
Abstract
Mycobacterium abscessus (Ma) is an emerging human pathogen that causes both soft tissue infections and systemic disease. We present the first comparative whole-genome study of Ma strains isolated from patients of wide geographical origin. We found a high proportion of accessory strain-specific genes indicating an open, non-conservative pan-genome structure and clear evidence of rapid phage-mediated evolution. Although we found fewer virulence factors in Ma compared to M. tuberculosis, our data indicated that Ma evolves rapidly and therefore should be monitored closely for the acquisition of more pathogenic traits. This comparative study provides a better understanding of Ma and forms the basis for future functional work on this important pathogen.
Similar content being viewed by others


Introduction
Rapidly growing mycobacteria (RGM) are found in soil, water and as contaminants on medical equipment1. Recent decades have witnessed increasing association of RGMs and human disease. Although this trend is partly explained by better methods of detection in clinical samples, the RGM species Ma (discovered in 1953)2 may be actively evolving into a more virulent pathogen. Ma causes skin and soft tissue infections and contributes to the respiratory pathology of cystic fibrosis, chronic lung diseases and lung transplantation3, where treatment is complicated because Ma is resistant to many antibiotics. Patterns of drug resistance differ among the three recognized Ma subspecies2,4: M. abscessus (sensu stricto), M. massiliense and M. bolletii5,6,7. Here, we compare and report the sequence and analysis of 12 Ma genomes in the comparative context of an additional 28 genome sequences recently deposited in the public database.
Results
We sequenced on technology platform Illumina GA 2X, twelve Ma clinical isolates (M18, M24, M93, M94, M115, M139, M148, M152, M154, M156, M159, M172) cultured from patients attending the University Malaya Medical Centre (UMMC), Kuala Lumpur, Malaysia, between the years 2006 and 2011. All patients, Malaysian adults of Chinese, Malay and Indian ethnicity, except a Nepalese and a Myanmar national residing in Malaysia (isolates M139 and M172 respectively) presented with symptoms suggestive of lower respiratory tract pathology. Eleven isolates were from sputum or bronchoalveolar lavage; one from a cervical lymph node. For comparison, we downloaded from NCBI Genbank and analyzed the genome sequences of 28 strains isolated from different geographical locations in the United States, UK and France. Only the reference strain Ma ATCC 19977 is a complete genome, the rest being drafts.
General features of the Ma genome
We assessed completeness of the draft genomes by mapping all assemblies onto the complete reference genome using the NUCmer program. The percentage of the reference genome that was successfully covered by the 39 draft genomes is from 84.2–99.9% with an average of 91.1% while the identity is around 97.4–99.9% (Supplementary Table S1). The Ma genome has a size range of 4.8–5.5 Mbp and a G+C content of 63.8–64.3%. The number of rRNAs (3) was the same in all strains including the type strain ATCC 19977. The variation in genome identity (around 97.4–99.9%) reflects different subspecies or distinct lineages within Ma. (Supplementary Fig. S2). Our 12 new genome sequences were annotated using the Rapid Annotation using Subsystem Technology (RAST) pipeline8. For consistency, the database derived genomes were re-annotated with the same pipeline. We found the number of functional genes ranging from 4,709 to 5,605, whereas the number of tRNAs (45–82) was significantly higher in some strains.
Genome organization
We aligned all 40 genome sequences using the Mauve software9. The results showed Ma genomes to be mostly collinear apart from some prominent genome rearrangements. The total length of conserved regions was about 2.95 Mb, which accounts for 35% of the pan-genome. The multiple alignments of all 40 Ma genomes using Mauve showed many synteny blocks across genomes interrupted by prophages, deletions and rearrangements (data not shown). Manual inspection of multiple genome alignments in Mauve identified large inversions (Supplementary Fig. S3): a 350 kb inversion was observed in strain 4S0206, a 220 kb inversion was observed in 4S0726RA, 4S0303, 6G0728S and 5S0304 and a large 700 kb inversion is present in strains 4S0726RA and 5S1215.
Ma is not a conservative human pathogen
Grouping together all functional genes from the 40 Ma genomes, we identified 12,656 pangenomic gene clusters, of which, 3,354 were classified as core gene clusters (shared by all strains), 4,001 as dispensable (shared by 2–39 strains) and 5,301 as strain-specific genes. The core clusters accounted for approximately 26% of the total gene clusters. On comparison with other bacteria, the Ma genome structure appears to be non-conservative; for instance, Salmonella Paratyphi A, reported to be a conservative pathogen, has a pan-genome size of 4,252 gene clusters with 87.5% of clusters in the core genome10 and Streptococcus agalactiae has 2,571 genes with core genes accounting for 57.8% of the pan-genome11. The Ma genome may be more akin to the open pan-genome of Escherichia coli which has only 6% of its pan-genome in core gene clusters12.
Ma has a rapidly evolving ‘open’ pan-genome
Bacterial pan-genomes are described as open (infinite) or closed13. To predict the pan-genome and core genome sizes in Ma by extrapolation we calculated the gene clusters and core clusters for N genomes, where N is the number of Ma genomes (N = 1,2,3….38,39,40). For each N genome, the pan-genome size and core genome for each of the permutations of genome comparisons was predicted. Our results showed that the pan-genome size increased rapidly when the number of genomes increased (Figure 1A). The curve for the pan-genome size can be represented by the following mathematical function:

In this function, Y represents the pan-genome size while X represents the number of sequenced genomes. By using this model, we would expect the pan-genome size to be infinite when X- > ∞.

Size prediction for Ma pan- and core- genomes.
