
Abstract
Cdc42 regulates the initial establishment of cytoskeletal and junctional structures, but only little is known about its role at later stages of cellular differentiation. We studied Cdc42′s role in vivo in auditory supporting cells, epithelial cells with high structural complexity. Cdc42 inactivation was induced early postnatally using the Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice. Cdc42 depletion impaired elongation of adherens junctions and F-actin belts, leading to constriction of the sensory epithelial surface. Fragmented F-actin belts, junctions containing ectopic lumens and misexpression of a basolateral membrane protein in the apical domain were observed. These defects and changes in aPKCλ/ι expression suggested that apical polarization is impaired. Following a lesion at adulthood, supporting cells with Cdc42 loss-induced maturational defects collapsed and failed to remodel F-actin belts, a process that is critical to scar formation. Thus, Cdc42 is required for structural differentiation of auditory supporting cells and this proper maturation is necessary for wound healing in adults.
Similar content being viewed by others

Introduction
Hair cells and supporting cells of the auditory sensory epithelium, the organ of Corti of the cochlea, possess elaborate actin and microtubule cytoskeletons that account for the unique shapes, morphological specializations and functions of these cells. The whole three-dimensional (3D) structure of the cochlea is needed for auditory perception and transduction, but the sensory hair cells are the key cellular players in these events. In addition, the two types of auditory supporting cells, the pillar and Deiters' cells, are essential for normal hearing1,2,3. Apices of supporting cells possess circumferential F-actin belts and intercellular junctions of which adherens junctions between pillar cells are unusually wide and have unique structural features4,5,6,7. Pillar cells contain distinct apical-basal arrays of thousands of microtubules8. F-actin and microtubules in supporting cells are thought to contribute to hearing biomechanics by providing stiffness to the organ of Corti9,10,11. Further, in the lesioned sensory epithelium, F-actin in the apices of supporting cells is prominently remodeled upon the loss of adjacent hair cells, an event that importantly contributes to the wound healing process5,12,13,14. Recently, in supporting cells of mammalian vestibular organs, structural characteristics have been linked with the poor regenerative capacity of these cells: Thick F-actin belts and the abundance of junctional E-cadherin have been suggested to form barriers against cell cycle re-entry and supporting cell-to-hair cell transdifferentiation15,16,17. Barriers against regeneration are likely to be even more relevant in the case of supporting cells of the organ of Corti, because of the high structural complexity of these cells.
The present work focuses on the in vivo role of Rho GTPase Cdc42 in the organ of Corti. There is a wealth of studies on Cdc42′s role in cultured cells, demonstrating its involvement in processes such as adhesion, motility and polarity. This regulation is often mediated through modulation of F-actin dynamics and, as more recently shown, it is linked with membrane protein recycling18. Much less is known about the function of Cdc42 and other GTPases in the mouse in vivo, especially in postnatal tissues19,20. In rodents, morphological differentiation of auditory supporting cells is to a large extent a postnatal process. By analyzing the Cdc42loxP/loxP;Fgfr3-iCre-ERT2 transgenic mice (hereafter called the mutant mice), we provide evidence that Cdc42 is required for structural maturation of the apical adherens junctions and circumferential F-actin belts in auditory supporting cells and that atypical protein kinase C (aPKC), a component of the apical polarity complex, is likely involved in mediating the effects of Cdc42 on apical polarization. We also show that, upon the loss of hair cells of the organ of Corti, the properly developed apical actin cytoskeleton in supporting cells is required for scar formation. Structural dedifferentiation of adult cells has been shown to underlie natural regeneration is some non-mammalian tissues, e.g. in the case of cardiomyocytes of the lesioned zebrafish heart21. Structural barriers or the inability for structural dedifferentiation have been suggested to prevent regeneration in the mammalian inner ear15,16,17. An understanding of the molecular mechanisms underlying structural development and wound healing response of mammalian supporting cells has translational value, because this knowledge might contribute to the development of methods to stimulate regeneration or repair.
Results
Recombination characteristics in the organ of Corti of Fgfr3-iCre-ERT2 mice
To achieve temporally controlled and selective gene inactivation in postnatal auditory supporting cells, we studied characteristics of iCre-mediated recombination in the Fgfr3-iCre-ERT2 mouse line22. We crossed these animals with the ROSA26tm14(CAG-tdTomato) reporter mice that express tdTomato protein under the modified Rosa26 locus (hereafter called the Ai14(tdTomato) mice)23. In the Fgfr3-iCre-ERT2;Ai14(tdTomato) double transgenic mice, we first assayed for recombination following tamoxifen administration between P2 and P4. Recombination was revealed at P7 and P10 by red fluorescent protein (RFP) immunohistochemistry on paraffin sections (Fig. 1A,B) and direct visualization of tdTomato fluorescence in wholemount surface specimens (Fig. 1C–F). At both time points, in addition to the bony otic capsule and cochlear ganglion (Fig. 1A), Fgfr3-iCre-ERT2;Ai14(tdTomato) mice showed high recombination efficiency (above 95%) in pillar and Deiters' cells throughout the cochlea. Also some non-sensory cells located laterally to the organ of Corti proper, particularly Claudius cells, were recombined in the basal part of the cochlea (Fig. 1B,D,E,F). In addition, many outer hair cells in the apical turn and, surprisingly, a small number of these cells in the basal turn showed recombination (Fig. 1A,C,E,F). In the medial turn, used for the analysis of Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice, only scattered recombined outer hair cells were found (Fig. 1A,B,D,F). Cochlear inner hair cells (Fig. 1A–I) and cells of the vestibular organs (saccule; Fig. 1A) lacked recombination. Of note, when tamoxifen administration was initiated one day earlier, at P1, clearly higher numbers of outer hair cells were recombined in the lower half of the cochlear duct (data not shown).

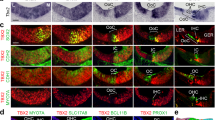
Recombination in the cochlea of Fgfr3-iCre-ERT2 mice. Cdc42 expression in the organ of Corti.
(A,B) Fgfr3-iCre-ERT2;Ai14(tdTomato) mice treated with tamoxifen between P2 and P4 show recombination in the organ of Corti, cochlear ganglion and surrounding bone, as revealed by RFP immunohistochemistry in paraffin sections at P7. The organ of Corti of the medial turn boxed in (A) is shown in higher magnification in (B). Hair cells are negative, as opposed to the adjacent supporting cells. (C–F) At P10, tdTomato fluorescence is found in supporting cells throughout the cochlea of Fgfr3-iCre-ERT2;Ai14(tdTomato) mice treated with tamoxifen between P2 and P4. Boxed areas in the 3D isosurface view of the cochlea (F) are shown in z-projections (C–E). DAPI marks nuclei. In addition to supporting cells, a part of hair cells, especially in the apical, but also in the most basal part of the cochlea are recombined. Note the single recombined outer hair cell (arrow) in the medial turn (D). (G–I) At P30, tdTomato fluorescence at different levels of the cochlea of Fgfr3-iCre-ERT2;Ai14(tdTomato) mice injected with tamoxifen between P16 and P18. Recombination is restricted to supporting cells. (J) Fgfr3 is expressed in supporting cells of the organ of Corti at P10. (K,L) Cdc42 is expressed throughout the cochlea at P0. Expression is more regionalized at P10. (M) Absence of Cdc42 expression in the organ of Corti of Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice at P10. Thin arrows mark outer hair cells and thick arrow an inner hair cell in (B,J-M). Abbreviations: cg, cochlear ganglion; ca, otic capsule; ip, inner pillar cell; op, outer pillar cell; d, Deiters' cell; p, pillar cell; Cl, Claudius cell; oh, outer hair cell; ih, inner hair cell; sa, saccule; WT, wildtype; Mut, mutant; TM, tamoxifen. Scale bar (in J): A,F, 160 μm; B, 20 μm; C–E, G–I, 40 μm; J–M, 50 μm.
In the second scheme of induction of recombination, Fgfr3-iCre-ERT2;Ai14 mice received tamoxifen between P16 and P18, followed by analysis at P30. Similar to tamoxifen administration early postnatally, wholemounts showed high recombination rate (about 90%) in the populations of pillar and Deiters' cells, based on the broad tdTomato fluorescence. Hair cells were negative (Fig. 1G–I). We confirmed the absence of recombination in these sensory cells by using antibodies against myosin VIIa, a cell type-specific marker (data not shown). A few Claudius cells were also recombined, but the amount of these cells was clearly less as compared to the early postnatal tamoxifen administration (Fig. 1G–I). Together, these results are consistent with prior data demonstrating that, around birth, Fgfr3 is downregulated in outer hair cells in a basal-to-apical turn gradient along the length of the cochlear duct and that the expression is thereafter restricted to supporting cells (Fig. 1J)24,25. Thus, the Fgfr3-iCre-ERT2 mouse line is a useful tool for inducible gene inactivation in postnatal auditory supporting cells. Fgfr3-iCre-ERT2;Ai14 mice injected with corn oil lacked recombination (data not shown).
