component of the mature cadherin-based junctional domain9. In hSCC cells, Lin-7C may act as a metastasis suppressor with a network consisting of 20 related genes including CASK, HTR2C and β-catenin5. Given that aberrant expression of the cellular adhesion molecules including β-catenin may contribute to metastasis and that the relative reagents regulating β-catenin have remained largely unknown, we wanted to characterize related genes and their potential chemicals that can restore the Lin-7C/β-catenin protein expression. The current study characterized mirtazapine, an effective reagent belonging to a novel metastasis-suppressing antagonist of HTR2C through the Lin-7C/β-catenin pathway in human metastatic cancer cells.
We initially focused on the differences in the status of Lin-7C expression and the related genes in a hSCC-derived cell line (SAS) compared with a metastatic cell line from SAS cells (SAS-H1). As expected, according to our previous report on other human oral SCC-derived cell lines5, the current series of target molecules, i.e., HTR2C, Lin-7C, CASK (also known as Lin-2) and β-catenin, had steady-state levels of mRNAs in the SAS cells, whereas SAS-H1 had significantly lower expression levels. These molecules, especially for β-catenin, are critical factors in human carcinogenesis10,11. In addition to Lin-7C, CASK is a cellular adhesion molecule with PDZ domains and thus can bind to the cellular surface11,12, indicating that there may be a relationship between CASK and cellular metastasis/invasion. The precise function of CASK in metastasis and its clinical relevance remain unclear. Several reports have shown that CASK may affect epidermal adhesion and wound healing together with cellular migration, indicating a potential clue13,14. In contrast, HTR2C seems to be altered in patients with oral SCC10, but the critical mechanism of human tumorigenesis remains inconclusive.
We next attempted to delineate the potential clinical use by chemical restoration of the Lin-7C/β-catenin pathway. Although IPA did not identify any reagents affecting Lin-7C expression, five different ligands specific for HTR2C, which is an upstream molecule of Lin-7C, were nominated. It is noteworthy that while caffeine was thought to be a promising reagent for enhancing cisplatin in human osteosarcoma cells15 and effective for treating patients with lung metastases16 and we thought that caffeine might be a strong candidate as an antimetastatic reagent with Lin-7C up-regulation, we did not observe a significant effect of caffeine or other reagents examined on Lin-7C expression in our metastatic cell lines and the non-metastatic cells. Despite evidence for an association between caffeine and apoptosis induction through the AKT/mTOR/S6K, NF-κB and MAPK pathways17, the reason why the effect of caffeine depends on cellular types is unclear. In contrast, based on the current study, mirtazapine is now the primary reagent of the HTR2C agonist leading to Lin-7C/β-catenin pathway induction. Mirtazapine is an antidepressant drug that induces antagonist activity at the 5HT2- and 5HT3-receptors including HTR2C18,19. It also has been suggested that there is a potential anticancer effect in human cancer cells with mirtazapine20. Several suggested mechanisms have been proposed by which mirtazapine may inhibit tumoral growth. Mirtazapine acts as cytotoxic agent in human osteosarcoma cells, while the ability to inhibit tumoral growth may require activation of the immune response and restoration of serotonergic system in a colon carcinoma-bearing animal model21. However, our work indicated that mirtazapine may affect cellular migration/invasion but not cellular proliferation in hSCC cells (Figure 5). Thus, it is likely that the anticancer effect of mirtazapine differs from cell to cell. Nevertheless, it is reasonable to speculate that this agent may at least enhance the beneficial effect of cancer treatment.
Finally, it is noteworthy that our in vitro results in hSCC cells were similar to those in a melanoma-derived cell line, G-361 and more importantly that our in vitro data also were reproduced in vivo. Whereas no visible metastatic lesions were observed in distant organs, such as the submandibular gland, lung, liver and kidney, the human-specific Alu sequence was detected only in the murine organs injected with the untreated metastatic cancer cells examined (Figure 7), indicating that mirtazapine may inhibit micrometastasis. This agrees with the results of our previous study in which we studied inhibition of metastasis in Lin-7C overexpressed in hSCC cell lines5. Although these findings provide new insight into the antimetastatic concept, a limitation of the current study was the relatively small number of cancer cell types. Further investigation is needed to reveal whether mirtazapine works with other types of human cancers. The strength of the current study is its clinical applicability in high-risk patients with malignant tumors, because mirtazapine has already been used to treat patients with depression, and, therefore, future clinical evaluation with appropriate doses can be performed.
