
Abstract
FUS is genetically and pathologically linked to amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). To clarify the RNA metabolism cascade regulated by FUS in ALS/FTLD, we compared the FUS-regulated transcriptome profiles in different lineages of primary cells from the central nervous system. The profiles of FUS-mediated gene expression and alternative splicing in motor neurons were similar to those of cortical neurons, but not to those in cerebellar neurons despite the similarity of innate transcriptome signature. The gene expression profiles in glial cells were similar to those in motor and cortical neurons. We identified certain neurological diseases-associated genes, including Mapt, Stx1a and Scn8a, among the profiles of gene expression and alternative splicing events regulated by FUS. Thus, FUS-regulated transcriptome profiles in each cell-type may determine cellular fate in association with FUS-mediated ALS/FTLD and identified RNA targets for FUS could be therapeutic targets for ALS/FTLD.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by selective motor neuron death in adulthood. The etiology of ALS remains obscure, although many pathomechanisms have been suggested including RNA metabolism and non-cell autonomous toxicity1,2. While most ALS cases are sporadic, approximately 10–20% of ALS patients have a family history. In this regard, several genes have been identified as the cause or risk-factors for ALS, such as FUS and TAR DNA-binding protein (TDP-43), that are RNA binding proteins known to regulate RNA splicing and transcription3,4. Interestingly, accumulation of FUS and TDP-43 is observed in the cytoplasmic inclusions in sporadic ALS and frontotemporal lobar degeneration (FTLD), which is the second most common cause of presenile dementia5,6. Together with the evidence of FUS and TDP-43, which accumulate in cytoplasmic inclusions, the above studies strongly suggest that FTLD, ALS with dementia and classical ALS are all parts of a clinicopathological spectrum of diseases characterized by disruption of RNA metabolism, which causes neurodegeneration in a subset of neurons in the central nervous system (CNS)7.
How does loss-of-function and/or gain-of-toxicity cause neuronal cell death in diseases? Several lines of evidence indicate the involvement of loss-of-function in neurodegeneration observed in ALS/FTLD associated with FUS. Pathological studies have demonstrated FUS redistribution into the cytoplasm from the nucleus in sporadic and familial ALS as well as FTLD8,9,10. Other studies also showed the distribution of mutant FUS proteins associated with familial ALS in the cytoplasm from the nucleus, in contract to nuclear localization of endogenous and wild-type FUS11,12,13,14. Furthermore, the loss of FUS directly leads to neuronal cell death in drosophila15 and zebrafish16.
The cell selectivity in ALS/FTLD has remained a long-standing mystery. As mentioned above, the pathology of FUS-associated ALS/FTLD involves major selective neuronal vulnerability in both motor neurons and cortical neurons. Glial cells such as astrocytes and microglial cells are also likely to be involved in ALS/FTLD with regard to non-cell-autonomous toxicity17,18,19. On the other hand, cerebellar neurons are typically spared in ALS/FTLD. The expression pattern of FUS cannot explain the cell and region specific-selectivity in ALS/FTLD, since they are expressed ubiquitously throughout the CNS8. Based on this background, it is important to define Fus-targeting RNA profiles in different cell lineages from the CNS for more precise interpretation of selective neuronal death in FUS-associated ALS/FTLD.
We reported recently that FUS regulates a subset of exon splicing events and gene expression in mouse primary cortical neurons in a position-dependent manner20. We also showed scattered binding of FUS to and around alternatively spliced exons, including those associated with neurodegeneration, such as Mapt, Camk2a and Fmr120. In the present study, we extend our research to the global roles of FUS on RNA metabolism in different cell lineages from the different CNS regions, including primary motor neurons, cortical neurons, glial cells and cerebellar neurons and describe a new pathomechanism of FUS-related ALS/FTLD.
Results
Lentivirus-mediated silencing of FUS in different cell lineages from the CNS
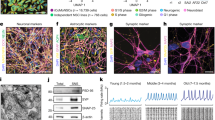
We have described recently the establishment of Fus-silenced primary cortical neurons20. In order to compare RNA profiles of FUS in different cell types of the CNS, we prepared Fus-silenced primary motor neurons, primary cerebellar neurons and primary glial cells by introducing lentivirus-expressing shRNA against Fus (shFUS) or scrambled control (Fig. 1A). The purity of each primary cell culture was confirmed by immunostaining for cell specific markers. We successfully established primary cultures of motor and cortical neurons with more than 95% purity. For primary glial cell cultures, we obtained astroctyes with purity of more than 95%, as confirmed by staining for GFAP. Both Purkinje cells and granule cells were the main cell populations among primary cerebellar neurons (Fig. 1B).

Experimental schema and characterization of four primary cells from the central nervous system.