Cdc42 expression in the organ of Corti
Radioactive in situ hybridization showed ubiquitous Cdc42 expression in the cochlea around birth (Fig. 1K). By the beginning of the second postnatal week, the expression became regionally more localized. Between P7 and P15, Cdc42 was expressed in pillar and Deiters' cells, whereas hair cells lacked a detectable signal (Fig. 1L). Thereafter, Cdc42 was downregulated in a basal-to-apical turn gradient along the length of the cochlear duct, so that expression was undetectable by in situ hybridization at P20. Cdc42 signal was absent from the supporting cell population of tamoxifen-challenged Cdc42loxP/loxP; Fgfr3-iCre-ERT2 mice, as shown at P10 (Fig. 1M). Together, although Cdc42 might be expressed in mature supporting cells at levels undetectable by in situ hybridization, our data show that the expression is concentrated to these cells during the early postnatal period of morphological differentiation.
Cdc42 depletion targets the apical domain of immature supporting cells
To elucidate the role of Cdc42 during supporting cell maturation, tamoxifen-based gene inactivation was induced in Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice between P2 and P4. At the period of tamoxifen administration, supporting cells, especially pillar cells, have immature shapes and still poorly-developed actin and microtubule cytoskeletons. Adult-like cell morphologies are achieved during the second postnatal week, concomitantly with the formation of the large, fluid-filled spaces within the organ of Corti (Fig. 2A,B)26,27,28,29. At P10, transverse paraffin sections through the cochleas of mutant and control mice showed no substantial differences in the cytoarchitecture of the organ of Corti. Based on β-tubulin staining, also the supporting cell's microtubule network was comparable in the two genotypes (Fig. 2A,C). However, sections showed that the lateral-to-medial width of the surface area of the organ of Corti, the so-called reticular lamina, was decreased in mutants (Fig. 2A,C). These findings were confirmed in wholemounts labeled with phalloidin, a marker for F-actin. Wholemounts revealed that the shrinkage of the reticular lamina resulted from shortened circumferential F-actin belts of Deiters' cells and outer pillar cells (Fig. 2D,E).

Impaired differentiation of the apical domain of supporting cells of Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice.
Tamoxifen treatment between P2 and P4. Paraffin sections (A,C,H–M) and wholemount confocal specimens (D–G′,N,O) represent the medial part of cochlea from wildtype control and mutant mice at P10. (A,B) β-tubulin staining and schematic representation of the cytoarchitecture of normal organ of Corti in transverse plane and in a surface view (light blue, outer pillar cells; dark blue, inner pillar cells; green, Deiters' cells; red, hair cells; asterisk, tunnel of Corti). Heads of outer pillar cells are covered by heads of inner pillar cells. The grey line in (B) marks the level of confocal z-projections used in (F–G′,N,O). (C) β-tubulin staining is unaltered in mutant mice. (D,E) Phalloidin labeling shows constriction of the reticular lamina of mutants. (F–G′) Adherens junctions and F-actin belts of outer pillar cells of mutants are abnormal, as revealed by double-labeling for F-actin and E-cadherin. (H,I) Progression of differentiation of outer pillar cells during the first postnatal week, as shown by CD44 staining. Arrows mark adherens junctions. (J,K) At P10, outer pillar cells of mutant mice display CD44-positive ectopic lumens at apical junctions. (L,M) A paraffin section from a mutant cochlea cut in the plane parallel to the epithelial surface shows an abundance of CD44-positive ectopic lumens at outer pillar cells junctions. (N,O) In mutants at P10, F-actin belts are fragmented at the site of the lumens, as revealed by double-labeling for F-actin and CD44. Abbreviations: tub, β-tubulin; R, reticular lamina; ip, inner pillar cell; op, outer pillar cell; d, Deiters' cell; oh, outer hair cell; ih, inner hair cell, p, pillar cell; WT, wildtype; Mut, mutant; E-cad, E-cadherin. Scale bar (in O): A,C,H,I, 20 μm; D,E, 15 μm; F–G′,J–O, 8 μm.
In mutant mice at P10, while shortened adherens junctions and underlying F-actin belts comprised the primary alterations in Deiters' cells (Fig. 2D,E), outer pillar cells showed additional disturbances. At this stage, F-actin belts of outer pillar cells are still weakly phalloidin-positive (compare the immature and mature control specimens; Figs. 2D,F and 5E,G). Despite this immaturity, phalloidin labeling revealed conspicuous fragmentation of F-actin belts in mutant, but not in control specimens (Fig. 2F,G). Double-staining with E-cadherin, an adherens junction marker, showed ectopic lumens of variable size between outer pillar cells of mutant mice, specifically in the gap regions in the fragmented F-actin belts (Fig. 2F–G′). We penetrated into this altered membrane phenotype by studying the expression of the cell-surface glycoprotein CD44 that marks outer pillar cells in the organ of Corti30. At birth and during the first postnatal week, CD44 was strongly expressed in the lateral walls of these cells, while the expression in the apical adherens junctions was very weak (Fig. 2H–J). This expression was maintained until the end of the second postnatal week (data not shown). At P10, paraffin sections from mutant mice showed CD44 misexpression in the apical domain of outer pillar cells. Ectopic lumens were lined by CD44 expression, similar to E-cadherin expression (Fig. 2J–M). Double-immunofluorescence showed that these lumens were associated with gaps in F-actin belts (Fig. 2N,O). This misexpression of a lateral membrane protein in the apical domain, coupled with the presence of dysmorphic F-actin belts suggested that there are defects in apical-basal polarity of Cdc42-depleted supporting cells.
Subcellular defects in Cdc42-depleted supporting cells
Given the complexity of the cellular architecture of the organ of Corti and our need to penetrate into subcellular defects caused by Cdc42 inactivation, 3D structural information and resolution better than achieved by light microscopy were required. To this end, we applied the method of Serial Block-Face Scanning Electron Microscopy (SBF-SEM). The two basic features of this technique are a microtome mounted inside the chamber of the SEM to shave repeatedly fresh block faces of resin-embedded tissue for imaging and a sensitive back scatter electron detector resulting in a classic TEM like images31. With this technique we were able to collect datasets covering the whole organ of Corti to select snapshots of corresponding areas at defined orientation from control and mutant animals for comparison. At the surface of the organ of Corti, pillar cells and the three rows of both Deiters' cells and outer hair cells form a mosaic comprising heterotypic and homotypic cell-cell junctions32. The complexity of this mosaic is illustrated, for example, by the cellular contacts made by outer pillar cells. Apices of these cells extend outwards under the cover formed by the heads of inner pillar cells, reaching the lumenal surface in front of outer hair cells of the second row. In addition, apices of outer pillar cells contact the first row of Deiters' cells (Fig. 2B).
Analysis at P10 showed decreased width of the reticular lamina and decreased apical cell area of individual supporting cells of mutant mice, similarly as seen in phalloidin-labeled wholemounts. Contacts between Deiters' cells were abnormally short while the length of the heterotypic junctions between Deiters' cells and outer hair cells were unaltered (Fig. 3A,B). In Cdc42-depleted outer pillar cells, the analysis of series of SBF-SEM images at P10 confirmed the presence of lumens of variable size. These ectopic structures were lined by thin appendages and some of the lumens contained matrix inside, suggestive of secretory function (Fig. 3C–G). Interestingly, these apical abnormalities coincided with the normal formation of the large fluid-filled lumen, the tunnel of Corti that surrounds the basolateral walls of pillar cells. The tunnel of Corti is lined by short microvilli that have been suggested to possess secretory function26, thus resembling the lining of the ectopic lumens found in mutant mice. These results demonstrate that the lateral and apical cell membranes of Cdc42-depleted outer pillar cells are incorrectly segregated, suggestive of cell polarity defects. Together, subcellular analysis confirmed that Cdc42 depletion targets the apical adherens junctions and perijunctional F-actin in immature Deiters' and outer pillar cells. The heads of inner pillar cells did not show structural alterations similar to those seen in the other two supporting cell subtypes of the organ of Corti. Hair cells in the medial turn of the cochlea, the region used for phenotypic analysis of supporting cells, showed an unaltered morphology, including unaltered orientation of stereociliary bundles (Fig. 3H,I). This is consistent with the lack of recombination in hair cells at this level of the cochlea of the Fgfr3-iCre-ERT2;Ai14(tdTomato) mice (Fig. 1A–I).

