
Abstract
Over the past 30 years, we have performed many fundamental studies on two Oryza sativa subsp. indica varieties, Zhenshan 97 (ZS97) and Minghui 63 (MH63). To improve the resolution of many of these investigations, we generated two reference-quality reference genome assemblies using the most advanced sequencing technologies. Using PacBio SMRT technology, we produced over 108 (ZS97) and 174 (MH63) Gb of raw sequence data from 166 (ZS97) and 209 (MH63) pools of BAC clones, and generated ~97 (ZS97) and ~74 (MH63) Gb of paired-end whole-genome shotgun (WGS) sequence data with Illumina sequencing technology. With these data, we successfully assembled two platinum standard reference genomes that have been publicly released. Here we provide the full sets of raw data used to generate these two reference genome assemblies. These data sets can be used to test new programs for better genome assembly and annotation, aid in the discovery of new insights into genome structure, function, and evolution, and help to provide essential support to biological research in general.
Design Type(s) | strain comparison design • genome assembly |
Measurement Type(s) | reference genome data • whole genome sequencing |
Technology Type(s) | DNA sequencing |
Factor Type(s) | selectively maintained organism |
Sample Characteristic(s) | Oryza sativa Indica Group |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
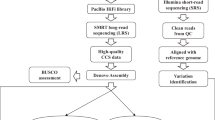
Rice is the leading staple crop for mankind and has been recognized as an important model organism for biological research, especially for monocot plants. Asian cultivated rice (Oryza sativa) is composed of two subspecies: O. sativa subsp. japonica and subsp. indica; indica rice accounts for over 70% of rice production worldwide1 and is genetically much more diverse2. The indica varieties Zhenshan 97 (ZS97) and Minghui 63 (MH63) represent two major varietal groups of indica rice3, contain a number of important agronomic traits and are the parents of Shanyou 63 (SY63), the most widely cultivated hybrid rice in China. The ZS97, MH63, SY63 hybrid system has been used as a model4–9 over the past 30 years, and concomitantly our lab has made a series of attempts to gain a fundamental understanding of the genetic basis of heterosis—a biological mystery that has puzzled the scientific community for more than 100 years. Hence, we initiated a joint collaborative project to generate two reference-quality genome assemblies for ZS97 and MH63 to be used as a fundamental tool to help us understand the underlying molecular genetic basis of heterosis10. In this descriptor, we report the resources and data sets that were generated and used to assemble the ZS97 and MH63 reference genomes: (1) two BAC libraries, (2) two improved physical maps and minimum tiling paths (MTP), (3) raw PacBio sequencing data of BAC pools and full clone sequence assemblies, (4) Illumina WGS sequence and assembly data, and (5) the first release of reference genome assemblies for ZS97 and MH63. With the resources and data generated in this study, we were not only able to de novo assemble two reference-quality genome sequences, but we were able to provide the scientific community with data to advance biological research at the genomic level, especially for further understanding of the genetic basis of heterosis.
Methods
BAC library construction and end-sequencing
The two BAC libraries used in this study were previously reported11. Briefly, partially digested (i.e., HindIII) and size-selected genomic DNA from each variety was cloned into the HindIII site of pAGIBAC1, and transformed into Escherichia coli DH10B T1-resistant competent cells11. Both libraries, named OSIZBa (ZS97) and OSIABa (MH63), contained 36,864 BAC clones, had average insert sizes were ~117 kb (ZS97) and ~125 kb (MH63), and covered ~10.0× (ZS97) and ~10.7× (MH63) of each genome11. Additionally, 33,969 (ZS97) and 35,549 (MH63) bi-directional BAC end sequences (BESs) were generated for the first half of each library11.
Physical maps
Two low coverage physical maps (PMs), using the SNaPshot fingerprinting method, were described previously11. We rebuilt the two PMs using KeyGene’s Whole Genome Profiling (WGP) method12. WGP FingerPrint Contig (FPC) PMs were built in four steps: (1) BAC DNA preparation, (2) WGP preparation of BAC plasmids with indexing and sequencing adaptors, (3) Illumina sequencing, and (4) bioinformatic processing. In step 4, using WGP deconvolution scripts, 99,996 (ZS97) and 103,609 (MH63) unique WGP tags were deconvoluted, and 32,829 (89.1%) and 30,749 (89.3%) BACs in the ZS97 and MH63 libraries were tagged, respectively. Using the WGP sequence tags for each BAC clone from each library, two new PMs were constructed with FPC software13 (Version 9.4). After manually editing and integration with the previous SNaPshot PMs11, the improved ZS97 and MH63 PMs consisted of 539 and 401 contigs, containing 28,372 and 24,519 clones, and had 4,457 and 6,230 clones as singletons, respectively. Total contig sizes were estimated at 342 Mb for ZS97 (N50=940 kb) and 349 Mb for MH63 (N50=1,270 kb).