In conclusion, our data supported a potential mechanism by which induction of the Lin-7C/β-catenin pathway in cancer cells is essential for eliminating the risk of metastasis and mirtazapine is a strong candidate for restoration of this pathway. This discovery may provide a novel strategy that expands the application of mirtazapine in the future treatment of human cancer metastasis.
Methods
Antibodies and reagents
The reagents used in the current study were goat antihuman Lin-7C polyclonal antibody (sc-11504, Santa Cruz Biotechnology, Santa Cruz, CA, USA), goat antihuman HTR2C polyclonal antibody (sc-17797, Santa Cruz Biotechnology), rabbit antihuman CASK polyclonal antibody (sc-10777, Santa Cruz Biotechnology), rabbit antihuman β-catenin (CTNNB1) monoclonal antibody (NB110-56924, Acris Antibodies Inc., San Diego, CA, USA) and mouse antihuman β-actin monoclonal antibody (364375, Abcam, Cambridge, UK). The following reagents were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA): an agonist for HTR2C (apomorphine) and HTR2C antagonists such as mirtazapine, caffeine, risperidone and quetiapine.
Cell lines
The hSCC-derived cell line SAS and the human malignant melanoma cell line G-361 were purchased from RIKEN BioResource Center (Tsukuba, Ibaraki, Japan). Kazuhiko Okumura (Health Science University of Hokkaido, Hokkaido, Japan) kindly provided the highly metastatic hSCC cell line SAS-H1 (derived from SAS)22. SAS and SAS-H1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (D6046, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) and antibiotics. The G-361 cells were grown in McCoy's 5A modified medium (M8403, Sigma-Aldrich) supplemented with 10% FBS and antibiotics. All cell lines were cultured at 37°C in a humidified atmosphere in the presence of 5% CO2-95% air.
IPA
The target gene lists focused on Lin-7C5 were overlaid on a cellular pathway map in the IPA software (http://www.ingenuity.com). We also carried out IPA to identify the candidate chemicals that may modulate Lin-7C and/or its interacting molecules.
mRNA expression analyses
Total RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Complementary DNA was generated from 5 μg of total RNA by using Ready-To-Go You-Prime First-Strand Beads (GE Healthcare, Buckinghamshire, UK) and oligo (dT) primer (Hokkaido System Science, Sapporo, Japan) according to the manufacturers' instructions. Quantitative real-time reverse transcription-PCR (qRT-PCR) was performed to evaluate the mRNA expression levels of Lin-7C, HTR2C, CASK and CTNNB1. The qRT-PCR analyses were carried out using primers and probes designed by the Universal Probe Library (Roche Diagnostics, Mannheim, Germany). The primer sequences used were Lin-7C, forward, 5′-TTC CAT TCC TAG AGA CAA ACC AG -3′; reverse, 5′-TTT TCC TTC TTT GCT TCA GCT AC -3′; and universal probe #77; HTR2C, forward, 5′- CCG AGT CCG TTT CTC GTC TA-3′; reverse, 5′- TCG CGG GTG TTA GCT GAT -3′; and universal probe #27; CASK, forward, 5′- TGG AAA ACT CCA AAA ATG GAA-3′; reverse, 5′- AAG CTA CTC GCC ATT CCT GA-3′; and universal probe #2; and CTNNB1, forward, 5′- GAA TCC ATT CTG GTG CCA CT-3′; reverse 5′- CCT CAG GAT TGC CTT TAC CA-3′; and universal probe #17. All qRT-PCR procedures were performed using the LightCycler® 480 PCR system (Roche). Amplifications were initiated by a 10-minute pre-incubation at 95°C, followed by 45 cycles of 10 seconds at 95°C for template denaturation and 30 seconds at 55°C for primer annealing/extension and a cooling step for 30 seconds at 40°C. The transcript amounts were estimated from the respective standard curves and normalized to the antiglyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-CAT CTC TGC CCC CTC TGC TGA-3′; reverse, 5′-GGA TGA CCT TGC CCA CAG CCT-3′; and universal probe #60) transcript amount determined in corresponding samples.