(A) Curves for the Ma, M. abscessus (sensu stricto) and M. massiliense pan-genomes and core genomes. M. bolletii is not included in this analysis because there is only one strain in this study. The light blue dots denote the Ma pan-genome size for each genome comparison. The dark blue dots denote the Ma core genome size for each genome comparison. The median values were connected to represent the relationship between number of genomes and gene clusters. (B) Curve for the number of expected new genes detected with every increase in the number of Ma genomes.
This is counter to the general case where we expect the number of new genes detected to converge to zero with an increase in the number of genomes analysed. Instead, here, the rate of new discovery stabilizes at about 100 new genes per additional genome (Figure 1B). For instance, in our 40 genomes, 595 new genes were detected when the second genome was added to the first Ma genome, but the number of new genes detected decreased to 113 when 39 genomes were added. By mathematical extrapolation, it is predicted that there would be about 112 new genes detected when each additional genome is added. We have also performed the pan-genome analysis at subspecies level for M. abscessus (sensu stricto) and M. massiliense. In line with our observation in the pan-genome analysis of Ma, both subspecies M. abscessus (sensu stricto) and M. massiliense have also an open pan-genome (Figure 1A). Ma's infinite or open pan-genome indicates that Ma is continuously gaining new genes, is actively evolving and thus capable of rapidly acquiring new phenotypes.
Comparative analysis of the Ma core genome
Functional enrichment analysis
To apportion distinct functions to the Ma core and accessory genes, we performed a classification using the RAST system8. As expected, the Ma core genes are significantly enriched in basic functions such as cofactors and vitamins (~15%), amino acids and derivatives (~18%). In contrast, Ma accessory genes are enriched in transposable elements such as plasmids, phages and prophages, indicating that phage/prophages have significantly played a role in the evolution and adaptation of Ma species in different environments (Figure 2). Other functional categories that are enriched in accessory genes are the fatty acids, lipids and isoprenoids and metabolism of aromatic compounds.

Functional classification of the core and accessory genes.
Distribution of core and accessory genes in the functional subsystem classified using RAST. Categories that show significant difference of distribution between the core and accessory genes are marked with a red asterisk on the top of the bar (p value after Bonferroni correction = 0.05).
Many Ma accessory genes are lineage-specific
We also studied the frequency of accessory genes across different numbers of genomes (Supplementary Fig. S4A). Genes present in a single genome represent the strain-specific genes; while at the opposite end of the scale, genes found in all 40 genomes represent the Ma core genome. Of 9,302 accessory genes, 5,301 (42%) genes are present in only one genome; therefore they are lineage-specific, suggesting that a large proportion of the accessory genes were recently acquired by Ma.
Comparisons between gene lists
The current collection of sequenced Ma genomes contains sequences from all three Ma subspecies. This provided an opportunity for us to identify subspecies-specific genes. We identified and compared the core genes in each subspecies (Supplementary Fig. S4B). Our analysis showed that 3,354 genes are shared by all three subspecies; 19 genes specific to M. massiliense, 24 genes specific to M. abscessus (sensu stricto) and 722 genes specific to M. bolletii (Supplementary Fig. S4B). The large number of M. bolletii–specific genes could be due to our inclusion of only one strain (M24) from this subspecies. Therefore most of these genes might be M. bolletii M24-specific, rather than M. bolletii–specific. We further reduced the 722 genes to 38 after filtering these genes by comparing with the M. bolletii BD genome which has been recently submitted to the public database. Thus, there are only 38 M-bolletii specific genes while the remaining 684 genes are M24-specific genes. These 38 M. bolletii-specific genes might be responsible for unique features of M. bolletii compared to the other two subspecies.
Mycobacteria are divided into fast growing mycobacteria and slow growing mycobacteria (SGM). To look at the genetic differences between these two main groups, we compared Ma, a fast growing mycobacterium with two slow growing mycobacteria, M. tuberculosis CCDC 5079 and M. avium K10. There are 1,608 genes shared by all three species, 1,413 genes in Ma but not in the slow growers and 785 genes that are found in both M. tuberculosis and M. avium (slow growers) but not in Ma (Supplementary Fig. 4C). Of these 785 possibly slow grower-specific genes, 17 are orthologous (with identity and coverage at least 50%) to the 89 genes reported by Beste to be potentially related to slow growth in mycobacteria63.
tRNA islands are observed in some Ma strains
Among the 12 strains from Malaysia, M. bolletii M24 stood out as the most resistant to antibiotics and M94 had the highest number of tRNAs. Examining these two genomes, we found two uncommonly large tRNA islands. The tRNA island in the M24 genome spanned the region from nt 11,282 to nt 20,728 in contig4. This 9,445 bp region has 35 putative tRNAs, 1 pseudo-tRNA-CGG and a small number of functional genes. The second, smaller tRNA island of M94 has a genomic length of 6,388. This region has 17 putative tRNAs and 3 genes. To rule out the possibility that the observed tRNA islands are assembly artifacts, we re-sequenced the genomes of M24 and M94 using the long read PacBio sequencing technology. After de novo assembly of the error-corrected PacBio reads we observed the same islands and could smoothly map the PacBio confirmed islands onto the assemblies of the respective strains. We conclude that the tRNA islands are unlikely to be assembly artifacts (Figure 3). Subsequently, we looked for tRNA islands in the remaining Ma genomes and found them in 12 other strains (Supplementary Table S11). The 5S strains have 36 tRNAs in their islands and our strain M24 has 35, the highest numbers among the observed tRNA islands. The densest islands are in 6G1108, 6G0728S, 6G0125S and 6G0728R with 55.9% of each island covered by tRNA sequences and an average distance of 59 bp between tRNAs. The order of the tRNAs in the island found in our M24 strain is similar to that in the other 5S strains from the US group, suggesting that these tRNAs might be from the same source (Supplementary Fig. S5). As these tRNA islands were observed in strains isolated from distinct geographic origins (Malaysia and United States) and belonging to distinct subspecies (M24 is M. bolletii while the rest of the strains are M. massiliense), the insertion of this genomic island could have occurred independently at least twice during the Ma genome evolution.