(A) Primary motor neurons were harvested from the spinal cords of C57BL/6 mouse embryos at embryonic E13. Primary cortical neurons and primary glial cells were obtained from the cerebrum of C57BL/6 mouse at E15. Primary cerebellar neurons were obtained from the cerebellum of C57BL/6 mouse at E15. Motor and cortical neurons, but not cerebellar neurons, are affected in ALS/FTLD. Glial cells are disease-modifiers in ALS/FTLD. Primary cells were infected with lentivirus expressing two different shRNAs against FUS (shFUS1 and shFUS2) and control shRNA (shCont). Total RNA was isolated and analyzed by the Affymetrix Mouse Exon Array. Each experiment was performed in triplicate. (B) The purity of primary motor neurons, primary cortical neurons, primary glial cells and primary cerebellar neurons was confirmed by immunostaining using specific antibodies: anti-neurofilament-H antibody (SMI32R) for primary motor neurons; anti-β tubulin antibody (TU20) for primary cortical neurons; anti-GFAP antibody for glial cells; anti-Zic1 antibody for granule cells in primary cerebellar neurons; and anti-calbindin antibody for Purkinje cells in primary cerebellar neurons. (C) The innate gene expression profiles of each cell type introduced with shCont were compared by principal component analysis (n = 3 for each cell-type). The correlation coefficients were calculated (Supplementary Material, Table S1) and summarized in the correlation plot (left). The 2D PCA scores and the loadings plots of the innate gene expression profiles of each cell-type introduced with shCont indicated that significant separation between the profiles of the three primary neurons and that of glial cells (right).
To exclude possible off-targeting effects, we used two different shRNAs, shFUS1 and shFUS2 in experiments performed in triplicate, as described in our recent study20. Correlation analysis between shFUS1 and shFUS2 showed a coefficient of determination (R2) of 0.93 in primary cortical neurons, indicating that these two shRNA have minimal off-targeting effects20. The expression levels of Fus mRNA were efficiently suppressed in all four types of cells by both shFUS1 and shFUS2 by real-time quantitative PCR (Supplementary Fig. S1A). The FUS protein levels were decreased in all four types of cells by both shFUS1 and shFUS2 by immunoblot (Supplementary Fig. S1B). Immunohistochemistry also showed markedly decreased FUS protein levels in all four primary cells infected with shFUS1 and shFUS2 compared to shRNA against control scrambled oligonucleotides (shCont) (Supplementary Fig. S1C).
Innate gene expression profiles in neurons and glial cells
The innate gene expression profiles of each cell-type infected with shCont were analyzed using the Affymetrix Mouse Exon 1.0 ST Array and compared by principal component analysis. The correlation coefficients were calculated (Supplementary Table S1) and summarized in the correlation plot and 2D-PCA scores (Fig. 1C). We found that the expression profiles of cerebellar neurons are very close to those of cortical neurons. The expression profiles of motor neurons are also similar to those of cortical neurons but with a lesser extent. On the other hand, the expression profiles of glial cells were divergent from those of the other three neuronal cells.
FUS regulates gene expressions in motor neurons, cortical neurons and glial cells, but to a lesser extent in cerebellar cells
In the next step, we analyzed the gene expression and alternative splicing events in Fus-silenced and control primary cells using the Affymetrix Mouse Exon 1.0 ST Array. To identify the common effects of Fus-silencing in different cell lineages from the CNS, we compiled a list of differentially expressed genes and differentially spliced exons in primary motor neurons, cortical neurons, glial cells and cerebellar neurons, in which shFUS was introduced (GEO accession numbers: GSE36153 for cortical neurons, GSE42421 for motor neurons, glial cells and cerebellar neurons). We first prepared a list of FUS-regulated genes which were differentially expressed by shFUS for each cell lineage by filtering the gene-level signal intensities with t-test p values of ≤0.1. Then, we identified differentially expressed genes shared among the four primary types of cells.
The results of plot analysis of gene expression in primary cells of the CNS after Fus-depletion are shown in Fig. 2A. The numbers of differentially expressed genes with more than 1.3-fold change were higher in primary cortical neurons, motor neuron and glial cells than in cerebellar neurons. Indeed, there were more than 2000 differentially expressed genes in these three cell types but only 494 genes in primary cerebellar neurons (Supplementary Table S2).

Comparison of gene expression and exon splicing profiles among different cell lineages from the central nervous system after Fus knockdown.
(A) Scatter plot analysis of gene expression using shCont and shFUS in different primary cells from the central nervous system. Genes whose expression levels changed by more than 1.3-fold after Fus-silencing with shFUS1 are indicated in red. (B) The profiles of gene expression and alternative splicing events were compared among the three different primary neurons. Venn diagrams indicate overlap in genes (top) and exons (bottom) whose expression levels are uniquely or concordantly regulated by FUS among motor, cortical and cerebellar neurons (p < 0.1, by t-test). (C) The fold-changes in overlapped genes filtered by the t-test (p < 0.1) were plotted for primary motor, cortical and cerebellar neurons. Scatter plots of fold-changes in gene expression levels (top) and alternative splicing events (bottom) after Fus knockdown. The R2 value was calculated for genes and exons with t-test p values < 0.1. (D) As in Fig. 2B, the profiles of gene expression and alternative splicing events in glial cells were compared with those of motor and cortical neurons. Venn diagrams indicate overlap in genes (top) and exons (bottom) whose expression levels are uniquely or concordantly regulated by FUS among motor, cortical and glial cells (p < 0.1, by t-test). (E) Scatter plots of fold-changes in gene expression levels (top) and alternative splicing events (bottom) after Fus knockdown. The R2 value was calculated for genes and exons with t-test p values < 0.1.