Subcellular abnormalities in the apical domain of supporting cells of Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice.
Tamoxifen treatment between P2 and P4. SBF-SEM analysis of wildtype control (A,C,E,H) and mutant (B,D,F,I) mice at P10. (A,B) Supporting cells of mutant specimens show unelongated adherens junctions and F-actin belts (arrows). (C–D) Ectopic lumens (arrows) are seen between outer pillar cells of mutant specimens. (G) Schematic representation of the organ of Corti shows the planes of images (A,B; solid), (C,D; dashed) and (E,F; dots). (H,I) 3D reconstruction of stereociliary bundles (arrows) of outer hair cells shows unaltered morphology in mutant mice. Abbreviations: op, outer pillar cell; ip, inner pillar cell; d, Deiters' cell; oh, outer hair cell; ih, inner hair cell; WT, wildtype. Scale bar (in I): A–F, 3.5 μm; H,I, 2.5 μm.
Changes in aPKC expression in Cdc42-depleted supporting cells
The Par6/aPKC complex is one of the several targets of Cdc42. Binding of Cdc42 to Par6 activates aPKC, triggering multiple downstream signaling events that promote apical polarity of epithelial cells33,34. We found that one of the mammalian aPKC isoforms, aPKCλ/ι, was expressed in supporting cells of the organ of Corti during the early postnatal period of structural differentiation. We documented this expression in sectioned material using antigen retrival to expose epitopes (Fig. 4A–E). The antibody used did not reliably work in wholemount specimens. As a positive staining control, we used developing kidney glomeruli where aPKCλ/ι expression has been characterized (Fig. 4A, inset)35. aPKCλ/ι was not expressed in the organ of Corti at birth (Fig. 4A). The expression in the apical junctional domain of Deiters' and pillar cells was initiated at the end of the first postnatal week. aPKCλ/ι expression was especially prominent and sharp in pillar cell's adherens junctions (Fig. 4B,C). Adult organ of Corti did not anymore show this expression (data not shown). The possible expression in hair cell junctions could not be reliably determined by immunohistochemistry on sections. In response to Cdc42 depletion, aPKCλ/ι expression was partly delocalized from the junctional area of supporting cells, resulting in diffuse cytoplasmic expression close to the cell cortex (Fig. 4D,E), consistent with the findings made in some other mammalian epithelial cells36. This delocalization was clearly seen in the broad adherens junctions between pillar cells. Together, these results suggest that Cdc42 targets aPKCλ/ι, likely within the Par6/aPKCλ/ι complex, in auditory supporting cells and that this might be a mechanism by which Cdc42 regulates apical polarity of these cells.

Expression of the aPKCλ/ι polarity protein in the organ of Corti.
Tamoxifen treatment between P2 and P4. Paraffin sections from the medial part of the cochlea. (A) aPKCλ/ι expression is absent from the organ of Corti at birth. The sensory epithelial surface is marked by a line. (B,C) In wildtype mice at P10, apices of supporting cells show aPKCλ/ι expression. Note the sharp expression in the adherens junction of pillar cells, shown in a high-magnification view (C). (D,E) In Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mutant mice, aPKCλ/ι expression is delocalized from the junctions and has a diffuse pattern. The three parallel arrows mark the heads of Deiters' cells and the single arrow the adherens junction of pillar cells. Asterisks mark the phalangeal processes of Deiters' cells located between outer hair cells. Abbreviations: oh, outer hair cell; op, outer pillar cell; WT, wildtype; Mut, mutant. Scale bar (in E): A,B,D, 160 μm; C,E, 10 μm.
Persistence of structural defects in Cdc42-depleted adult supporting cells
We next investigated the outcome of supporting cells depleted for Cdc42 from developmental stages onward (tamoxifen treatment between P2 and P4). At P20, the medial part of the cochlea of control and mutant mice showed comparable overall cytoarchitecture of the organ of Corti. Also the global organization of the microtubule cytoskeleton in supporting cells was comparable (Fig. 5A–D). Cdc42 inactivation did not cause cellular loss at this stage. However, phalloidin-labeled wholemounts (Fig. 5E–H′) and SBF-SEM preparations (Fig. 5I–N) displayed maintained shrinkage of the reticular lamina and of the apical cell area of individual supporting cells similar to that seen during the immature stage. Quantification at P20 (Fig. 6A–D) showed a highly significant (p < 0.001) difference in the width of the reticular lamina between control and mutant specimens, based on phalloidin-labeled wholemounts (controls: 47.7 ± 1.5 μm, n = 4; mutants: 30.6 ± 0.3 μm, n = 4). There were also highly significant (p < 0.001) differences in junctional lengths between individual supporting cells, based on SBF-SEM analysis (Outer pillar cell–1st row Deiters' cell junction: controls 4.3 ± 0.6 μm, n = 15; mutants 0.5 ± 0.4 μm, n = 28) (1st and 2nd row Deiters' cell junction: controls 2.5 ± 0.8 μm, n = 13; mutants 0.7 ± 0.5 μm, n = 18). Interestingly, the conspicuous fragmentation of the short F-actin belts of outer pillar cells was maintained in adults (Fig. 5G–H′,K–N). However, the ectopic lumens between outer pillar cells that were abundant at the immature stage were not anymore seen at P20 (Fig. 5G–H′,K–N).

Adult phenotype of the organ of Corti of Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice.
Tamoxifen was administered between P2 and P4. Paraffin sections (A–D, Q,R), confocal views (E–H′) and SBF-SEM images (I–P) represent the medial part of cochlea at P20. Paraffin sections (S,T) from the basal part at P51. (A–D) Hematoxylin- and β-tubulin stainings show largely comparable histology in wildtype control and mutant mice. (E,F) Phalloidin staining reveals constricted organization of the reticular lamina of mutant mice. (G-H′) Double-staining for phalloidin and E-cadherin shows discontinuous F-actin belts of pillar cells of mutants. (I,J) Junctions and F-actin belts between supporting cells (arrows) are unelongated in mutant mice, as shown in surface plane. (K–N). Outer pillar cells of mutant mice display fragmented F-actin belts (arrows), as revealed in transverse (K,L) and surface planes (M,N). Note that the anchoring of microtubule bundles (arrow) follows F-actin fragmentation in mutants (N). (O,P) The centrosome (arrows) has a comparable localization in the heads of Deiters' cells of wildtype and mutant mice. (Q,R) Hematoxylin staining shows that, in the cochleas of mutant mice at P51, cell numbers and the cytoarchitecture are preserved in the medial turn, but there is cellular degeneration in the basal turn. Abbreviations: tub, β-tubulin; ih, inner hair cell; oh, outer hair cell; ip, inner pillar cell; op, outer pillar cell; p, pillar cell; d, Deiters' cell; E-cad, E-cadherin; WT, wildtype; Mut, mutant; med, medial turn. Scale bar (in T): A–D, Q–T, 60 μm; E,F, 25 μm; G–H′ 10 μm; I–N, 4 μm; O,P, 3 μm.

Quantification of the width of the reticular lamina and the length of supporting cell junctions in wildtype control and Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice at P20.
Both the reticular lamina (A) and the junctions between outer pillar cells and the 1st row of Deiters' cells and between the 1st and 2nd rows of Deiters' cells (B) show highly significant shortening (p < 0.001) in mutants. The former quantification was based on phalloidin-labeled wholemounts, the latter on SBF-SEM specimens. Mean ± standard deviation (s.d.) and the number of junctions (n) measured are shown. (C,D) Examples of the lines drawn to perform quantification of junctional lengths, shown both in wildtype and mutant specimens (see Methods). Abbreviations: op, outer pillar cell; d, Deiters' cell; oh, outer hair cell; WT, wildtype.
Analysis of serial SBF-SEM images at P20 confirmed that, similar to control animals, supporting cells of mutants had thick apical-basal arrays of microtubules whose minus-ends were anchored to the area of adherens junctions and cortical F-actin. This anchoring followed the fragmented pattern of F-actin belts in mutants (Fig. 5N). Apical localization of the centrosome was comparable in supporting cells of both genotypes (Fig. 5O,P). Taken together, these findings suggest that Cdc42 is necessary for the maturation of the F-actin rather than microtubule cytoskeleton in the heads of auditory supporting cells and that the dysmorphic actin belts are maintained at adulthood.