PacBio BAC clone sequencing
Minimum tiling path (MTP) BAC clones from each PM were selected automatically with a customized script and re-arrayed manually into MTP library plates designated OSIZBzz (ZS97) and OSIABzz (MH63), and were stored at −80 °C. A total of 4,714 and 4,751 BAC MTP clones were picked for ZS97 and MH63, respectively. The full lists of MTP clones are available in Supplementary Table 1a–b.
For PacBio BAC clone sequencing, MTP BAC clones were inoculated into 96-well deep-well growth blocks, grown overnight at 37 °C, centrifuged to pellet the cells and then stored at −80 °C until use. BAC pools were then created by combining wells from the frozen blocks into one of six configurations: i.e., single row pools (12 BACs per pool), two row pools (24 BACs per pool), four column pools (32 BACs per pool), six column pools (48 BACs per pool), eight column pools (64 BACs per pool), or full plate pools (96 BACs per pool). DNA was then extracted from each pool using a standard alkaline lysis plasmid DNA isolation protocol14. In total, 166 (ZS97) and 209 (MH63) pools were sequenced (see our detailed pooling schema in Supplementary Table 2a–b). Using 16 ug of pooled plasmid DNA, PacBio sequencing libraries were produced following manufacturers protocols as described for the 20 kb template preparation with Blue Pippin size selections. SMRT sequencing was performed on a PacBio RSII instrument using P5/C3 sequencing chemistry and 3 h movies.
Raw read production with PacBio
Subread analysis for both ZS97 and MH63 BAC pool sequences was performed using the PacBio SMRT Portal (Version 2.3.0). For ZS97, data from 227 SMRT cells (which counts redo reactions) was separated and filtered (i.e., using the RS_Subreads protocol, minimum polymerase read length=50 bp, minimum polymerase read quality=75, and minimum subread length=50 bp) resulting in a total of 107.5 Gb of useable sequence data (total number of polymerase reads=11.6 M, polymerase read N50=12.8 kb; total number of subreads=17.7 M reads, mean subread length=5.7 kb, subread N50=8.0 kb). For MH63, data from 317 SMRT cells were treated as above (174 Gb of useable data; 18.2 M polymerase reads, polymerase read N50=12.1 kb; 26.8 M subreads, mean subread length=5.5 kb, subread N50=7.8 kb).
PacBio data assembly and BAC sequence identification
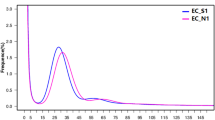
Sequence data was collected for each BAC pool and assembled independently with PacBio HGAP software (version 2 or 3)15 to recover circularized plasmids or BAC specific sequences. In total, 501 HGAP assemblies were run (including assemblies with multiple data sets of the same pool if redos were required) for all sequence pools. Associations between pools and assembly job Ids are shown in Supplementary Table 2a–b. We used a custom pipeline called ‘postHGAP’16 to automatically perform circularization and identification of BAC sequences. After running ‘postHGAP’ for each HGAP assembly, we were able to identify 4,571 and 4,488 BAC sequences (4,415 and 4,320 of these were fully circularized) from ZS97 and MH63, respectively. The average lengths of sequenced BACs were 121 kb for ZS97 and 151 kb for MH63. On average, each BAC has about 74–83× PacBio sequence coverage (ranging from 18× to 1747×, Fig. 1a,b), with an overall consensus accuracy of 99.94% (calculation based on all HGAP unitigs, ranging from 99.58 to 99.99%). We also determined the average identity of overlapping regions between two neighboring BAC sequences, which was 99.9787% for ZS97 and 99.9749% for MH63, indicating a high sequence accuracy in this study10. Notably, the overall full-circularization rates for BAC pool sequencing were 94% (ZS97) and 91% (MH63). Due to some unavoidable errors or contaminations in MTP clone re-arraying, we identified 125 (ZS97) and 200 (MH63) circularized sequences from non-MTP clones and assigned potential BAC IDs to them with available WGP tag data and/or BES information (Supplementary Table 3a–b).

Base coverage distributions of ZS97 (a) and MH63 (b) BAC sequences.
Plant material, DNA library construction for illumina sequencing
We also employed Illumina short-read sequencing technology to sequence the ZS97 and MH63 genomes using a whole genome shotgun (WGS) approach. Plant materials were grown in a greenhouse, and 4 week-old seeding leaves were used to extract genomic DNA using standard procedures. Paired-end libraries, including small-insert (~300 bp) and two large-insert libraries (5 kb, 10 kb), were prepared with Illumina’s paired-end and mate-pair kits, respectively (Table 1). At least 5 μg of genomic DNA was fragmented by nebulization with compressed nitrogen gas for the short-insert paired-end libraries. A larger amount of high-quality genomic DNA (10–30 ug) was required for the long-insert mate-pair library construction. Illumina sequencing libraries were prepared following the manufacturer’s protocol. The libraries were sequenced on an Illumina HiSeq 2000 following standard Illumina protocols (Illumina, San Diego, CA). The total amount of raw sequence data generated for each variety was ~97.5 Gb data for ZS97 and ~74.0 Gb data for MH63. After a series of data filtering steps were used to remove artificial reads caused by PCR duplication and adapter contamination, a total ~87.4 Gb of high-quality reads (>200×) for ZS97, and ~67.9 Gb (>170×) for MH63 were obtained (Table 1). Library quality was checked by determining the distribution of insertion sizes and sequence depths. Actual insert lengths were determined by mapping paired-end reads to the O. sativa subsp. japonica cv. Nipponbare reference genome (henceforth termed the Nipponbare RefSeq)17.