FUS-mediated gene expression profiles are similar between cortical and motor neurons and are less similar when these neuronal cells are compared to cerebellar neurons
We investigated the similarity in differential gene expression profiles regulated by FUS among primary motor, cortical and cerebellar neurons. Venn diagrams indicated overlap in genes or exons whose expression was regulated in the same direction by FUS (t-test, p < 0.1). Both motor and cortical neurons which are destined to die in ALS/FTLD, shared appreciable proportions of gene expression profiles (775/2321, 33.4% of genes in motor neurons; 775/2470, 31.4% of genes in cortical neurons). Cerebellar neurons, which are considered to be spared in ALS/FTLD, contained fewer numbers of FUS-regulated genes (t-test, p < 0.1) than motor and cortical neurons and showed small overlapping rate with other neurons (58/494, 11.7% of genes with motor neurons; and 50/494, 10.1% of genes with cortical neurons) (Fig. 2B). The fold-changes in overlapped genes filtered by the t-test (p < 0.1) were plotted for primary motor, cortical and cerebellar neurons. Comparison of gene expression profiles showed significantly high correlations between motor and cortical neurons (R2 > 0.80, Fig. 2C: p ≤ 0.1, by t-test, Supplementary Fig. S2A: p ≤ 0.075 and 0.05, by t-test).
FUS-mediated alternative splicing profiles are similar in cortical and motor neurons but largely divergent compared with cerebellar neurons
We next investigated the similarity in differential alternative splicing profiles regulated by FUS among primary motor, cortical and cerebellar neurons. We filtered the exon-level signal intensities with t-test p values ≤ 0.1 and prepared a list of FUS-regulated exons for each cell lineage and identified shared FUS-regulated exons. In alternative splicing profiles, about 10% (965/10727, 9.0% of genes in motor neurons; and 965/8183, 11.8% of genes in cortical neurons) of genes showed overlap between motor and cortical neurons at the exon levels, whereas cerebellar neurons showed only overlap in about 4% (434/9907, 4.4% of genes with motor neurons; and 325/9907, 3.3% of genes with cortical neurons) of genes at the exon levels compared with the other two neurons (Fig. 2B). The overlapped exons filtered by the t-test were also plotted for primary motor, cortical and cerebellar neurons. Comparisons of exon splicing profiles showed a significant correlation only between motor and cortical neurons (R2 > 0.7), but not between motor and cerebellar neurons, or cortical and cerebellar neurons (R2 < 0.1, each) (Fig. 2C: p ≤ 0.1, by t-test, Supplementary Fig. S2B: p ≤ 0.075 and 0.05, by t-test).
Glial cells are similar to motor and cortical neurons in FUS-regulated gene expression profiles but not in alternative exon profiles
Glial cells, which are thought to be the modifier in ALS/FTLD, were also investigated with regard to similarity to motor and cortical neurons in differential gene expression and alternative splicing profiles regulated by FUS. Venn diagrams showed that the gene expression profiles of glial cells shared certain similarities with motor and cortical neurons (422/2074, 20.3% of genes with motor neurons; 453/2074, 21.8% of genes with cortical neurons, Fig. 2D). Comparison of gene expression profiles in the plot analysis showed positive correlations between glial cells and both motor and cortical neurons (R2 = 0.58 and 0.42, respectively). On the other hand, comparisons of exon splicing profiles did not show a significant correlation between motor neurons and glial cells and cortical neurons and glial cells (R2 < 0.1, each, Fig. 2E). Thus, FUS regulates the expression levels and alternative splicing of the largest number of shared genes between cortical and motor neurons. Similarly, though less concordantly, FUS regulates the expression levels of the same genes among the cortical/motor, cerebellar and glial cells, but not alternative splicing of these cells.
Characterization of genes with altered gene expression and spliced alternative exon regulated by FUS in different cell lineages of the CNS
We also analyzed Gene Ontology (GO) terms of genes shown in the Venn diagrams using DAVID 6.721,22. The GO terms of genes regulated by FUS in primary motor neurons were mainly those involved in signaling cascades and metabolic processes that were similar to those in primary cortical neurons (Table 1). The GO terms of those in glial cells are mainly involved in the regulation of the immune system. The GO terms of those in cerebellar neurons were not available since the number of differentially expressed genes was too small. The GO terms of genes involved in FUS-related regulation of alternative splicing events in both motor and cortical neurons were mainly involved in various neuronal functions such as synapse, nerve impulse and neuronal projection. In contrast, none of the neuronal function terms emerged in the list of alternative splicing events in cerebellar neurons. Alternative splicing events in glial cells were mostly categorized into immunohematological functions (Table 1).