tRNA islands and validation by a single molecule and long read PacBio data.
(A) A putative tRNA island in the contig4 of M24 strain. (B) A putative tRNA island in the contig33 of M94 strain. Both islands are supported by PacBio data. PacBio reads track: blue subtrack shows the coverage of reads; red line = forward strand and green = reverse strand.
The occurrence of tRNA anticodon types in Ma genomes
We examined the frequency of tRNA anticodon types in Ma genomes and compared it with those in other bacterial species (Supplementary Fig. S6 & S7). Strikingly, there are large numbers of “methionine” tRNAs in these Ma strains. For instance, M24 has six copies of tRNAs containing CAU anticodons (methionine tRNAs), which is the highest number of copies found in the Ma genomes, followed by five copies in strains 5S0304, 5S1212, 5S0921, 5S1215, 5S0817, 5S0708, 5S0422 and 5S0421 and four copies in M94, 6G0728R, 6G0125S, 6G0728S and 6G1108 (Supplementary Fig. S6). This is interesting because almost all bacteria have at least three “methionine” tRNAs (ie genetically encoded with the methionyl anticodon ‘CAU’): at least one initiator tRNA-Met (“tRNAMet”); at least one elongator tRNA-MET (“tRNAfMet”); and at least one (“tRNAMet->Ile”) that is the substrate for tRNA modification by the enzyme TilsS (Supplementary Fig. S6). TilS modifies the ‘CAU’ anticodon by the covalent addition of lysine to one of the anticodon bases to form the modified nucleotide “lysidine” rendering the lysinylated tRNA capable of decoding the isoleucine codon ‘AUA’. The cognate isoleucine uau anticodon is generally absent from bacterial genomes, thus, these extra methionine tRNAs could be interesting in that they might boost expression of AUA codon-rich gene subsets, as appears to occur in the LEE pathogenicity islands of E. coli (Mitchell, unpublished data).
Prophage identification in sequenced Ma genomes
Phage-mediated horizontal gene transfer events have been reported to play an important role in increasing bacterial virulence and promoting antimicrobial resistance, for example, by bringing in virulence and resistance genes14. Here we predicted the prophages in all Ma genomes using PHAST (Phage Search Tool)15. The integration of these phages into Ma genomes have contributed to the diversity of this emerging pathogen. Interestingly, 9 different putative intact prophages were predicted in 18 out of 40 Ma genomes (Supplementary Fig. S8 & S9; Supplementary Table S12). In addition to many phage protein orthologs, we also noticed att sites (attL and attR) and ancillary enzymes such as integrase, in some of the prophages. att sites direct site-specific integration and recombination of phage DNA into the bacterial genome. Of the 9 putative intact prophages that we found, 6 prophages are present in more than one genome and 3 prophages are strain-specific (Supplementary Fig. S8). The 6 shared prophages contributed around 1,588 dispensable genes and the other 3 unique prophages resulted in 150 strain-specified genes. When mapped onto the phylogenetic tree derived from SNP analysis, the prophage distribution further subdivides the clade containing Ma ATCC 19977 into three branches (Figure 5B). This means that rate of prophage invasion is faster than the rate of mutation, implying rapid evolution of Ma on this branch. We hoped to define the provenance of these prophage sequences, but found no good matches in Genbank. Their origin remains obscure.

Comparison between virulence genes in Ma and M. tuberculosis.
In the last column is the M. vaccae used as a reference for the comparison. There are only a few virulence genes found in this non-pathogenic species compared to Ma and M. tuberculosis which are known pathogens.

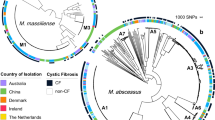
Phylogenetic relationship of 40 Ma strains and intact prophage distribution.
(A) The phylogenetic tree was generated using core genome SNPs and the maximum likelihood method. Bootstrap numbers were generated in 1,000 runs. Nodes with bootstrap support values of 100 are indicated. (B) Mapping of putative intact prophages onto the SNP-based phylogenetic tree. The intact prophage distribution further subdivides the clade into branches. “I” means no intact prophage.
Virulence genes in Ma genomes
Like other pathogens, Ma deploys virulence factors to escape from the host's immune system, to promote growth in the hostile environment of the host cell and to enhance binding to the host cell for invasion. The main virulence factor has been attributed to Ma's ability to persist inside phagocytes to cause prolonged infections with many recurrences16. This ability for intracellular persistence has been observed mostly with rough colony types that are deficient in certain surface glycopeptidolipids17 and are capable of cord formation as well as escaping from phagocytosis18. In Ma cultures from clinical material, rough colonies are not infrequently recovered alongside smooth colonies (authors' observation). This suggests that, in vivo, a subpopulation of cells of increased virulence are present in most infecting strains. To better understand the pathogenic potential of Ma, we searched for virulence genes in the 40 Ma genomes by performing a BLAST search against the Virulence Factor Database (VFDB) using stringent criteria (see Method section)19,20,21. Around ninety non-redundant virulence genes were predicted across the 40 Ma genomes, divided into functional categories including proteins inhibiting antimicrobial factors, cell envelope proteins, metal transporter proteins and other virulence proteins with unknown function22.
Cell envelope proteins
To cause disease, Ma must attach to and invade the host cell. Ma surface proteins mediate these interactions. We found that the 40 Ma genomes contain conserved virulence genes erp, fbp, mce and hbhA that encode surface proteins, The erp gene, also known as P36, encodes a cell wall associated surface protein. Although erp's function is unknown, M. tuberculosis erp mutants are not able to replicate in the cultured macrophage23. fbp is another virulence gene that assists the adhesion of mycobacteria to the surface and promotes their entry into the host cell24,25. Like other mycobacteria, Ma possesses a large group of surface exported Mce proteins organized in large operons. These proteins enable mycobacteria to enter into mammalian cells and survive in the macrophages26.