The possible long-term effects of Cdc42 inactivation induced between P2 and P4 were studied at P51. Paraffin sections from the medial part of cochleas of mutant mice did not display changes in supporting cell numbers, despite structural defects in the heads of these cells. Also the erected position of these cells was preserved (Fig. 5Q,R). However, in the basal part of cochleas of mutant animals, cellular collapse and degeneration was evident at P51, in contrast to age-matched control specimens (Fig. 5S,T). At the earlier time point, P20, the basal region of mutants did not show these defects (data not shown), suggesting that Cdc42 has a maintenance role in adult supporting cells at this location. These findings may be coupled with the well-established fact that the basal turn is the most vulnerable region of the cochlea e.g. in response to aging.
Cdc42 depletion-induced developmental defects impair F-actin remodeling in supporting cells following trauma
To investigate the functional significance of Cdc42 in supporting cells, we challenged mice with an ototoxic lesion. We hypothesized that the abnormally developed actin cytoskeleton in Cdc42-depleted supporting cells might affect wound healing. Mice were treated with the aminoglycoside kanamycin and the loop diuretic furosemide. The synergistic effect of these drugs leads to rapid and selective loss of outer hair cells37,38. Using the Fgfr3-iCre-ERT2;Ai14 double-transgenic mice, we first confirmed the identity of the cells in the lesioned organ of Corti, taking into account the possibility that cells from outside areas might occupy this site. Fgfr3-iCre-ERT2;Ai14 mice received tamoxifen between P2 and P4 and ototoxic drugs at P20. RFP-stained cochlear sections were analyzed at days 3 and 28 post-lesion. At both time points, the organ of Corti was entirely stained, except for the remaining inner hair cells and sparse outer hair cells in the uppermost part of the cochlea (Fig. 7A,B; data not shown). These results demonstrate that, during the post-lesion time period of one month, pillar and Deiters' cells are the key cellular players contributing to the recovery process.

Cdc42 depletion leads to collapse of the organ of Corti upon ototoxic insult.
Tamoxifen treatment between P2 and P4. Transverse sections from the medial part of the cochlea between days 2 to 28 post-lesion. (A,B) RFP-stained pillar and Deiters' cells in the organ of Corti from Fgfr3-iCre-ERT2;Ai14(tdTomato) mice, as revealed at days 3 (A) and 28 (B) post-lesion. The arrow marks the inner hair cell. (C–J) Hematoxylin-stained sections at days 2, 7, 14 and 28 post-lesion show gradual collapse of the organ of Corti of Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice, in contrast to wildtype control animals. In mutants, note the disappearance of the tunnel of Corti (asterisk). Outer hair cells are absent at all post-lesion time points in both genotypes. Abbreviations: ip, inner pillar cell; op, outer pillar cell; ds, Deiters' cells; He, Hensen cell; d.pl, days post-lesion; WT, wildtype. Scale bar (in J): A–J, 20 μm.
Cdc42 inactivation was induced between P2 and P4, ototoxins were administered at P20 and the medial part of the cochlea was analyzed at days 2, 7, 14 and 28 post-lesion. In hematoxylin-stained transverse sections, the lesioned organ of Corti of control and mutant mice had a very different appearance, mutants showing a general collapse of the epithelium and disappearance of the tunnel of Corti. Loss of the tunnel was seen at day 2 post-lesion in a subset of mutant specimens, but at later time points in all these specimens. In contrast to Cdc42-depleted outer pillar cells, these cells from control animals stayed erect with a widely open tunnel, consistent with previous studies (Fig. 7C–J)38,39. To find out if Cdc42 inactivation affected the survival of supporting cells following lesion, supporting cell nuclei in the organ of Corti were counted in transverse paraffin sections from mutant and control specimens at day 28 post-lesion (see Methods). The two genotypes showed a highly significant difference (p < 0.001) in the amount of cell nuclei (control nuclei: 4.3 ± 0.5, n = 3 cochleas; mutants: 3.1 ± 0.7, n = 3 cochleas).These findings suggest that the dysmorphic actin cytoskeleton of supporting cells is associated with progressive death of these cells.
Rapidly after hair cell loss, apices of supporting cells expand and occupy the area of lost hair cells. This process, termed scar formation, contributes to the preservation of an intact reticular lamina. It is based on F-actin remodeling5,12,13,14. We next asked whether the collapse of the organ of Corti is coupled with defects in scar formation. Phalloidin-labeled wholemounts from control mice at day 2 post-lesion showed prominent F-actin remodeling; the formation of actin-rich bridge-like structures at the site of lost hair cells (Fig. 8A,B). It has been described that these bridges comprise actin filaments of two neighboring supporting cells that have laterally expanded to the site of a lost hair cell13. In contrast, in the heads of supporting cells of mutant mice, F-actin network had a strikingly wrinkled appearance (Fig. 8C,D) and actin bridges were absent (Fig. 8E,F), except in a few cases where weak phalloidin labeling was detected at the site of a lost hair cell (Fig. 8G,H). E-cadherin was colocalized to F-actin bridges in control animals, indicating that the lateral plasma membranes comprising adherens junctions expanded to the site of lost hair cells together with the cortical actin cytoskeleton (Fig. 8E′). In mutants, consistent with the lack of F-actin bridges, E-cadherin staining was obscure or absent at the sites previously occupied by hair cells (Fig. 8F′), demonstrating that supporting cells did not extend their plasma membranes across the site of missing hair cells. Abnormal scar formation was also seen at day 7 post-lesion, indicating that normal scar formation was not activated in a delayed fashion in mutant mice (data not shown). To confirm that impaired F-actin remodeling was not a consequence of supporting cell collapse in mutant specimens, we took advantage of the fact that non-lesioned cochleas often show a loss of a few hair cells. Whereas the few lost hair cells were replaced by F-actin scars in control specimens, scar formation was not seen in non-collapsed mutant specimens (Fig. 8I,J). Interestingly, despite the defects in F-actin remodeling, sections showed that the lesioned epithelium of mutant mice was covered by cellular material (Fig. 7C,D). This was also evidenced by the expression of the tight-junction-marker ZO-1 in the reticular lamina of mutants, although this staining was disorganized (Fig. 8K,L). These findings suggest for at least partial sealing of the epithelial surface, a process that is important to prevent leakage of the cytotoxic endolymph into the epithelium. Taking together, in auditory supporting cells, Cdc42-dependent proper development of the apical F-actin cytoskeleton is required for a normal wound healing response.

Effects of Cdc42 depletion on wound healing in the organ of Corti.
Wildtype control and Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mutant mice were treated with tamoxifen between P2 and P4 (A–L) or between P16 and P18 (M,N) and the medial cochlear turn was analyzed. Ototoxically lesioned animals were analyzed at day 2 post-lesion. Asterisks mark examples of the sites of lost outer hair cells. Phalloidin-labeled (red) images are partially represented as isosurface views in (A–D). Outlines of Deiters' cells at the site of lost outer hair cells are shown in (A–D). (A) The normal organization of hair cells and supporting cells in the organ of Corti of wildtype non-lesioned mice. (B) Following lesion, lost outer hair cells are replaced by supporting cell scars in wildtype mice. (C) In mutant non-lesioned mice, the width of the reticular lamina is decreased due to shortening of supporting cell junctions. (D) In mutant lesioned mice, the F-actin network appears shrunken and no signs of scars at the sites of lost hair cells can be seen. (E–F′) Double-labeling for F-actin and E-cadherin shows the expansion of the plasma membranes and cortical F-actin of neigbouring Deiters' cells to the site of a lost outer hair cell, in the form of a bridge-like structure. This remodeling process is not seen in the mutant specimen. (G) A higher magnification view shows a well-formed F-actin bridge in a control specimen. (H) Mutant specimens occasionally show weak attempts (arrow) towards formation of this structure. (I,J) Supporting cell scars are formed at the sites of sporadic outer hair cell loss, marked by the absence of nuclear DAPI staining (blue), in non-lesioned control, but not in mutant animals. (K,L) Sections show ZO-1 staining (arrows) at the reticular lamina of both control and mutant animals after the lesion. The staining has an irregular pattern in mutants. (M) Tamoxifen treatment at a mature stage does not affect the cytoarchitecture of the organ of Corti of mutant mice (compare to Fig. 8A,C). (N) Scar formation is unaffected in mutant mice treated with tamoxifen at a mature stage (compare to Fig. 8B,D). Abbreviations: d, Deiters' cell; op, outer pillar cell; ph, phalloidin; E-cad, E-cadherin; WT les., wildtype lesioned; Mut les., mutant lesioned; TM, tamoxifen. Scale bar (in N): A–D,M,N, 8 μm; E–H, 5 μm; I,J, 6 μm; K,L, 30 μm.