Supplementary Table S3 lists the top 10 genes that were differentially expressed in Fus-silenced motor neurons, together with the fold-change values in cortical neurons, glial cells and cerebellar neurons. Similar to the global profile comparison shown in Fig. 2, the expression patterns of genes in the three different primary cells were similar except for cerebellar neurons, whose profile was less altered by Fus-depletion than other cell types.
After filtering exons with genes that were differentially expressed by both shFUS1 and shFUS2 with t-test p value of ≤0.1 and with fold-changes ≥ 1.3-fold in each primary cell, we categorized alternative splicing exons into category “A” to “E”. In total, 44 exons were validated by RT-PCR and shown in Table 2, Fig. 3A–C and Supplementary Fig. S3A–E. Alternative splicing exons A were specific to primary cortical neurons and primary motor neurons; B, specific to primary motor neurons; C, specific to primary cortical neurons; D, specific to primary glial cells, E, were common among primary motor neurons, cortical neurons, glial cells and/or cerebellar neurons.

Validation of representative altered splicing events in different cell lineages from the central nervous system after Fus-silencing.
(A–C) Three representative alternative splicing events regulated by Fus are shown in each category: (A) both motor and cortical neuron-specific, (B) motor neuron-specific and (C) cortical neuron-specific. All the validation of altered splicing events including category D and E, are shown in Supplementary Fig. S3. The top panels represent schematic splicing changes mediated by FUS. shCont and shFus resulted in splicing events shown in the top and bottom rows, respectively. The second panels show representative RT-PCR of the indicated exons in primary motor neurons. Similarly, the third, fourth and fifth panels show representative RT-PCR of the indicated exons in primary cortical neurons, primary glial cells and primary cerebellar neurons, respectively. The experiments were repeated four times using four independent sets of samples. The results of densitometric quantification of RT-PCR are shown in bar graphs (n = 4; mean ± SD). *p < 0.05, between shCont and shFUS (by Student's t-test).
We identified 18 alternative splicing events that were motor- and cortical neuron-specific, including Mapt, Dlgap4 and Snap25 (Fig. 3A). All the validated splicing events that were motor- and cortical neuron-specific are shown in Supplementary Fig. S3A. We observed motor-neuron-specific alternative splicing events in Synj1, Scn8a and Rims1 (Fig. 3B). We also identified several alternative spliced events that were differentially expressed in Fus-silenced cortical neurons. Fig. 3C shows representative cortical-neuron-specific alternative splicing events in Kcnip, Stxbp1 and Fmr1. All the validated events are shown in Supplementary Fig. S3C. Furthermore, Glial-cell-specific alternative splicing events were seen in Sgce, Wdr35 and Fip1l1 (Supplementary Fig. S3C). Exons in Fxr1 and Tsc22d2 were alternatively spliced in all primary cell types. Map4 exon14 inclusion was also observed in primary cortical neurons, motor neurons and glial cells; but not in cerebellar neurons (Supplementary Fig. S3D).
Interestingly, we also identified many neurological disease-associated genes among the profiles of gene expression and alternative splicing events. We list such representative genes in Table 3.
The protein levels of representative genes with altered spliced events and gene expression were validated by immunoblot in all four primary cells in the CNS (Fig. 4). The expression of 4-repeat Tau (RD4) which corresponds to the exon10 (+) isoform of Mapt gene was increased by shFUS in primary cortical neurons but was undetectable in motor neurons. On the other hand, the expression of 3-repeat Tau (RD3) which corresponds to the exon10 (-) isoform was decreased in primary cortical and motor neurons. The 89kD form of Braf protein encoded by the exon12 (+) variant of Braf gene was decreased in Fus-silenced primary cortical neurons. The protein expression level of Syntaxin-1A was upregulated in Fus-silenced cortical and motor neurons as observed in its mRNA levels.

Protein expressions of genes with altered splicing events and gene expression in different cell lineages from the central nervous system after Fus-silencing.
Protein levels of representative genes with altered splicing events (Tau and Braf) and gene expression (Syntaxin-1A) were evaluated in different primary cells in the CNS with Fus-silencing. Expression levels of FUS, Syntaxin-1A (Stx1a), 3-repeat Tau (RD3), 4-repeat Tau (4RD), Braf and actin were measured by immunoblot.