Protein inhibiting antimicrobial factors
Pathogenic mycobacteria are able to counteract and escape macrophage killing by increasing the resistance to host-produced toxic compounds, by supressing the induction of apoptosis and by inhibiting phagosome maturation23. Host cells produce reactive oxygen species (ROS) and reactive nitrogen species (RNS) that damage mycobacteria. In our analysis, we observed the virulence genes hspX, ahpC, sodC and katG in all Ma genomes examined. These genes encode enzymes that detoxify ROS and RNS, thus avoiding bacterial lysis. Also present are phoP and secA2, genes that may allow Ma to arrest phagosomal killing and digestion23,27. Finally, Ma contains the anti-apoptotic factors nuoG, secA and sodA which are essential for the full virulence of this species. NuoG, a subunit of type 1 NADH dehydrogenase, has been recently reported as able to suppress neutrophil apoptosis. The absence of this gene accelerates CD4 T cell priming and induces apoptosis28. SecA2 which is also required for phagosome maturation arrest encodes a preprotein translocase ATPase that translocates superoxide dismutase (SodA). Hinchey et al. (2007) indicated that SodA secretion is the major SecA2-dependent process that inhibits cell apoptosis29 and that M. tuberculosis with mutant sodA has proved more susceptible to the bactericidal activity of hydrogen peroxide30.
Metal transporter proteins
Metals like iron, magnesium, zinc, cobalt and manganese are essential for intracellular prokaryotic growth, for example, as enzyme co-factors. At the same time, the accumulation of these metals is potentially toxic. Microorganisms have evolved import and export pumps to regulate the concentration of metals23. mbtB, irtA,irtB and ide, present in all 40 Ma genomes, encode proteins involved in iron acquisition. Ma possesses a cluster of mbt genes including mbtA to mbtJ that encode for siderophores, the most important group of iron-chelating compounds. Magnesium is another important metal required, in particular, for DNA and RNA synthesis23. The mtgC gene, ubiquitous in the 40 genome set, encodes a transmembrane P-type ATPase Mg++ uptake protein. The concentration of Mg2+ is especially low in macrophages and Ma requires active Mg++ importer to survive.
Virulence factor distribution among Ma subspecies
There are, in general, no salient differences in the assortment of virulence genes observed among the three Ma subspecies except for some gene deletions occurring in specific strains (Supplementary Fig. S10). hbhA, a virulence gene, although consistently present, contains a subspecies specific, missense mutation in our data set (Thr165 in M. abscessus (sensu stricto), Ala165 in M. bolletii and M. massiliense).
Comparative pathogenomics
We compared the virulence genes found in Ma with those in various M. tuberculosis strains (H37Rv, H37Ra, CDC1551, F11 and KZN1436) and in M. vaccae ATCC 25954, a non-pathogenic mycobacterium (Figure 4). We found many more virulence genes in M. tuberculosis (196) than in Ma (90) and M. vaccae (4). Ma appears to lack virulence factors related to lipid and fatty acid metabolism and, unlike M. tuberculosis, does not have virulence genes such as kasB, mmaA4, pcaA and mymA involved in the synthesis of mycolic acid. Mycolic acid is important in the preservation of the mycobacterium cell wall structure and functionality and also plays a role in the modulation of the interaction with host immune system. Disruption of mycolic acid synthesis has been shown to impair the ability of M. tuberculosis to survive in macrophages31. The lack of some mycolic acid synthesis genes in MA could indicate a decreased ability for intracellular survival compared to M. tuberculosis.
Ma may also lack the mmpL8 gene which encodes a gene product required for the synthesis of sulfolipid-1(SL-1), a compound that is able to prevent the fusion of phagosome with lysosome to form the phagolysosome in macrophages. It also blocks oxidative phosphorylation and inhibits the production of reactive oxygen. We did not find genes which encode the phospholipase C-type enzyme (plcA, plcB, plcC and plcD). These genes allow some intracellular pathogens to escape from phagosomal vacuoles by disrupting the host membrane32. Also absent are genes that encode for proteins involved in the biosysnthesis of PDIM (phthiocelrol dimycocerosate) and PGL (phenolic glycolipid). PDIM participates in the prevention of phagosomal acidification33, an important requirement for the enzymic digestion of internalized bacteria. PGL in M. tuberculosis has been reported to affect cytokine production in the host; more specifically to reduce pro-inflammatory cytokine release.
The lack of these virulence genes again suggests a decreased ability in Ma to protect itself against phagosomal digestion and cytotoxic T cell activity. PE (proline-glutamate) and PPE (proline-proline-glutamate) protein families are unique for mycobacteria. Although the function of these proteins is still unclear, some members are believed to be involved in antigenic variation and immune avoidance34,35,36. In M. tuberculosis there are about 200 pe/ppe genes45 closely linked with five esx gene clusters (ESX1-5) that encode proteins in the ESX secretion system37. In all our 40 Ma genomes, we found only two ESX gene clusters (ESX-3 and ESX-4), only one gene (espR) in ESX-1 and only one pair of pe/ppe genes (pe5 and ppe4) located in the ESX-3 gene cluster. The ESX-1 cluster is required for full virulence in M. tuberculosis38; it is responsible for the secretion of the ESAT-6 (early secreted antigenic target, 6 kDa) and CFP-10 (culture filtrate protein, 10 kDa) proteins which are potent T-cell antigens that are believed to play a role in the survival of M. tuberculosis in the host39,40. The ESX-5 cluster encodes several membrane proteins, PE/PPE proteins, a putative cytochrome P450 and a mycobacteriophage protein in a secretion system that is only found in slow-growing pathogenic mycobacteria like M. tuberculosis but not in fast-growing mycobacteria like M. smegmatis37. The absence of almost all of ESX-1 as well as all of ESX-2 and ESX-5 gene clusters with their associated pe/ppe genes makes Ma appear to be a lesser pathogen than M. tuberculosis. It has to be remembered, however, that the expression of virulence is affected by the host environment and host-parasite interactions. In patients with cystic fibrosis, for instance, pulmonary tuberculosis is rare but Ma infections are common and frequently associated with poor clinical outcome43,44.