Acute Cdc42 depletion at adulthood does not lead to phenotypic changes
To study whether Cdc42 regulates the maintenance of apical structures of fully differentiated supporting cells, independently of its maturational role, mice were treated with tamoxifen between P16 and P18 and the analysis was performed at P25. Two doses of tamoxifen, 50 and 200 μg/g body weight were used. Phalloidin-labeled wholemounts did not display alterations in the organization of the apical F-actin cytoskeleton in supporting cells (Fig. 8M), as opposed to specimens in which Cdc42 was depleted from the early postnatal stages onward (Fig. 8C). To determine if acute Cdc42 inactivation at a mature stage affects lesion-induced F-actin remodeling, tamoxifen was administered between P16 and P18, ototoxins at P22 and cochleas were analyzed at days 2 and 7 post-lesion. Surprisingly, this late Cdc42 inactivation did not prevent normal F-actin remodeling, based on the presence of well-formed actin scars at the sites of lost hair cells (Fig. 8N), similarly as in control specimens (Fig. 8B). On the other hand, these findings are in agreement with the fact that we could not detect Cdc42 expression in supporting cells of control mice at adulthood, neither in normal conditions nor in response to ototoxic challenge, as analyzed by in situ hybridization at days 2 and 7 post-lesion (data not shown).
Lesion-induced cyclin D1 upregulation in non-sensory cells flanking the organ of Corti
Finally, we asked whether the defects in the apical domain of Cdc42-depleted supporting cells and the collapse of these cells following ototoxic insult affect phenotypic characteristics and cell cycle status. Cdc42 inactivation was induced between P2 and P4, ototoxins were administered at P20 and the medial part of cochleas was analyzed 2, 7, 14 and 28 days post-lesion. At day 28 post-lesion, β-tubulin immunostaining revealed prominent collapse of supporting cells of mutant animals, as opposed to controls (Fig. 9A,B). Expression of the cell type-specific markers Sox940 and Sox2 persisted in Deiters' and pillar cells of both genotypes (Fig. 9C,D; data not shown). Prox1, a transcription factor expressed in developing supporting cells41,42, was not upregulated in either genotype (data not shown). Pillar and Deiters' cells did not show induction of Ki-67 (Fig. 9E,F) or cyclin D1 (Fig. 9G,H) or downregulation of p27Kip1, the main negative cell cycle inhibitor in these cells (data not shown). Thus, following ototoxic insult, the normal transcriptional phenotype and postmitotic status are maintained in supporting cells of both control and mutant mice. Moreover, no signs of induction of the hair-cell-marker myosin VIIa were found in these animals (data not shown). In non-lesioned normal cochleas, cyclin D1 is found in scattered non-sensory cells located laterally to the organ of Corti, mostly in Claudius cells, as previously described (Fig. 9H)43. Following ototoxic trauma, these postmitotic cells showed cyclin D1 upregulation, both in control and mutant mice (Fig. 9G,H). Although the lateral non-sensory cells did not show cell cycle re-entry based on the absence of Ki-67 expression, cyclin D1 induction suggests that increased proliferative potential of these cells exists.

The phenotype of auditory supporting cells of Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mice challenged with ototoxins.
Tamoxifen treatment between P2 and P4. Paraffin sections from the medial part of the cochlea at day 28 post-lesion. Ototoxic drugs induce outer hair cell loss while inner hair cells (large arrows) are maintained. (A,B) In mutant mice, β-tubulin staining shows the collapse of outer pillar cells and Deiters' cells and the absence of the tunnel of Corti (asterisk). (C,D) Both in wildtype and mutant mice, Sox9 is expressed in supporting cells of the organ of Corti and in non-sensory cells of flanking regions. The inset shows Sox9 expression in supporting cells of a wildtype, non-lesioned cochlea. Large arrow marks the inner hair cell, thin arrows outer hair cells. (E,F) Ki-67 is absent from the organ of Corti of wildtype and mutant mice. (G,H) Cyclin D1 is expressed in Claudius cells (arrows) of wildtype, non-lesioned mice (inset) as well as in ototoxically challenged animals of both genotypes. Abbreviations: ip, inner pillar cell; ip, outer pillar cell; Ds, Deiters' cells; WT, wildtype; tub, β-tubulin; cD1, cyclin D1. Scale bar (in H): A–H, 10 μm.
Discussion
The current study focuses on mammalian auditory supporting cells, the cells that have been under intensive research during recent years, because of their potential to serve as a platform for therapeutic regeneration of new sensory hair cells. Supporting cells and hair cells have common precursors and, thus, understanding the regulation of cell fate decisions and early differentiation during embryogenesis is instrumental for the attempts to stimulate regeneration. What has received very little attention, however, is the regulation of maturation of supporting cells, specifically the generation of their unique shapes and structural specializations. This knowledge is important, because induced dedifferentiation may be an essential step in regeneration attempts. In the mammalian inner ear sensory epithelia, regeneration is replaced by a permanent scar. The current study shows that the Rho-family GTPase Cdc42 is necessary for postnatal differentiation of auditory supporting cells and that Cdc42′s developmental expression is critical for proper wound healing in adult cells. We show the role of Cdc42 by inducible and conditional gene inactivation. Our results emphasize the importance of this approach, allowing exclusion of early developmental effects, when studying functional significance of Cdc42 in later-developing and mature tissues.
Cdc42 is a well-known regulator of the actin and microtubule cytoskeletons. Most in vivo studies have investigated the effects of Cdc42 inactivation on early-developing, proliferating cells. In Drosophila and in mouse embryos, Cdc42 depletion has been shown to impair the establishment and maintenance of new cell-cell junctions and the underlying F-actin network44,45,46,47,48,49. In the present study, we passed the apparent effects of Cdc42 on the early stages of formation of these structures and focused on postnatal, non-proliferating auditory supporting cells that are undergoing structural differentiation to a highly specialized epithelial cell type. We found that Cdc42 is required for the maturation of the apical adherens junction/F-actin belt complexes in these cells. We did not find evidence for its participation in the global development of the microtubule cytoskeleton. Consistent with the fact that phenotypic alterations were built up in Cdc42-depleted supporting cells during the early postnatal maturation period, Cdc42 expression in the normal organ of Corti was most distinct in these cell types and at this period of life.
Cdc42-depleted supporting cells possessed short adherens junctions and F-actin belts, resulting in a constricted appearance of the reticular lamina. The main part of the elongation of the reticular lamina occurs during the first 10 days of postnatal life50, at the period when Cdc42 depletion was induced in our study. The short F-actin belts of Cdc42-depleted Deiters' cells were very thick, a finding that was readily seen in SBF-SEM images. These results suggest that limitations in actin stability or turnover are not direct reasons for the altered phenotype. Our data show that, in the case of outer pillar cells, Cdc42 is required not only for the elongation of adherens junctions and F-actin belts, but also for the integrity of these belts, as evidenced by their distinct fragmentation. This defect was associated with the formation of ectopic lumens between outer pillar cells. The fact that F-actin belts were discontinuous at the site of the lumens illustrates the close relationship between the cortical actin cytoskeleton and adherens junctions at the plasma membrane. Interestingly, ectopic lumens have been described in the liver following hepatocyte-specific Cdc42 inactivation51, thus displaying a partly similar cellular phenotype as in the current study.
The alterations in Cdc42-inactivated auditory supporting cells also partly resemble the phenotype of early-developing, Cdc42-deficient epithelial cells of Drosophila47,48,49. In those studies, defects in the early establishment of apical structures were shown to result from disturbances in the Cdc42-dependent Par/aPKC polarity complex, leading to defects in endocytosis-mediated turnover of junctional proteins. Also our data reveal disturbed polarity of Cdc42-depleted supporting cells undergoing structural development, as evidenced by misexpression of a lateral membrane protein, CD44, in the apical domain. The fact that this abnormal CD44 expression lined ectopic lumens is interesting, because these apical defects coincided with the normal formation of the basolateral large lumen, the tunnel of Corti, which is also lined by CD44 expression. These results suggest that defects in Cdc42 influence apical-basal polarization. Connected to these findings, our data show that aPKCλ/ι is delocalized from adherens junctions, suggesting that aPKCλ/ι activation, probably through Par6, is involved in mediating the effects of Cdc42 on structural maturation of the heads of supporting cells. However, the signaling events downstream of the Cdc42 and aPKCλ/ι remain to be elucidated. Our study demonstrates for the first time the involvement of aPKCλ/ι in the auditory sensory epithelium. Further, as most previous studies in the mouse have used embryonic tissues and linked Cdc42 and the components of the apical polarity complex to primordial cell-cell junctions and the early-developing cortical F-actin cytoskeleton19,20, the current results provide novel in vivo evidence that this complex functions at later stages of maturation of epithelial cells.