Direct binding of FUS to target mRNA was not tisseue-type specific in CNS
To investigate the direct binding of FUS to mRNA of the genes and exons with altered expression, RNA immunoprecipitation (RIP) using differenet tissues of CNS was performed. We prepared the spinal cord (E13), the cerebrum (E15) and the cerebellum (E15) from the mouse embryos and FUS protein was immunoprecipitated from each tissue (Fig. 5A). FUS-associated mRNA levels were evaluated by RT-PCR using specific primers for Mapt, Dlgap1 and Stxbp1, of which alternative splicing events were regulated by FUS. Primers for Gapdh and intergenic region were used as negative controls. The RIP results showed that the interaction levels between FUS and mRNA of Mapt, Dlgap1 and Stxbp1 among three different CNS tissues were comparable, whereas these three genes showed cell-type specific splicing patterns in Fig 3 (Fig. 5B). There was no apparent binding of FUS to Gapdh mRNA or intergenic region (Fig. 5B). We next analyzed position dependence of FUS-binding to splicing targets and their effects on alternative splicing through comprehensive analysis of the exon array using primary glial cells and HITS-CLIP of mouse brain (Fig. 5C–D). We analyzed the positions of CLIP-tags of 121 FUS-responsive exons (29 exon skipped and 92 exon included by shRNA) that were filtered by t-test p value ≤ 0.1 and fold-change of ≤0.67 or ≥1.5 for shFUS1. We combined these exons into a single composite pre-mRNA and prepared integrated RNA maps from our HITS-CLIP reads mapped to the corresponding genomic regions, as described in more detail previously23,24,25. The analysis showed that scattered FUS binding sites mainly around the alternatively spliced exons. Conspicuous binding of FUS was observed at ~500 nt upstream of the 3′ end of the downstream intron in skipped exons (arrows in Fig. 5C). This finding was similar to the complexity map of primary cortical neurons that was reported previously by our group (Fig. 5D)20.

The binding of FUS to mRNA is not tissue and cell type specific in the CNS.
(A–B) RNA immunoprecipitation (RIP) was performed to detect the interaction between FUS and mRNA of genes with altered splicing events using mouse cerebrum, cerebellum and spinal cord. (A) Anti-FUS immunoblot of 1% input, FUS-IP and control IP (IgG) from mouse cerebrum, cerebellum and spinal cord at E15 embryos was shown. (B) FUS-associated RNA was reverse transcribed and evaluated by semi-quantative PCR with specific primers for three representative FUS-target genes with altered alternative splicing events, Mapt, Dlgap1 and Stxbp1. Primers for Gapdh and intergenic region (intergenic) were used as controls. (C–D) We compared normalized complexity map of FUS-dependent splice sites of primary glial cells and cortical neurons. shFUS-mediated alternative splicing events in primary glial cells (C) and primary cortical neurons (D) are compiled. Blue arrows point to conspicuous peaks at ~500 nt upstream of the 3′ end of the downstream intron. The complexity map of primary cortical neuron (D) is identical to that shown in the Supporting Information/Fig. S3 in our previous report20, which is shown for comparison with that of primary glial cells (C).
Discussion
In the present study, we determined the gene expression profiles and alternative splicing events in four different primary cells of the CNS, with silenced Fus induced by lentivirus encoding shRNA against Fus. By comparing the gene expression profiles of the four primary cells, we found that the genes altered by Fus-silencing were fewer in cerebellar neurons than in the other three cell types, although the innate gene expression profiles of motor, cortical and cerebellar neurons were similar. These findings suggest that gene expression profile reflects cellular response affected by Fus-silencing in each cell type, given that ALS/FTLD-relevant cell types showed more alterations than non- ALS/FTLD-relevant cell type. On the other hand, the profiles of alternative splicing events were only similar in motor and cortical neurons. Alternative splicing events in these two types of neurons were far different from glial cells and cerebellar neurons, suggesting that alternative splicing events are uniquely fine-tuned in a cell-specific manner (Fig. 2). Alternative splicing contributed to brain development in mammals during the evolution process26,27. In the adult human, the brain expresses more alternatively spliced transcripts than any other tissues28 and the majority of splicing events are regulated in a tissue-specific manner29. Our finding that FUS-regulated alternative splicing events were more region- and cell-type-specific than those of gene expression suggests that the profiles of alternative splicing events may reflect direct phenomenon caused by Fus-depletion whereas those of gene expression include both indirect phenomenon as well as direct suppressive effects on transcription by binding to promoter antisense transcripts20. Indeed, we observed more frequent FUS-tagged sites on genes with altered spliced alternative exons than genes with altered gene expression (Supplementary Fig. S4). In this context, the alternative splicing events regulated by FUS could explain the cell vulnerability in ALS/FTLD associated with FUS, whereas the gene expression regulated by FUS could represent the size of cellular impact in FUS-associated ALS/FTLD. The fact that cell death broadly occurs in the cerebral cortex but not in the cerebellum and that cell death occurs only in neurons but not in glial cells in ALS/FTLD also supports this notion30,31. Although we found cell-type-specific profiles of FUS-regulated alternative splicing events, the RIP experiments showed that direct interaction between FUS and target RNA was comparable in different CNS tissues. The complexity map showed similar patterns for FUS-binding positions around the alternatively spliced exons in cortical neurons and glial cells. These findings indicate that FUS binding to mRNA is not dependent on cell/tissue type. Instead, other molecules that associate with FUS in the spliceosome are likely to dictate cell/tissue type-specificity of FUS-mediated alternative splicing events. FUS protein-interaction analysis may provide more detail information about cell specific machinery of FUS on alternative RNA splicing.