Immunoblotting
The cells were washed twice with cold phosphate-buffered saline (PBS) and centrifuged briefly. The cell pellets then were incubated at 4°C for 30 minutes in a lysis buffer (7 M urea, 2 M thiourea, 4% w/v CHAPS and 10 mM Tris (pH 7.4) with a proteinase inhibitor cocktail (Roche). The protein concentration was measured using a Bio-Rad Protein Assay, (Bio-Rad Laboratories, Hercules, CA, USA). Protein extracts (20 μg) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis in 4–12% gel, transferred to nitrocellulose membranes and blocked for 1 hour at room temperature in Blocking One (Nacalai Tesque, Tokyo, Japan). The membranes were incubated with primary antibodies for 4 hours at room temperature. The membranes were washed with 0.1% Tween-20 in Tris-buffered saline, incubated with secondary antibody and coupled to horseradish peroxidase-conjugated antirabbit, antigoat, or antimouse IgGs (Promega, Madison, WI, USA) for 1 hour at room temperature. The proteins were detected by SuperSignal Chemiluminescent substrate (Thermo, Waltham, MA, USA). Finally, immunoblotting analysis results were visualized by exposing the membrane to a cooled CCD camera system (ATTO, Tokyo, Japan). Signal intensities were quantitated using the CS Analyzer software version 3.0 (ATTO).
Effects of HTR2C ligands on Lin-7C expression
Five HTR2C ligands, mirtazapine, apomorphine, caffeine, risperidone and quetiapine, were studied. To examine the effect of these reagents on Lin-7C mRNA expression, we performed qRT-PCR analysis in SAS and SAS-H1 cells. The effective concentrations of exposure to reagents were chosen based on previous reports23,24,25,26,27. Cells were treated with each reagent and kept in a CO2 incubator for 24 hours. After treatment, the cells were washed twice with PBS and total RNA was isolated for further experiments. The control cells were treated with dimethyl sulfoxide. Values also were normalized with respect to control cells.
Proliferation assay
To evaluate the effect of mirtazapine on cellular proliferation, we analyzed cellular growth in SAS, SAS-H1 and G-361 cells. The cells were seeded in 6-well plates at a density of 1 × 104 viable cells/well. The experiments were carried out for 7 days by counting the cells every 24 hours. At the indicated time points, the cells were trypsinized and counted using a hemocytometer in triplicate samples. We compared the numbers between the cells treated with mirtazapine and control cells.
Invasiveness assay
A total of 2.5 × 105 cells (SAS, SAS-H1 and G-361) were treated with vehicle or mirtazapine (10 μM) for 24 hours. The cells then were resuspended in the serum-free medium and seeded on a polyethylene terephthalate membrane insert with a pore size of 3 μm in a trans-well apparatus (Becton Dickinson Labware, Franklin Lakes, NJ, USA). In the lower chamber, 2 ml of serum-free medium with vehicle or mirtazapine (10 μM) was added as a chemoattractant. After the cells were incubated for 72 hours at 37°C, the insert was washed with PBS and the cells on the top surface of the insert were removed with a cotton swab. Cells adhering to the lower surface of the membrane were stained with crystal violet/methanol and the numbers of cells entering the pores in five random fields were counted using a light microscope at ×100 magnification.
Migration assay
To investigate the antimigration potential of mirtazapine, the cells (SAS, SAS-H1 and G-361) were seeded in a 6-well plate with 10% FBS/DMEM until a confluent monolayer formed. One wound was created in the middle of each well using a micropipette tip. After treatment with mirtazapine, we replaced the medium with a free-serum medium and incubated it at 37°C at 5% CO2. The results were visualized by measuring the wound area that was free of cells using Lenaraf 220 b software (Vector Japan Co, Ltd, Tokyo, Japan) (http://www.vector.co.jp/soft/dl/win95/art/se312811.html). The mean value was calculated from data obtained from three separate chambers.
Mirtazapine administration in vivo
To investigate the antitumor activity of mirtazapine, we used xenograft models in the SAS, SAS-H1 and G-361 cell lines. The cells (2 × 106) were injected directly into the tongues of BALB/cAnNcrj-nu/nu mice (Charles River Japan Inc., Yokohama, Japan). The mice were assigned randomly into three groups: no treatment (n = 5), control (n = 5) and mirtazapine treated (n = 5). Mirtazapine (10 mg/kg) was suspended in a 1% aqueous solution of Tween 80 (Sigma) and administered orally once daily for 4 weeks. The control groups received the vehicle through the entire experimental period. The animals were sacrificed after 4 weeks. The care and treatment of experimental animals were performed in accordance with institutional guidelines.