Importantly, when Cdc42 was acutely inactivated in supporting cells at a mature stage, no phenotypic alterations were seen, in contrast to acute inactivation during early postnatal development. However, as our study did not include long-term follow-up of this former phenotype, we cannot exclude the possibility of gradually developing defects in the maintenance of cytoskeletal and junctional structures in adult cells. In any case, the current results point to a major role of Cdc42 in supporting cells undergoing structural maturation.
With a model of ototoxically lesioned cochlea, causing loss of most of outer hair cells, we studied the functional significance of Cdc42 in supporting cells. As a background for this analysis, we performed cell tracing using the Fgfr3-iCre-ERT2;Ai14(tdTomato) mice and found that the lesioned area is principally occupied by Deiters' and pillar cells, at least during the one-month-long post-lesion period. Following lesion, mutant cochleas showed prominent collapse of supporting cells, this being most distinct in the case of outer pillar cells. The collapse led to the disappearance of the tunnel of Corti. The fact that the apical F-actin belts of outer pillar cells had abnormal morphologies suggests that these structures and the associated junctions provide rigidity and adhesive support to supporting cells and to the entire organ of Corti.
Additional evidence of the importance of Cdc42-dependent properly formed apical domain of supporting cells came from the observations of impaired scar formation in mutant mice. As opposed to control mice, the heads of supporting cells of mutants did not expand to the sites of lost hair cells, as evidenced by the lack of F-actin belts and adherens junctions at these sites. This perturbed F-actin remodeling might account to the collapse of tissue architecture, also these considerations pointing to the role of apical F-actin in structural support of the lesioned sensory epithelium. Importantly, F-actin remodeling was impaired only when Cdc42 was inactivated from the immature stage onward. Acute Cdc42 inactivation at the mature stage did not block scar formation. A previous in vitro study with ototoxically lesioned vestibular organs from adult bullfrogs showed blockade of scar formation in response to pharmacological inhibitors of actin polymerization52. Connected to the current results, it appears that Cdc42 as such does not regulate scar formation in the adult inner ear. Our conclusion is that, rather than fully formed structures, the primary targets of Cdc42′s action are the apical adherens junctions and cortical actin cytoskeleton in immature supporting cells and that these developmental defects underlie the inability for normal wound healing.
The Cdc42loxP/loxP;Fgfr3-iCre-ERT2 mouse line generated in the current study represents to our knowledge the first in vivo mouse model with impaired scar formation following lesion to the organ of Corti. With this model we asked whether scarring activity antagonizes regenerative plasticity of supporting cells. We found that this was not the case, based on the lack of cell cycle reactivation and changes in the expression of transcription factors in supporting cells of mutant animals. Based on these results, regenerative plasticity is not an absolute alternative to stable scar formation. However, we cannot rule out the possible direct, inhibitory effects of Cdc42 depletion on regenerative plasticity, such as the ability to re-enter the cell cycle. It has been suggested that the stout actin cytoskeleton and cell-cell junctions in supporting cells of the mammalian utricle antagonize true regeneration15,16,17. With the Cdc42 mutant mouse model and the cochlea, we could not address this possibility, because, despite abnormal in length and integrity, these structures were prominently present in supporting cells.
It is likely that the complex F-actin and microtubule cytoskeletons in pillar and Deiters' cells constitute physical barriers to cell cycle progression. Therefore, the findings, made both in control and mutant animals, of lesion-induced cyclin D1 upregulation in non-sensory cells located laterally to the organ of Corti proper are intriguing. These data suggest that cells such as Claudius cells have proliferative potential, likely in conjunction with exogenous mitogens. For successful regeneration, proliferation of cochlear non-sensory cells needs to be followed by the conversion of these cells into hair cells. This was not found to be the case in a study where proliferation of non-sensory cells was documented following severe ototoxic lesion53. However, based on the less complex structural features of the non-sensory cells flanking the organ of Corti, these cells might serve as a useful platform for future regeneration attempts.
In conclusion, auditory supporting cells serve as excellent models to understand the molecular regulation of the development of epithelial cells with high structural complexity. The present data show that Cdc42 participates in apical polarization of supporting cells and that it is required for proper organization of F-actin belts of these cells, thereby regulating the characteristics of the reticular lamina of the organ of Corti. Our data suggest that Cdc42-dependent structural maturation of supporting cells provides rigidity and wound healing activity of adult supporting cells.
Methods
Animals
Mice homozygous for the floxed Cdc42 allele (Cdc42loxP/loxP)54 were crossed with mice carrying the Fgfr3-iCre-ERT2 transgene19. Subsequently, Cdc42loxP/wt;Fgfr3-iCre-ERT2 mice were mated with Cdc42loxP/loxP mice to obtain Cdc42loxP/loxP;Fgfr3-iCre-ERT2 animals. From the same crossings, mice carrying one wild-type Cdc42 allele and mice lacking the iCre transgene served as control animals. Litters were genotyped by PCR as described in the original publications. To study the characteristics of iCre-mediated recombination, mice carrying the Fgfr3-iCre-ERT2 transgene were bred with the ROSA26tm14(CAG-tdTomato) Cre-conditional reporter mice (obtained from the Jackson Laboratory)20 to generate Fgfr3-iCre-ERT2;Ai14(tdTomato) animals. The Ai14(tdTomato) transgene was identified by PCR as described in the original publication or by direct visualization of the tdTomato native fluorescence in tails of the animals. The day of birth was assigned as postnatal day 0 (P0). Both females and males were used in the analysis. Mouse lines were maintained in a mixed background. NMRI mice were used for the detection of expression of Cdc42, CD44 and aPKCλ/ι at P0. All animal work has been conducted according to relevant national and international guidelines. Approval for animal experiments has been obtained from the National Animal Experiment Board.
Induction of iCre-mediated recombination
Preparation of tamoxifen (Sigma, catalog number T5648) stocks (10 mg/ml in a 1:7 mixture of ethanol and corn oil) and the administration technique were according to a previous publication55. Between P2 and P4 or between P16 and P18, animals received daily intraperitoneal injections of 50 μg/g body weight tamoxifen (50 and 150 μl/injection, respectively). In addition, a dose of 200 μg/g body weight tamoxifen (stock 15 mg/ml, 130 μl/injection) was administered to a subset of animals between P16 and P18.
Drug-lesioning paradigm
Hair cell loss was induced in control and mutant mice at P20 or P22 by a single subcutaneous injection of 1 mg/g kanamycin (Sigma) dissolved in phosphate-buffered saline (PBS), followed 45 min later by a single intraperitoneal injection of 0.4 mg/g furosemide (Fresenius Kabi). Animals were killed 2, 7, 14 and 28 days after challenge with the ototoxins.
Wholemount specimens
For confocal microscopy, cochleas between P7 and P30 were fixed by perilymphatic perfusion with 4% paraformaldehyde (PFA) in PBS, followed by immersion in the fixative for 5 h. The sensory epithelium was dissected from cochlear tissue and tectorial membrane was removed. Full-length organ of Corti was divided into upper, middle and basal parts. TdTomato fluorescence was directly visualized in wholemounts. For immunofluorescence, wholemounts were blocked for 30 min with 10% donkey serum in PBS containing 0.25% Triton-X-100 (PBS-T), followed by incubation overnight at +4°C with appropriate primary antibodies in PBS-T. As primary antibodies, rat monoclonal CD44 (BD Biosciences) and rat monoclonal E-cadherin (Sigma) were used. Secondary antibodies made in donkey and conjugated to Alexa 488 were used. Following antibody incubations, F-actin filaments were visualized using rhodamine-labeled phalloidin (1:1000, 30 min at room temperature). ProLong Gold anti-fade reagent with or without DAPI was used for mounting (all from Molecular Probes/Invitrogen). Confocal images were acquired using a Leica TCS SP5 laser scanning microscope with Plan Apochromat 63×/1.3 NA and 20×/0.7 NA glycerol objectives. The acquisition software was Leica LAS AF. Z-projections were processed with Imaris 7 (Bitplane Scientific Software) and ImageJ (NIH). Blind 3D deconvolution was made with AutoQuant X2 (Media Cybernetics). Absolute intensity-based 3D isosurfaces from confocal stacks were rendered with Imaris 7. An isosurface shows a voxel-based 3D model of the imaged fluorescence signal. The correct representativeness of the generated isosurface was confirmed by parallel comparison with the original data. A minimum of 5 cochleas per genotype, per age, per tamoxifen dosing regimen and per post-lesion time point were prepared for wholemount analyses.