The results showed concordant regulation of many alternative splicing events by FUS in both motor and cortical neurons, which underscores the notion that FUS-mediated ALS and FTLD could be considered the same disease entity based on clinicopathological and genetic findings7,32. The cell-type specific transcriptome profiles we established were embryonic; however many of identified FUS-mediated splicing events were likely to be seen in adult tissues. Indeed, 34% (15/44) of alternative splicing events in Table2 were also seen in the list of another report33 using adult mouse brain with Fus-silencing (data not shown). Among them, Mapt is the most notable gene whose splicing was affected by Fus-depletion (Fig. 3A and Table 2). The inclusion exon 10 yields 4-repeat Tau (RD4), whereas skipping of exon10 generates 3-repeat Tau (RD3). We reported previously the increase of exon 10 inclusion in Mapt in Fus-silenced primary cortical neurons, a finding also reported by several other groups20,33,34,35. In this study, we also found the increase of exon 10 inlusion in primary motor neurons, although the protein level of RD4 was only dectable in cortical neurons. In this regard, it is intriguing that previous studies reported the presence of high RD4/RD3 ratio in various neurodegenerative disorders, including FTLD36,37. However, little is known about the involvement of Tau pathology in motor neuron degeneration in ALS. Further studies are necessary to clarify the association between the FUS-Tau pathway and pathogenesis of ALS and FTLD.
As described above, the entire profiles of alternative splicing events in motor and cortical neurons were almost identical. Nonetheless, we identified only motor neuron- or cortical neuron-specific alternative splicing events with close observation of region- and cell-type specific profiles of alternative splicing. Those splicing events may potentially represent differences in cell fate in each clinical subtype of ALS/FTLD. We identified some channel-associated genes, such as Synj1, Scn8a and Rims1 as motor neuron-specific alternative splicing targets regulated by FUS (Fig. 3B and Table 2), indicating that synaptic dysfunction provoked by Fus-silencing seems to be one of causes of motor neuron degeneration.
On the other hand, cortical neuron-specific alternative exons might affect cerebral neurons, leading to cortex pathology in FTLD. Stxbp1 is the causative gene for epileptic encephalopathy early infantile type 4 (EIEE4)38 and participates in the regulation of synaptic vesicle docking and fusion by associating with the SNARE complex, which is essential for fusion of opposing cellular membranes necessary for neurotransmission39. It is noteworthy that two other SNARE complex components; Syntaxin-1A and Snap25, were also identified as FUS-regulated gene and alternative exon, respectively (Fig. 3C and Table 2). Fmr1 is essential for normal cognitive development and its mutation can lead to fragile X syndrome characterized by mental retardation, autism, Parkinson's disease and other cognitive deficits40. The other fragile X syndrome-related gene, Fxr1, is also included among the list of alternative exons in which Fxr1 exon 16 is skipped by Fus-silencing in all cell types (Supplementary Fig. S3D and Table 3).
Involvement of glial cells, such as astrocytes and microglia, likely modifies and exaggerates the ALS/FTLD disease process. Although it is not clear whether non-cell autonomous mechanism is also relevant to FUS-associated ALS/FTLD, we showed here that primary glial cells showed more altered differential genes by Fus-silencing than cerebellar neurons (Fig. 2A), suggesting that knock-down of FUS gene could have certain impact on cellular homeostasis of glial cells, in addition to motor and cortical neurons.
Our global analysis also identified several genes relevant to various neurological diseases (Table 3). The alteration of gene expression and/or alternative splicing of these genes may have a large impact on neuronal function. It is unlikely that only one of these genes or exons is solely responsible for neurodegeneration in ALS/FTLD. FUS silencing would have a partial effect instead of total loss of function by altering isoforms or down/up regulation of these genes; therefore, it is possible that accumulation of altered genes affected by FUS-depletion could cause neurodegeneration after reaching a critical threshold level even when the individual alternative splicing event and gene expression are not critical by themselves. Investigation of FUS-targeting molecules, especially those relevant to neurological diseases may provide mechanistic insights into selective neuronal degeneration in FUS-associated ALS/FTLD.
Methods
Primary cells from the central nervous system
Primary motor neurons were harvested from the spinal cords of C57BL/6 mouse embryos at embryonic (E) day 13. Primary cortical neurons and primary glial cells were obtained from the cerebri of C57BL/6 mice at E15. Primary cerebellar neurons were obtained from cerebelli of C57BL/6 mice at E15. The procedure for culture of each primary cell and lentivirus infection are described in the online supplementary information. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and under the approval of the Nagoya University Animal Experiment Committee (Nagoya, Japan).