Quantitative detection of human tumor cell metastasis
Four weeks after transplantation, several organs including the tongue, submandibular gland, lung, liver and kidney were dissected from the mice implanted with the tumor cells. The body weight was measured throughout the 4 weeks of treatment. The genomic DNA was extracted immediately from each of the homogenates using the Wizard SV Genomic purification system (Promega). To determine the tumor cell-derived DNA in each tissue, 20 ng of DNA was subjected to qRT-PCR to detect the human Alu sequence. To correlate the amount of the Alu PCR signal (and thereby the amount of human tumor cell metastasis) with the amount of amplified genomic DNA (and thereby the amount of murine tissue), we used the murine GAPDH gene as an internal control. The primers specific for the human Alu sequences were: forward, 5′- ACG CCT GTA ATC CCA GCA CTT -3′ and reverse, 5′- TCG CCC AGG CTG GAG TGC A -3′. qRT-PCR was performed using the SYBR® Green qRT-PCR Master Mix Kit according to the manufacturer's protocol (Roche). The PCR conditions included polymerase activation at 95°C for 3 minutes followed by 40 cycles at 95°C for 30 seconds, 63°C for 30 seconds and 72°C for 30 seconds. Each assay included a negative control (RNase free water), a positive control (human genomic DNA) and the experimental samples in triplicate. A quantitative measurement of murine DNA was obtained through amplification of the murine GAPDH genomic DNA sequence with murine GAPDH primers (forward, 5′- AAC GAC CCC TTC ATT GAC -3′ and reverse, 5′- TCC ACG ACA TAC TCA GCA C -3′) using the same PCR conditions described for Alu. The threshold cycle (Ct) of each sample was recorded as a quantitative measure of the amount of PCR product in the sample using the LightCycler 480 PCR system. The Alu signal was normalized against the relative quantity of murine GAPDH and expressed as ΔCt = (CtAlu − CtGAPDH). The changes in the Alu signal relative to the total amount of murine genomic DNA were expressed as ΔΔCt = ΔCttreatment − ΔCtcontrol. The relative changes in metastasis then were calculated as 2−ΔΔCt.
Statistical analysis
Statistical analyses were performed using SPSS 17.0 software (SPSS Inc., Chicago, IL, USA) or Microsoft Excel (Microsoft, Redmond, WA, USA). Single-group data were assessed using the Mann-Whitney U test. The statistical significance of the metastatic levels of each tissue was evaluated using one-way analysis of variance with the post hoc Tukey-Kramer test for multiple comparisons. Two-tailed P values < 0.05 were considered significant. The data are expressed as the mean ± standard error of the mean.
References
Xu, L. et al. Aberrant expression of beta-catenin and E-cadherin is correlated with poor prognosis of nasopharyngeal cancer. Hum Pathol 44, 1357–1364 (2013).
Arozarena, I. et al. In melanoma, beta-catenin is a suppressor of invasion. Oncogene 30, 4531–4543 (2011).
Takahashi, Y., Nishikawa, M., Suehara, T., Takiguchi, N. & Takakura, Y. Gene silencing of beta-catenin in melanoma cells retards their growth but promotes the formation of pulmonary metastasis in mice. Int J Cancer 123, 2315–2320 (2008).
Ogoshi, K. et al. Dickkopf-1 in human oral cancer. Int J Oncol 39, 329–336 (2011).
Onda, T. et al. Lin-7C/VELI3/MALS-3: an essential component in metastasis of human squamous cell carcinoma. Cancer Res 67, 9643–9648 (2007).
Polakis, P. Wnt signaling and cancer. Genes Dev 14, 1837–1851 (2000).
Bankfalvi, A. et al. Gains and losses of adhesion molecules (CD44, E-cadherin and beta-catenin) during oral carcinogenesis and tumour progression. J Pathol 198, 343–351 (2002).
Saito, Y., Takazawa, H., Uzawa, K., Tanzawa, H. & Sato, K. Reduced expression of E-cadherin in oral squamous cell carcinoma: relationship with DNA methylation of 5′ CpG island. Int J Oncol 12, 293–298 (1998).
Perego, C. et al. Invasive behaviour of glioblastoma cell lines is associated with altered organisation of the cadherin-catenin adhesion system. J Cell Sci 115, 3331–3340 (2002).
Cha, J. D., Kim, H. J. & Cha, I. H. Genetic alterations in oral squamous cell carcinoma progression detected by combining array-based comparative genomic hybridization and multiplex ligation-dependent probe amplification. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 111, 594–607 (2011).
Wang, Q. et al. CASK and its target gene Reelin were co-upregulated in human esophageal carcinoma. Cancer Lett 179, 71–77 (2002).
Martinez-Estrada, O. M. et al. Association of junctional adhesion molecule with calcium/calmodulin-dependent serine protein kinase (CASK/LIN-2) in human epithelial caco-2 cells. J Biol Chem 276, 9291–9296 (2001).