Histological sections
Cochleas between P0 and P51 were perilymphatically fixed with 4% PFA and decalcified in 0.5 M EDTA (pH 8.0). Thereafter, specimens were embedded into paraffin and cut to 5-μm-thick sections. Epitopes were unmasked by microwave heating (800 W) in 10 mM citrate buffer (pH 6.0) for 10 min. Sections were blocked for 30 min with 10% goat serum or commercial blocking agent (from Vectastain Mouse-On-Mouse kit) in PBS-T. Sections were incubated overnight at +4°C with an appropriate primary antibody in PBS-T. The following primary antibodies were used: rabbit RFP (Rockland Immunochemicals); rabbit polyclonal β-tubulin (Abcam); rabbit monoclonal cyclin D1, rabbit monoclonal Ki-67 (LabVision/ThermoScientific); rabbit monoclonal cleaved caspase-3 (Cell Signaling Technology); rabbit polyclonal myosin VIIa (a kind gift from Tama Hasson, University of California, San Diego); mouse monoclonal ZO-1 (Molecular Probes/Invitrogen); mouse monoclonal p27Kip1, rat monoclonal CD44 (BD Biosciences); rabbit polyclonal Sox9 (Millipore); goat polyclonal Prox1 (R&D Systems); goat polyclonal Sox2, rabbit polyclonal aPKCλ/ι (H-76) (both from Santa Cruz Biotechnology). Detection was done with Vectastain Elite ABC kit or Vectastain Mouse-On-Mouse kit and the diaminobenzidine substrate (DAB Detection kit, all from Vector Laboratories). Sections were counterstained with 3% methyl green and mounted in Permount (Fisher Scientific). A subset of sections was stained with hematoxylin only (Shandon Instant Hematoxylin, Thermo Scientific). The antibodies used have been applied to the inner ear tissue in previous publications30,40,42,43,50. The RFP antibody has previously been used in immunofluorescence and immunohistochemistry in other mouse tissues56. Cochleas of Fgfr3-iCre-ERT2;Ai14 mice not treated with tamoxifen lacked RFP immunoreactivity (data not shown). In situ hybridization was performed with35S-labelled Cdc42 and Fgfr3 riboprobes on PFA-fixed, paraffin-embedded sections. Hematoxylin was used for counterstaining. Sections were analyzed with a BX61 microscope (Olympus) using bright- and darkfield optics. Images were acquired through the DP70 CCD colour camera and cell‘F software (Olympus) and processed using Adobe Photoshop CS4 (Adobe Systems). For visualization of in situ hybridization signals, autoradiographic silver grains in the darkfield image were selected, colored red and superimposed onto the brightfield image. For all antibodies and probes, a minimum of 4 cochleas per genotype, per age and per post-lesion time point were prepared for histological analyses.
Serial block-face scanning electron microscopy
Cochleas were perilymphatically fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) and immersed overnight in the fixative. The sensory epithelium of the medial part of the cochlear duct was prepared for SBF-SEM as previously described28. Shortly, after dehydration and resin (Durcupan ACM, Fluka) infiltration steps, dissected tissue pieces were embedded in hand-made silicone holders filled with 100% resin and clamped between two objective glasses. Images were acquired with an FEG-SEM Quanta 250 (FEI Company) using a backscattered electron detector (Gatan Inc.). Block was imaged with 2.5 kV beam voltage, spot size 3 and 0.3 Torr pressure. The microscope was equipped with a microtome (3View, Gatan Inc.), which allowed serial imaging of block faces with increments of 40 nm. The images were first processed and segmented using Amira (Visage Imaging, Inc.) and Microscopy Image Browser, a self-developed program written under Matlab environment and further rendered in Amira. Three cochleas per genotype were used for SBF-SEM analysis at P10 and P20.
Quantification
The length of junctions between Deiters' cells was measured from SBF-SEM image stacks in Amira with the length measurement tool. Randomly selected junctions between outer pillar cells and Deiters' cells of the 1st row and between Deiters' cells of the 2nd and 3rd row were measured from three control and mutant cochleas each. Cochlear medial turn was used for analysis. Correct imaging plane for measurements was selected with an oblique slice in orthographic view mode. Junctional length was defined as the distance, measured along the border of the adherens junction, between neighbouring supporting cells, connecting two hair cells from different rows. Alpha level of 0.05 was used for statistical tests. Normality of the data was tested with Shapiron-Wilk test. Two-tailed Student's t test was used for statistical analysis. Data are shown as averages with s.d. Values were regarded as highly significant at p < 0.001. The lenght of the reticular lamina was measured as the planar distance from the apical junction between an inner pillar cell and an inner hair cell to the apical junction between the 3rd row Deiters' cell and Hensen's cell. Measurements were made in Imaris 7 from confocal stacks of phalloidin-labeled cochlear medial turns.
For quantification of supporting cell numbers in lesioned cochleas, mid-modiolar (transverse) paraffin sections immunostained for cyclin D1 and counterstained with methyl green were analyzed. Preserved inner hair cells and cyclin D1-positive Claudius cells formed the medial and lateral borders of the region analyzed, respectively. Both from control and mutant animals at day 28 post-lesion, three cochleas, all from different individuals and seven to eight sections per cochlea were used for counting the numbers of supporting cell nuclei. Nuclei with well-defined outlines were counted. Data are shown as averages with s.d. Two-tailed Student's t test was used for statistical analysis. Values were regarded as highly significant at p < 0.001.
References
Forge, A. & Wright, T. The molecular architecture of the inner ear. Br. Med. Bull 63, 5–24 (2002).
Raphael, Y. & Altschuler, R. A. Structure and innervation of the cochlea. Brain Res Bull 60, 397–422 (2003).
Colvin, J. S., Bohne, B. A., Harding, G. W., McEwen, D. G. & Ornitz, D. M. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet 12, 390–397 (1996).
Gulley, R. L. & Reese, T. S. Intercellular junctions in the reticular lamina of the organ of Corti. J. Neurocytol 5, 479–507 (1976).
Leonova, E. V. & Raphael, Y. Organization of cell junctions and cytoskeleton in the reticular lamina in normal and ototoxically damaged organ of Corti. Hear. Res. 113, 14–28 (1997).
Slepecky, N. & Chamberlain, S. C. Distribution and polarity of actin in inner ear supporting cells. Hear. Res. 10, 359–370 (1983).
Tucker, J. B., Mackie, J. B., Bussoli, T. J. & Steel, K. P. Cytoskeletal integration in a highly ordered sensory epithelium in the organ of Corti: response to loss of cell partners in the Bronx waltzer mouse. J. Neurocytol. 28, 1017–1034 (1999).
Henderson, C. G. et al. Reorganization of the centrosome and associated microtubules during the morphogenesis of a mouse cochlear epithelial cell. J. Cell Sci 107, 589–600 (1994).
Tolomeo, J. A. & Holley, M. Mechanics of microtubule bundles in pillar cells from the inner ear. Biophys. J. 73, 2241–2247 (1997).
Furness, D. N., Katori, Y., Mahendrasingam, S. & Hackney, C. M. Differential distribution of beta- and gamma-actin in guinea-pig cochlear sensory and supporting cells. Hear. Res. 207, 22–34 (2005).
Szarama, K. B., Gavara, N., Petralia, R. S., Kelley, M. W. & Chadwick, R. S. Cytoskeletal changes in actin and microtubules underlie the developing surface mechanical properties of sensory and supporting cells in the mouse cochlea. Development 139, 2187–2197 (2012).
Forge, A. Outer hair cell loss and supporting cell expansion following chronic gentamicin treatment. Hear. Res. 19, 171–182 (1985).
Raphael, Y. & Altschuler, R. A. Scar formation after drug-induced cochlear insult. Hear. Res. 51, 173–184 (1991).
Taylor, R. R., Jagger, D. J. & Forge, A. Defining the cellular environment in the organ of Corti following extensive hair cell loss: a basis for future sensory cell replacement in the cochlea. PLoS One 7, e30577 (2012).
Meyers, J. R. & Corwin, J. T. Shape change controls supporting cell proliferation in lesioned mammalian balance epithelium. J. Neurosci. 27, 4313–4325 (2007).
Burns, J. C. et al. Reinforcement of cell junctions correlates with the absence of hair cell regeneration in mammals and its occurrence in birds. J. Comp. Neurol. 511, 396–414 (2008).