Lentivirus
We designed two different shRNAs against mouse Fus as well as a control shRNA. The targeted sequences were 5′-GCAACAAAGCTACGGACAA-3′ for shRNA/FUS1 (shFUS1); 5′-GAGTGGAGGTTATGGTCAA-3′for shRNA/FUS2 (shFUS2); and 5′-AATTCTCCGAACGTGTCACGT-3′ for shRNA/control (shCont). These were cloned into a lentiviral shRNA vector (pLenti-RNAi-X2 puro DEST, w16-1, a kind gift from Dr. Eric Campeau at Resverlogix Corp., Calgary, Alberta, Canada). Lentivirus was prepared using the protocol described by Campeau et al41. Briefly, lentiviral particles were produced in HEK293T cells by transfection using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The lentivirus-containing supernatant was collected at 48 hours after transfection and stored at −80°C. Lentivirus titer was measured using NucleoSpin RNA Virus kit (Clontech, Mountain View, CA).
Microarray analysis
Total RNA was extracted from primary motor neurons, cortical neurons, cerebellar neurons and glial cells by the RNeasy Mini kit (Qiagen, Hilden, Germany). We confirmed that the RNA integrity numbers (RIN) were all above 7.0. cDNA fragments were synthesized and labeled from 100 ng of total RNA using the GeneChip WT cDNA Synthesis Kit (Ambion, Austin, TX). Hybridization and signal acquisition of the GeneChip Mouse Exon 1.0 ST exon array (Affymetrix, Santa Clara, CA) were performed according to the instructions provided by the manufacturer. Each array experiment was performed in triplicate. The exon-level and gene-level signal intensities were normalized by the RMA and iterPLIER methods, respectively, using the Expression Console 1.1.2 (Affymetrix). We followed the gene annotation of the ENSEMBL version e!61, which is based on the mouse genome assembly NCBI build 37.1/mm9. All microarray data were registered in the Gene Expression Omnibus with accession number GSE36153 for cortical neurons, GSE42421 for motor neurons, glial cells and cerebellar neurons.
The principal component analysis of innate gene profiles in each cell type was conducted using the GeneSpring software (Agilent Technologies). For each cell type, the gene-level signal intensities of three controls treated with shCont were compared with those of three samples treated either with shFUS1 or shFUS2, using the Student's t-test. The gene expression profiles for each cell-type were established with or without statistical filtration of the t-test p value ≤ 0.1. We also obtained alternative splicing profiles by filtering the exon-level signal intensities of the probe sets on internal exons with t-test p value ≤ 0.1. Comparison analysis of the profiles was completed using shCont and shFUS1 subsets for each cell-type profile.
RT-PCR for alternative splicing analysis
Total RNA was isolated from cells using RNeasy Mini Kit (Qiagen, Hilden, Germany) followed by treatment with DNaseI (Qiagen). cDNA was synthesized from 1 μg of total RNA with the Oligo-dT primer (Promega, Madison, WI). The primers for each candidate exon were designed using Primer3 software (http://frodo.wi.mit.edu/primer3/input.htm). The sequences of primers are shown in Supplementary Table S4. Semi-quantitative RT-PCR was performed using Ex Taq (Takara Bio Inc, Otsu, Japan) at 25–30 cycles at 98°C for 10 sec, 60°C for 30 sec and 72°C for 1 min. PCR products was electrophoresed on 15% acrylamide gel and stained with ethidium bromide. The intensity of each band was measured by Multi Gage software (Fujifilm, Tokyo).
Immunoblot
Cells were lysed in TNE buffer containing protease inhibitors for 15 min on ice. The lysates were then cleared by centrifuging the cells at 13,000 g for 15 min at 4°C. Lysates were normalized for total protein (10 μg per lane), separated using a 4%–20% linear gradient SDS-PAGE and electroblotted. For immunoblot, we used anti-FUS antibodies (A300-293A, Bethyl Laboratories, Montgomery, TX and 4H11, Santa Cruz Biotechnology, Santa Cruz, CA), anti-Syntaxin-1A antibody (abcam, Cambridge, MA), anti-RD3 antibody (Millipore, Billerica, MA), anti-RD4 antibody (Millipore), anti-Braf antibody (Thermo Scientific, South Logan, UT) and anti-actin antibody (Sigma, St. Louis, MO).
RNA imunoprecipitation (RIP)
Extracts were taken from mouse cerebrum, cerebellum and spinal cord at E15, normalized for total protein (1.6 mg) and applied for RIP using anti-FUS antibody (A300-293A, Bethyl Laboratories) and RIP-Assay Kit (MBL, Nagoya Japan). Immunoprecipitation using rabbit IgG was used as a control. Semi-quantitative RT-PCR was performed using Ex Taq (Takara Bio Inc, Otsu, Japan) and random primers at 22–25 cycles at 98°C for 10 sec, 60°C for 30 sec and 72°C for 1 min. Based on the HITS-CLIP analysis information in our previous study, we designed primers for Mapt at exon6, Dlgap1 at exon11 and Stxbp1 at exon20, respectively. Primers for Gapdh and an intergenic region were used as controls.