Phylogenetic relationship and classification of sequenced Ma strains
To infer the phylogenetic relationship between sequenced Ma strains, we extracted all SNPs in the core genome. These core genome SNPs were identified by aligning all 40 genome sequences and two Ma subspecies reference genomes, using Panseq (see method section)12. We reconstructed a phylogenetic tree based on these identified SNPs (Figure 5A).
In the mid-point phylogenetic tree, all strains are clearly grouped into three distinct subgroups corresponding to the three known Ma subspecies: M. abscessus (sensu stricto), M. bolletii and M. massiliense. The largest subgroup is M. abscessus (sensu stricto), with 22 isolates. There are only two isolates (including the reference M. bolletii BD) in the subgroup of M. bolletii. The rest of the isolates are clustered into the subgroup of M. massiliense. All three subgroups are supported by a strong bootstrap value of 100. M. abscessus (sensu stricto) and M. bolletii strains from Malaysia are generally similar to those in the United States and Europe (e.g. the UK and France), but M. massiliense is splintered into distinct sister groups. M139, for instance, is in a sister group by itself, apart from the other M. massiliense isolates. This strain which was isolated from a Nepalese working in Malaysia, appears to be from a more ancient lineage than other M. massiliense strains from Southeast Asian, North American and European populations. Hence, we speculate that the Nepalese patient was probably infected in his home country, Nepal, before he entered Malaysia. In another sister group is M172 from a Myanmar national who had been staying in Malaysia for more than 2 years before he presented to our hospital. This isolate shares genomic similarity with other isolates from Malaysia and belongs to the same VNTR genotype as two other Malaysian strains, M148 and M15641, indicating that it is likely to be a local strain.
Discussion
We present here the first comparative analysis of multiple Ma genomes from different geographical regions, ethnic groups and isolation sites. We characterised and compared the whole-genome of all 40 Ma clinical isolates. We also identified the core and accessory genomes of Ma and compared their functions. Our results reveal that the rapidly evolving Ma genomes are very plastic with many recently introduced insertion sequences such as prophages and novel genes. We have also shown that Ma has an open pan-genome, suggesting that this pathogen will continue to acquire new genetic material. These results are consistent with Ma's rapid doubling time and the ease with which the species adapts to divergent environmental conditions.
Interestingly, we revealed an uncommon tRNA island in the highly antibiotic resistant strain M. bollettii M24 and also in some M. massiliense strains from the United States. BLAST analysis of this sequence against the NCBI database discovered no significant clusters of tRNA similarly ordered and matched. Therefore, this cluster of tRNAs might be a horizontal transfer of a region from another organism not yet sequenced. The biological role of this tRNA cluster is still unclear, but large tRNA gene clusters have been reported to be widespread in low-G+C gram-positive bacteria and could be associated with elevated, possibly modulated levels of protein synthesis, possibly tied-in with pathogenicity. Analysis of the expression patterns of these tRNA clusters in context cell invasion assays may illuminate this issue. The observation of the island of tRNAs is reminiscent of pathogenicity islands (PI's) in E.coli. PI's are horizontally transferred gene clusters, the expression of which is associated with pathogenic processes. These PI's often insert at tRNA sites (which are recombinagenic due to their secondary structure). PI's also carry extra tRNAs to aid high level expression.
Our analysis reveals many putative virulence factors found in Ma genomes. Importantly, these genes may facilitate the ability of Ma to interact with mammalian hosts. Information about these genes, as well as the strain differences present in Ma will not only help us to understand the strain's evolution and pathogenesis, but can potentially reveal more specific targets for vaccine development and drug design.
The whole-genome based phylogenetic classification presented clearly delineates the two M. bolletii isolates from the other two subspecies. This raises the question of whether the M. bolletii and M. massiliense should be classified together into a subspecies named Ma subspecies bolletii comb. nov as suggested by Leão and his colleagues42 or if they should be maintained as two different subspecies. It is noteworthy that the M. abscessus (sensu stricto) branch is prone to phage infection; sufficient independent phage invasion has occurred to allow tree refinement at a scale not supported by SNP divergence. In other words, phage-mediated horizontal gene transfer is driving rapid evolution on this branch.
As shown in the Figure 5, the M. abscessus (sensu stricto) isolates are very similar although they were obtained from different geographical locations; but for the M. massiliense isolates, there are different sister groups. Based on the phylogenetic tree, M139 which was isolated from a Nepalese patient appeared as the most ancient strain, followed by isolates from Malaysia and Europe; the most recent are the isolates from the US. Acknowledging the limited number of isolates in this study, we hypothesize that the M. massiliense migrated from Nepal or the Indian subcontinent to Southeast Asia, before spreading to Europe and then to the United States. Further support awaits the sequencing of more isolates especially from the Indian subcontinent. Advances in high-throughput sequencing technologies make this feasible.
We conclude with a warning. Ma belongs on the public health “watch list”. Although at present, this species appears to evince lower virulence than the important pathogen M. tuberculosis, it is rapidly evolving and has a low threshold for acquisition of foreign DNA by horizontal transfer. Not surprisingly, it has already acquired antibiotic resistance. Its respiratory habitat places it in close proximity to very serious pathogens which can act as donors of additional virulence genes. We suggest that Ma should be aggressively monitored by routine sequencing of clinical isolates. Given this emerging pathogen's high evolutionary potential, careful surveillance is only prudent.