Collado, M. S. et al. The postnatal accumulation of junctional E-cadherin is inversely correlated with the capacity for supporting cells to convert directly into sensory hair cells in mammalian balance organs. J. Neurosci. 31, 11855–11866 (2011).
Baum, B. & Georgiou, M. Dynamics of adherens junctions in epithelial establishment, maintenance and remodeling. J. Cell Biol. 192, 907–917 (2011).
Heasman, S. J. & Ridley, A. J. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell. Biol. 9, 690–701 (2008).
Pedersen, E. & Brakebusch, C. Rho GTPase function in development: How in vivo models change our view. Exp. Cell. Res. 318, 1779–1787 (2012).
Jopling, C. et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464, 606–609 (2010).
Young, K. M. et al. An Fgfr3-iCreER(T2) transgenic mouse line for studies of neural stem cells and astrocytes. Glia 58, 943–953 (2010).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Hayashi, T., Ray, C. A., Younkins, C. & Bermingham-McDonogh, O. Expression patterns of FGF receptors in the developing mammalian cochlea. Dev. Dyn. 239, 1019–1026 (2010).
Pirvola, U. et al. The site of action of neuronal acidic fibroblast growth factor is the organ of Corti of the rat cochlea. Proc. Natl. Acad. Sci. USA 92, 9269–9273 (1995).
Ito, M., Spicer, S. S. & Schulte, B. A. Cytological changes related to maturation of the organ of Corti and opening of Corti's tunnel. Hear. Res. 88, 107–123 (1995).
Kuhn, B. & Vater, M. The early postnatal development of F-actin patterns in the organ of Corti of the gerbil (Meriones unguiculatus) and the horseshoe bat (Rhinolophus rouxi). Hear. Res. 99, 47–70 (1996).
Souter, M., Nevill, G. & Forge, A. Postnatal maturation of the organ of Corti in gerbils: Morphology and physiological responses. J. Comp. Neurol. 386, 635–651 (1997).
Whitlon, D. S., Zhang, X., Pecelunas, K. & Greiner, M. A. A temporospatial map of adhesive molecules in the organ of Corti of the mouse cochlea. J. Neurocytol. 28, 955–968 (1999).
Hertzano, R. et al. CD44 is a marker for the outer pillar cells in the early postnatal mouse inner ear. J. Assoc. Res. Otolaryngol 11, 407–418 (2010).
Denk, W. & Horstmann, H. Serial block-face scanning electron microscopy to reconstruct three-dimensional tissue nanostructure. PLoS Biol. 2, e329 (2004).
Nunes, F. D. et al. Distinct subdomain organization and molecular composition of a tight junction with adherens junction features. J. Cell Sci. 119, 4819–4827 (2006).
Iden, S. & Collard, J. G. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell. Biol. 11, 846–59 (2008).
Yamanaka, T. et al. PAR-6 regulates aPKC activity in a novel way and mediates cell-cell contact-induced formation of the epithelial junctional complex. Genes Cells 6, 721–731 (2001).
Huber, T. B. et al. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J. Am. Soc. Nephrol 20, 798–806 (2009).
Martin-Belmonte, F. et al. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 128, 383–397 (2007).
Oesterle, E. C., Campbell, S., Taylor, R. R., Forge, A. & Hume, C. R. Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J. Assoc. Res. Otolaryngol 9, 65–89 (2008).
Taylor, R. R., Nevill, G. & Forge, A. Rapid hair cell loss: a mouse model for cochlear lesions. J. Assoc. Res. Otolaryngol 9, 44–64 (2008).
Oesterle, E. C. & Campbell, S. Supporting cell characteristics in long-deafened aged mouse ears. J. Assoc. Res. Otolaryngol 10, 525–544 (2009).
Loponen, H., Ylikoski, J., Albrecht, J. & Pirvola, U. Restrictions in cell cycle progression of adult vestibular supporting cells in response to ectopic cyclin D1 expression. PLoS One 6, e27360 (2011).
Bermingham-McDonogh, O. et al. Expression of Prox1 during mouse cochlear development. J. Comp. Neurol. 496, 172–186 (2006).
Kirjavainen, A. et al. Prox1 interacts with Atoh1 and Gfi1 and regulates cellular differentiation in the inner ear sensory epithelia. Dev Biol 322, 33–45 (2008).
Laine, H., Sulg, M., Kirjavainen, A. & Pirvola, U. Cell cycle regulation in the inner ear sensory epithelia: role of cyclin D1 and cyclin-dependent kinase inhibitors. Dev. Biol. 33, 134–146 (2010).
Cappello, S. et al. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat. Neurosci 9, 1099–1107 (2006).
Chen, L. et al. Cdc42 deficiency causes Sonic hedgehog-independent holoprosencephaly. Proc. Natl. Acad. Sci. USA 103, 16520–16525 (2006).
Wu, X. et al. Cdc42 is crucial for the establishment of epithelial polarity during early mammalian development. Dev. Dyn. 236, 2767–2778 (2007).
Georgiou, M., Marinari, E., Burden, J. & Baum, B. Cdc42, Par6 and aPKC regulate Arp2/3-mediated endocytosis to control local adherens junction stability. Curr. Biol. 18, 1631–1638 (2008).
Harris, K. P. & Tepass, U. Cdc42 and Par proteins stabilize dynamic adherens junctions in the Drosophila neuroectoderm through regulation of apical endocytosis. J. Cell. Biol. 183, 129–143 (2008).
Leibfried, A., Fricke, R., Morgan, M. J., Bogdan, S. & Bellaiche, Y. Drosophila Cip4 and WASp define a branch of the Cdc42-Par6-aPKC pathway regulating E-cadherin endocytosis. Curr. Biol. 18, 1639–1648 (2008).
Etournay, R. et al. Cochlear outer hair cells undergo an apical circumference remodeling constrained by the hair bundle shape. Development 137, 1373–1383 (2010).
van Hengel, J. et al. Continuous cell injury promotes hepatic tumorigenesis in cdc42-deficient mouse liver. Gastroenterology 134, 781–92 (2008).
Hordichok, A. J. & Steyger, P. S. Closure of supporting cell scar formations requires dynamic actin mechanisms. Hear. Res. 232, 1–19 (2007).
Kim, Y. H. & Raphael, Y. Cell division and maintenance of epithelial integrity in the deafened auditory epithelium. Cell Cycle 6, 612–619 (2007).
Wu, X. et al. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes Dev. 20, 571–585 (2006).
Pitulescu, M. E., Schmidt, I., Benedito, R. & Adams, R. H. Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 5, 1518–1534 (2010).
Rock, J. R. et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 108, E1475–1483 (2011).
Acknowledgements
This work was supported by the Academy of Finland, Biocenter Finland and Jane and Aatos Erkko Foundation. We thank Sanna Sihvo and Antti Salminen for excellent technical assistance. We are grateful to Heidi Loponen for the assistance with in situ hybridization, Magdalena Götz for the Cdc42 probe and Kimmo Tanhuanpää and Mika Molin for assistance with confocal imaging.
Author information
Authors and Affiliations
Contributions
U.P., T.A. and A.K. conceived and designed the experiments; T.A., A.K., M.L., I.B. and U.P. performed experiments; T.A., A.K., M.L. and U.P. analyzed data; W.R. and C.B. generated Fgfr3-iCre-ERT2 and Cdc42loxP/loxP mutant mice, respectively; E.J. contributed to SBF-SEM analysis; U.P. directed the project and wrote the paper; T.A. and A.K. assisted in writing. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Anttonen, T., Kirjavainen, A., Belevich, I. et al. Cdc42-dependent structural development of auditory supporting cells is required for wound healing at adulthood. Sci Rep 2, 978 (2012). https://doi.org/10.1038/srep00978
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00978
This article is cited by
-
Various Three-Dimensional Culture Methods and Cell Types for Exosome Production
Tissue Engineering and Regenerative Medicine (2023)
-
Exocyst Complex Member EXOC5 Is Required for Survival of Hair Cells and Spiral Ganglion Neurons and Maintenance of Hearing
Molecular Neurobiology (2018)
-
Indispensable role of Mdm2/p53 interaction during the embryonic and postnatal inner ear development
Scientific Reports (2017)
-
Survival of human embryonic stem cells implanted in the guinea pig auditory epithelium
Scientific Reports (2017)
-
Molecular cloning and functional characterization of the sex-determination gene doublesex in the sexually dimorphic broad-horned beetle Gnatocerus cornutus (Coleoptera, Tenebrionidae)
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