Giangreco, A., Jensen, K. B., Takai, Y., Miyoshi, J. & Watt, F. M. Necl2 regulates epidermal adhesion and wound repair. Development 136, 3505–3514 (2009).
Marquez-Rosado, L., Singh, D., Rincon-Arano, H., Solan, J. L. & Lampe, P. D. CASK (LIN2) interacts with Cx43 in wounded skin and their coexpression affects cell migration. J Cell Sci 125, 695–702 (2012).
Xie, X. K., Yang, D. S., Ye, Z. M. & Tao, H. M. Enhancement effect of adenovirus-mediated antisense c-myc and caffeine on the cytotoxicity of cisplatin in osteosarcoma cell lines. Chemotherapy 55, 433–440 (2009).
Kimura, H. et al. Caffeine-potentiated chemotherapy for metastatic osteosarcoma. J Orthop Sci 14, 556–565 (2009).
Miwa, S. et al. Caffeine induces apoptosis of osteosarcoma cells by inhibiting AKT/mTOR/S6K, NF-kappaB and MAPK pathways. Anticancer Res 32, 3643–3649 (2012).
Anttila, S. A. & Leinonen, E. V. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev 7, 249–264 (2001).
Millan, M. J. et al. Mirtazapine enhances frontocortical dopaminergic and corticolimbic adrenergic, but not serotonergic, transmission by blockade of alpha2-adrenergic and serotonin2C receptors: a comparison with citalopram. Eur J Neurosci 12, 1079–1095 (2000).
Pan, C. C. et al. The antidepressant mirtazapine-induced cytosolic Ca2+ elevation and cytotoxicity in human osteosarcoma cells. Chin J Physiol 49, 290–297 (2006).
Fang, C. K. et al. Mirtazapine inhibits tumor growth via immune response and serotonergic system. PLoS ONE 7, e38886 (2012).
Muramatsu, H. et al. Superoxide dismutase in SAS human tongue carcinoma cell line is a factor defining invasiveness and cell motility. Cancer Res 55, 6210–6214 (1995).
Chanrion, B. et al. Inverse agonist and neutral antagonist actions of antidepressants at recombinant and native 5-hydroxytryptamine2C receptors: differential modulation of cell surface expression and signal transduction. Mol Pharmacol 73, 748–757 (2008).
Newman-Tancredi, A. et al. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. III. Agonist and antagonist properties at serotonin, 5-HT(1) and 5-HT(2), receptor subtypes. J Pharmacol Exp Ther 303, 815–822 (2002).
Conde, S. V. et al. Caffeine inhibition of rat carotid body chemoreceptors is mediated by A2A and A2B adenosine receptors. J Neurochem 98, 616–628 (2006).
Pacchioni, A. M., Gabriele, A., Donovan, J. L., DeVane, C. L. & See, R. E. P-glycoprotein inhibition potentiates the behavioural and neurochemical actions of risperidone in rats. Int J Neuropsychopharmacol 13, 1067–1077 (2010).
Megens, A. A. et al. Survey on the pharmacodynamics of the new antipsychotic risperidone. Psychopharmacology (Berl) 114, 9–23 (1994).
Acknowledgements
We thank Ms. Lynda C. Charters for editing this manuscript. This work was supported partly by a Grant-in-Aid for Exploratory Research from the Ministry of Education, Science, Sports and Culture of Japan (24659885) and in part by The 21st Century Center of Excellence (COE) Program grant.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: K.U., A.K., H.T. Performed the experiments: K.U., A.K., T.S., Y.S., T.B. Analyzed the data: K.U., A.K., K.S., K.F. Contributed reagents/materials/analysis tools: K.U., A.K., T.S., Y.S., T.B., Y.S., K.O., M.S., H.T. Wrote the manuscript: K.U., A.K., T.B., H.T.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Uzawa, K., Kasamatsu, A., Shimizu, T. et al. Suppression of metastasis by mirtazapine via restoration of the Lin-7C/β-catenin pathway in human cancer cells. Sci Rep 4, 5433 (2014). https://doi.org/10.1038/srep05433
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05433
This article is cited by
-
Repurposing the serotonin agonist Tegaserod as an anticancer agent in melanoma: molecular mechanisms and clinical implications
Journal of Experimental & Clinical Cancer Research (2020)
-
Active repurposing of drug candidates for melanoma based on GWAS, PheWAS and a wide range of omics data
Molecular Medicine (2019)
-
Evidence for Critical Role of Lymphocyte Cytosolic Protein 1 in Oral Cancer
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