Methods
Bacterial strains and DNA extraction
The 12 clinical Ma strains (M18, M24, M93, M94, M115, M139, M148, M152, M154, M156, M159 and M172) from our laboratory were identified as Ma by either a PCR-RFLP assay or the GenoType Mycobacterium CM/AS test (Hain Lifescience GmbH, Germany)53,54,55,56,57,58,59,60,61,62. They were stocked in Middlebrook 7H9 broth (Difco) with 15% glycerol, kept at −80°C. When required for DNA extraction, the strains were subcultured on 7H10 agar and incubated at 37°C for up to 7 days. DNA extraction was carried out as described previously41.
The accession numbers for these 12 strains together with those for the 28 strains from the NCBI database are NC_010397, AJSC00000000, AJLY00000000, AJGF00000000, AJLZ00000000, AKVR00000000, AKVV00000000, AKVT00000000, AJMA00000000, AKVU00000000, AJSD00000000, AJSE00000000, AJGG00000000, AGQU00000000, AKUX00000000, AKUP00000000, AKUY00000000, AKUZ00000000, AKVD00000000, AKVE00000000, AKVA00000000, AKVB00000000, AKVC00000000, AKUT00000000, AKTU00000000, AKTV00000000, AKTW00000000, AKTX00000000, AKUC00000000, AKUQ00000000, AKUD00000000, AKUB00000000, AKUA00000000, AKTZ00000000, AKTY00000000, AKUE00000000, AKUR00000000, AKUS00000000, AKUF00000000, AKUG00000000, AKUH00000000.
The genome sequences of these Ma strains are downloadable from NCBI FTP.
Next-generation sequencing
The genomes of our 12 isolates were shotgun sequenced by using Illumina Genome Analyzer 2X technology. Paired-end libraries were prepared from 5 μg of isolated gDNA using the TruSeq DNA sample prep kit – Kit A (Illumina Inc., San Diego) according to the manufacturer's description. Genomic paired-end libraries were sequenced with a read length of 2 × 101 nucleotides using an Illumina GAIIx instrument according to the manufacturer's description. Image analysis and base calling were done by the standard Illumina pipeline.
For single molecule PacBio sequencing, the covaris G-tube was used to generate either 8 Kb or 12 Kb fragments by shearing genomic DNA according to the manufacturer's recommended protocol, using the AMpureXP bead purification system to remove all small fragments. A total of 5 μg for each sample was used as input into library preparation using standard Pacific Biosciences version C2 chemistry. The samples were sequenced on the PacBio RS running version 1.3.3 software with the standard 1 × 90 minute collection protocol and one SMRT Cell was used for each sample. Continuous long reads (CLR) were generated from zero mode waveguides (ZMW).
Genome assembly and annotation
The raw Illumina sequencing reads were trimmed at a threshold of 0.01 (Phred score of 20) and the sequences obtained were de novo assembled using CLC Genomics Workbench version 4.9 (CLC bio, Denmark). All Ma genome sequences were annotated using the Rapid Annotation using Subsystem Technology (RAST) pipeline8, which is a fully automated annotation engine for complete or draft archaeal and bacterial genomes.
PacBio data has high sequencing error rate46. The PacBioToCA was used to correct errors in sequencing reads of M24 and M93 yielded from PacBio Single Molecule Real Time (SMRT) sequencing platform using the Illumina sequencing reads above. At the minimum length threshold of 2,500 bp, error-corrected reads were generated by the PacBioToCA and these reads were mapped to the M24 or M94 genome assemblies. The mapping of these reads onto the tRNAs islands were manually examined in the genome browser provided in Avadis NGS software.
Ortholog identification
Functional ortholog clustering was performed using PGAP (pan-genome analysis pipeline)10. The protein sequences of 40 strains were used as the input. Orthologs among 40 strains were searched using the Gene Family (GF) method. The protein sequences of each strain were mixed together and marked with the strain identifiers. BLASTALL was first performed among the protein sequences with the minimum score value and E-value in BLAST as 50 and 1e-8 respectively47. The filtered BLAST results were clustered by MCL algorithm48. In order to group the same genes into the same cluster, the global match region must have at least 50% of the longer gene protein sequence and 50% sequence identity.
Whole-genome alignment of 40 Ma strains
Whole-genome alignment of the 40 Ma strains was performed using the aligner Progressive Mauve from the software Mauve version 2.09. The following parameters were used for alignment: default seed weight (11 for 1 MB genome size and around 15 for 5 MB), use seed families, determine LCB, full alignment and iterative alignment using MUSCLE 3.749 and finally sum of pair LCB scoring.
Virulence gene and prophage prediction
All protein sequences in the 40 Ma genomes were BLASTed against the virulence factor database (VFDB)19,20,21. The genes that were orthologous to virulence genes must have at least 96% identity and at least 80% sequence coverage in query and subject.
Prophages in Ma were identified using PHAST (Phage Search Tool) web server15. Before the submission of genome sequences to PHAST, the contig sequences of the 39 Ma genomes were reordered using the Ma ATCC 19977 as reference genome through a function in Mauve. Putative protein sequences in the Ma genome were BLAST searched against two prophage sequence databases to identify phage-related proteins. The two databases are the NCBI phage database and the prophage database that contains 159 prophage regions and 9,061 proteins that are not found in the NCBI phage database. After the prophages have been identified, the completeness for each prophage was calculated by PHAST. An intact prophage was defined as having at least a score of 90 by PHAST.