Bioinformatics analysis
The detail was described in Supplementary experimental procedures.
Change history
22 November 2013
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Rothstein, J. D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol 65 Suppl 1, S3–9 (2009).
Saxena, S. & Caroni, P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71, 35–48 (2011).
Strong, M. J. & Volkening, K. TDP-43 and FUS/TLS: sending a complex message about messenger RNA in amyotrophic lateral sclerosis? FEBS J 278, 3569–77 (2011).
Lagier-Tourenne, C. & Cleveland, D. W. Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–4 (2009).
Munoz, D. G. et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118, 617–27 (2009).
Neumann, M. et al. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–31 (2009).
Van Langenhove, T., van der Zee, J. & Van Broeckhoven, C. The molecular basis of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum. Ann Med 44, 817–28 (2012).
Kwiatkowski, T. J., Jr et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–8 (2009).
Vance, C. et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–11 (2009).
Deng, H. X. et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67, 739–48 (2010).
Bosco, D. A. et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19, 4160–75 (2010).
Dormann, D. et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J 29, 2841–57 (2010).
Ito, D., Seki, M., Tsunoda, Y., Uchiyama, H. & Suzuki, N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol 69, 152–62 (2011).
Kino, Y. et al. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res 39, 2781–98 (2011).
Wang, J. W., Brent, J. R., Tomlinson, A., Shneider, N. A. & McCabe, B. D. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Invest 121, 4118–26 (2011).
Kabashi, E. et al. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet 7, e1002214 (2011).
Boillee, S. et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–92 (2006).
Yamanaka, K. et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci U S A 105, 7594–9 (2008).
Yamanaka, K. et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11, 251–3 (2008).
Ishigaki, S. et al. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2, 529 (2012).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 (2009).
Dennis, G., Jr et al. DAVID: Database for Annotation, Visualization and Integrated Discovery. Genome Biol 4, P3 (2003).
Licatalosi, D. D. et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–9 (2008).
Masuda, A. et al. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci Rep 2, 209 (2012).
Xue, Y. et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 36, 996–1006 (2009).
Keren, H., Lev-Maor, G. & Ast, G. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11, 345–55 (2010).
Grabowski, P. Alternative splicing takes shape during neuronal development. Curr Opin Genet Dev 21, 388–94 (2011).
Yeo, G., Holste, D., Kreiman, G. & Burge, C. B. Variation in alternative splicing across human tissues. Genome Biol 5, R74 (2004).
Clark, T. A. et al. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol 8, R64 (2007).
Geser, F. et al. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol 65, 636–41 (2008).
Ravits, J. M. & La Spada, A. R. ALS motor phenotype heterogeneity, focality and spread: deconstructing motor neuron degeneration. Neurology 73, 805–11 (2009).
Da Cruz, S. & Cleveland, D. W. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol 21, 904–19 (2011).
Lagier-Tourenne, C. et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15, 1488–97 (2012).
Rogelj, B. et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep 2, 603 (2012).
Orozco, D. et al. Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep 13, 759–64 (2012).
Yoshida, M. Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology 26, 457–470 (2006).
Hong, M. et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282, 1914–7 (1998).
Saitsu, H. et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 40, 782–8 (2008).
Tian, J. H., Das, S. & Sheng, Z. H. Ca2+-dependent phosphorylation of syntaxin-1A by the death-associated protein (DAP) kinase regulates its interaction with Munc18. J Biol Chem 278, 26265–74 (2003).
Verheij, C. et al. Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature 363, 722–4 (1993).
Campeau, E. et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529 (2009).
Acknowledgements
Part of this study represents the results of “Integrated Research on Neuropsychiatric Disorders” carried out under the Strategic Research Program for Brain Sciences by the Ministry of Education, Culture, Sports, Science and Technology of Japan. This work was also supported by Grants-in-Aid from the CREST/JST, MEXT and MHLW of Japan.
Author information
Authors and Affiliations
Contributions
Y.F., S.I., A.M., T.U. and Y.I. performed the experiments. Y.F., S.I., A.M., H.W., K.O. and G.S. analyzed the data. S.I., M.K., K.O. and G.S. prepared the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Accession codes Microarray data were uploaded to the Gene Expression Omnibus database: GSE36153 for cortical neurons, GSE42421 for motor neurons, glial cells and cerebellar neurons.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Fujioka, Y., Ishigaki, S., Masuda, A. et al. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Sci Rep 3, 2388 (2013). https://doi.org/10.1038/srep02388
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02388
This article is cited by
-
Lack of Fgf18 causes abnormal clustering of motor nerve terminals at the neuromuscular junction with reduced acetylcholine receptor clusters
Scientific Reports (2018)
-
Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS
Scientific Reports (2017)
-
The neurogenetics of alternative splicing
Nature Reviews Neuroscience (2016)
-
FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis
Acta Neuropathologica Communications (2015)
-
FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization
Nature Communications (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