Phylogenetic analysis
To clarify the phylogenetic relationship between all Ma strains, the core genome was identified using Panseq 2.050. Two reference strains were added for this analysis which are M. bolletii BD and M. massiliense CCUG 48898. The SNPs within each core genome were identified, extracted and concatenated into one large sequence. The sequences were multiple-aligned using ClustalW from European Bioinformatics Institute (EBI)51. Phylogenetic calculation was performed to understand the relationship among the strains by using MEGA version 552. The phylogenetic tree was constructed using the Maximum Likelihood algorithm with Tamura-Nei model based on the lowest scoring based on Bayesian Information Criterion. The confidence levels of the phylogenetic trees were supported by 1,000 times bootstrapping replication.
References
Covert, T. C., Rodgers, M. R., Reyes, A. L. & Stelma, G. N., Jr Occurrence of nontuberculous mycobacteria in environmental samples. Appl Environ Microbiol. 65, 2492–2496 (1999).
Moore, M. & Frerichs, J. B. An unusual acid-fast infection of the knee with subcutaneous, abscess-like lesions of the gluteal region; report of a case with a study of the organism, Mycobacterium abscessus, n. sp. J Invest Dermatol. 20, 133–169 (1953).
Jonsson, B. E. et al. Molecular epidemiology of Mycobacterium abscessus, with focus on cystic fibrosis. J Clin Microbiol. 45, 1497–1504 (2007).
Kim, H. Y. et al. Mycobacterium massiliense is differentiated from Mycobacterium abscessus and Mycobacterium bolletii by erythromycin ribosome methyltransferase gene (erm) and clarithromycin susceptibility patterns. Microbiol Immunol. 54, 347–353 (2010).
Adekambi, T., Berger, P., Raoult, D. & Drancourt, M. rpoB gene sequence-based characterization of emerging non-tuberculous mycobacteria with descriptions of Mycobacterium bolletii sp. nov., Mycobacterium phocaicum sp. nov. and Mycobacterium aubagnense sp. nov. Int J Syst Evol Microbiol. 56, 133–143 (2006).
Adekambi, T. et al. Amoebal coculture of “Mycobacterium massiliense” sp. nov. from the sputum of a patient with hemoptoic pneumonia. J Clin Microbiol. 42, 5493–5501 (2004).
Macheras, E. et al. Inaccuracy of single-target sequencing for discriminating species of the Mycobacterium abscessus group. J Clin Microbiol. 47, 2596–2600 (2009).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 5, e11147 (2010).
Liang, W. et al. Pan-genomic analysis provides insights into the genomic variation and evolution of Salmonella Paratyphi A. PLoS One. 7, e45346 (2012).
Tettelin, H. et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 102, 13950–13955 (2005).
Rasko, D. A. et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 190, 6881–6893 (2008).
Medini, D., Donati, C., Tettelin, H., Masignani, V. & Rappuoli, R. The microbial pan-genome. Curr Opin Genet Dev. 15, 589–594 (2005).
Iyer, A. et al. Bacteriophages in Escherichia coli antimicrobial resistance. Advance in Biosciense and Biotechnology. 4, 469–476 (2013).
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J. & Wishart, D. S. PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–352 (2011).
Pang, Y. K., Ngeow, Y. F., Wong, Y. L. & Liam, C. K. Mycobacterium abscessus – to treat or not to treat? Respirology Case Reports. 1, 31–33 (2013).
Howard, S. T. et al. Spontaneous reversion of Mycobacterium abscessus from a smooth to a rough morphotype is associated with reduced expression of glycopeptidolipid and reacquisition of an invasive phenotype. Microbiology. 152, 1581–1590 (2006).
Jönsson, B., Ridell, M. & Wold, A. E. Phagocytosis and cytokine response to rough and smooth colony variants of Mycobacterium abscessus by human peripheral blood mononuclear cells. APMIS. 121, 45–55 (2013).
Chen, L., Xiong, Z., Sun, L., Yang, J. & Jin, Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40, D641–645 (2012).
Yang, J., Chen, L., Sun, L., Yu, J. & Jin, Q. VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res. 36, D539–542 (2008).
Chen, L. et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–328 (2005).
Forrellad, M. A. et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 4, 3–66 (2013).
Berthet, F. X. et al. Attenuation of virulence by disruption of the Mycobacterium tuberculosis erp gene. Science. 282, 759–762 (1998).
Wiker, H. G. & Harboe, M. The antigen 85 complex: a major secretion product of Mycobacterium tuberculosis. Microbiol Rev. 56, 648–661 (1992).
Wiker, H. G., Nagai, S., Harboe, M. & Ljungqvist, L. A family of cross-reacting proteins secreted by Mycobacterium tuberculosis. Scand J Immunol. 36, 307–319 (1992).
Arruda, S., Bomfim, G., Knights, R., Huima-Byron, T. & Riley, L. W. Cloning of an M. tuberculosis DNA fragment associated with entry and survival inside cells. Science. 261, 1454–1457 (1993).
Ferrer, N. L., Gomez, A. B., Neyrolles, O., Gicquel, B. & Martin, C. Interactions of attenuated Mycobacterium tuberculosis phoP mutant with human macrophages. PLoS One. 5, e12978 (2010).
Blomgran, R., Desvignes, L., Briken, V. & Ernst, J. D. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe. 11, 81–90 (2012).
Hinchey, J. et al. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest. 117, 2279–2288 (2007).
Edwards, K. M. et al. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 164, 2213–2219 (2001).
Singh, A. et al. Requirement of the mymA operon for appropriate cell wall ultrastructure and persistence of Mycobacterium tuberculosis in the spleens of guinea pigs. J Bacteriol. 187, 4173–4186 (2005).
Titball, R. W. Bacterial phospholipases C. Microbiol Rev. 57, 347–366 (1993).
Astarie-Dequeker, C. et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog. 5, e1000289 (2009).
Burts, M. L., Williams, W. A., DeBord, K. & Missiakas, D. M. EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci U S A. 102, 1169–1174 (2005).
Mostowy, S., Cleto, C., Sherman, D. R. & Behr, M. A. The Mycobacterium tuberculosis complex transcriptome of attenuation. Tuberculosis (Edinb). 84, 197–204 (2004).
Tan, T., Lee, W. L., Alexander, D. C., Grinstein, S. & Liu, J. The ESAT-6/CFP-10 secretion system of Mycobacterium marinum modulates phagosome maturation. Cell Microbiol. 8, 1417–1429 (2006).
Gey van Pittius, N. C. et al. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol. 6, 95 (2006).
Lewis, K. N. et al. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J Infect Dis. 187, 117–123 (2003).
Guinn, K. M. et al. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol. 51, 359–370 (2004).
Brodin, P. et al. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun. 74, 88–98 (2006).
Wong, Y. L., Ong, C. S. & Ngeow, Y. F. Molecular typing of Mycobacterium abscessus based on tandem-repeat polymorphism. J Clin Microbiol. 50, 3084–3088 (2012).
Leao, S. C., Tortoli, E., Euzeby, J. P. & Garcia, M. J. Proposal that Mycobacterium massiliense and Mycobacterium bolletii be united and reclassified as Mycobacterium abscessus subsp. bolletii comb. nov., designation of Mycobacterium abscessus subsp. abscessus subsp. nov. and emended description of Mycobacterium abscessus. Int J Syst Evol Microbiol. 61, 2311–2313 (2011).
Morand, C. et al. Mediastinal Tuberculosis in an Adult Patient with Cystic Fibrosis. J Clin Microbiol. 49, 750–751 (2011).
Charles, R. et al. Chronic Mycobacterium abscessus infection and lung function decline in cystic fibrosis. J Cyst Fibros. 9, (2010).
Cole, T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 393, 537–544 (1998).
Koren, S. et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature Biotech. 30, 693–700 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol. 215, 403–410 (1990).
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 5, 113 (2004).
Laing, C. et al. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics. 11, 461 (2010).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 23, 2947–2948 (2007).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol. 28, 2731–2739 (2011).
Wee, W. Y. et al. MabsBase: A Mycobacterium abscessus Genome and Annotation Database. PLoS One. 8, e62443 (2013).
Choo, S. W. et al. Genome Sequence of the Mycobacterium abscessus Strain M93. J Bacteriol. 194, 3278 (2012).
Ngeow, Y. F. et al. Genome Sequence of Mycobacterium massiliense M18, Isolated from a Lymph Node Biopsy Specimen. J Bacteriol. 194, 4125 (2012).
Wong, Y. L. et al. Draft Genome Sequence of Mycobacterium bolletii Strain M24, a Rapidly Growing Mycobacterium of Contentious Taxonomic Status. J Bacteriol. 194, 4475 (2012).
Choo, S. W. et al. Annotated genome sequence of Mycobacterium massiliense strain M154, belonging to the recently created taxon, Mycobacterium abscessus subspecies bolletii comb. nov. J Bacteriol. 194, 4778 (2012).
Choo, S. W. et al. Genome Analysis of Mycobacterium massiliense strain M172 with a putative mycobacteriophage. J Bacteriol. 194, 5128 (2012).
Ngeow, Y. F. et al. Genomic analysis of Mycobacterium massiliense strain M115, an isolate from human sputum. J Bacteriol. 194, 4786 (2012).
Choo, S. W. et al. Analysis of the genome of Mycobacterium abscessus strain M94 reveals an uncommon cluster of tRNAs. J Bacteriol. 194, 5724 (2012).
Ngeow, Y. F. et al. Genomic analysis of Mycobacterium abscessus strain M139 with an ambiguous subspecies taxonomic position. J Bacteriol. 194, 6002–3 (2012).
Ngeow, Y. F. et al. Genome Sequence of the Mycobacterium abscessus strain M152. J Bacteriol. 194, 6662 (2012).
Beste, D. J. et al. The genetic requirements for fast and slow growth in mycobacteria. PLoS One. 4, e5349 (2009).
Acknowledgements
We would like to thank all members of Genome Informatics Research Group (GIRG) for contributing to this research. Funding: This research is supported by UM High Impact Research Grant UM-MOHE UM.C/HIR/MOHE/08 from the Ministry of Higher Education Malaysia.
Author information
Authors and Affiliations
Contributions
S.W.C. conceived the project and supervised the genomic and bioinformatics studies. S.W.C., W.Y.W., Y.F.N., W.M., J.L.T., G.J.W., Y.Z. and J.X. performed the experiments and analysed the data. S.W.C., W.M., W.Y.W. and Y.F.N. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary info
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Choo, S., Wee, W., Ngeow, Y. et al. Genomic reconnaissance of clinical isolates of emerging human pathogen Mycobacterium abscessus reveals high evolutionary potential. Sci Rep 4, 4061 (2014). https://doi.org/10.1038/srep04061
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04061
This article is cited by
-
Genomic characterization of sporadic isolates of the dominant clone of Mycobacterium abscessus subspecies massiliense
Scientific Reports (2021)
-
Phenotypic and genomic hallmarks of a novel, potentially pathogenic rapidly growing Mycobacterium species related to the Mycobacterium fortuitum complex
Scientific Reports (2021)
-
Comparative genomics of Mycobacterium mucogenicum and Mycobacterium neoaurum clade members emphasizing tRNA and non-coding RNA
BMC Evolutionary Biology (2019)
-
Extended insight into the Mycobacterium chelonae-abscessus complex through whole genome sequencing of Mycobacterium salmoniphilum outbreak and Mycobacterium salmoniphilum-like strains
Scientific Reports (2019)
-
PGAP-X: extension on pan-genome analysis pipeline
BMC Genomics (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