Illumina reads preprocessing and de novo assembly
We employed a new hybrid approach by combining de novo assembly and reference-based methods18 to assemble Illumina reads for each genome. All sequenced reads from ZS97 and MH63 were corrected with Jellyfish19 and Quake20. Corrected reads were trimmed at their termini if their sequence quality was below 20 using the fastx_tool_kit (http://hannonlab.cshl.edu/fastx_toolkit/index.html), and reads adapters were removed using the cutadpat tool21 (https://github.com/marcelm/cutadapt/). The processed reads were then mapped to the Nipponbare RefSeq using BWA22. All reads that mapped to a continuous region were collected respectively, and the continuous covered region was defined as a block. The definition of blocks and superblocks are the same as previously described23 except that our minimum superblock length was 20 kb, and the overlap between superblocks was 2 kb. We locally de novo assembled all the collected reads in a superblock using SOAPdenovo24. A series of different k-mers was tested, and the assembled contigs with the largest N50 values were retained. The resulting contigs were assembled by AMOS25 using their respective reference chromosomes as a guide. Unmapped reads were re-mapped to the indica 9311 genome26, and assembled using the same procedure.
Nucmer27 was then used to align all supercontigs to the Nipponbare RefSeq. We then checked the coverage and mapping depth of the gaps between the contiguously aligned regions. Mapped reads were selected that bridged gaps in the MH63 and ZS97 genomes which were extended 200 bp on both sides of each gap. All selected reads in these regions were assembled using cap328. The assembled contigs were aligned to the two continuous supercontigs and connected based on their sequence alignments.
To incorporate MH63 and ZS97 specific sequences that were absent in both the Nipponbare and 9311 genomes, we performed whole genome de novo assembly using all processed reads with SOAPdenovo24, and then aligned the de novo assembled scaffolds to the combined supercontigs, and further linked the corresponding supercontigs. Lastly, scaffolding was performed using SSPACE29, and gaps were filled with gapCloser (http://sourceforge.net/projects/soapdenovo2/files/GapCloser/).
The final statistics of the WGS Illumina assemblies of ZS97 and MH63 are listed in Tables 2 and 3, and were used to fill gaps between neighboring PacBio contigs.
Building pseudomolecules in two steps
In the first step, all BAC sequence data was loaded into Genome Puzzle Master16 (GPM) to build PacBio-based sequence contigs using physical maps and the Nipponbare RefSeq as reference guides17. GPM is a semi-automated pipeline that was developed to integrate logical relationship data (e.g., physical maps) to scaffold sequence contigs into chromosome-scale sequences. As a result, 318 (ZS97) and 216 (MH63) assembled contigs were ordered and oriented, as well as anchored to their appropriated chromosomes, after manual checking, editing and removing redundancies. The final PacBio-based assemblies were composed of sequences from 3862 (ZS97, including 57 non-MTP) and 3254 (MH63, including 77 non-MTP) unique BACs.
Since we used a physical map-based sequencing strategy in this study, the gaps in our WGP physical maps are the main causes of the breaks in the PacBio-based assemblies. Hence, our second step was to fill gaps by integration of Illumina WGS assembly data. However, to minimize the impact of potential misassembles in Illumina data, we only used Illumina contigs that could fully connect two neighboring PacBio contigs in this step. In total, 81 gaps in ZS97 (8,988,328 bp) and 35 in MH63 (3,127,191 bp) were filled with 76 (ZS97) and 35 (MH63) Illumina contig sequences (Supplementary Table 4a–b). As a result, the final hybrid pseudomolecules (ZS97RS1 and MH63RS1) contained 237 (including 2 unanchored) and 181 (including 2 unanchored) contigs for ZS97 and MH63, respectively.
Data Records
All non-sequence data are available at the iPlant Datastore (Table 4). Both the OSIZBa and OSIABa BAC libraries, or individual BAC clones, can be obtained from AGI’s BAC/EST resource center at www.genome.arizona.edu/orders. BAC end sequences were previously deposited in GenBank under accession numbers KG737749–KG771717 (ZS97, Data Citation 1) and KG702200–KG737748 (MH63, Data Citation 2).
Raw PacBio sequencing data is available at NCBI’s Short Read Archive (SRA) under accession numbers SRP071597 (ZS97, Data Citation 3) and SRP071598 (MH63, Data Citation 4). All Illumina sequencing data can be found under accession number SRP071944 (ZS97 and MH63, Data Citation 5). Due to an unexpected disc error, we lost raw PacBio sequencing data from 57 pools. Fortunately, however, all HGAP runs were archived in the iPlant Datastore under ‘smrt-jobs’ where the filtered subreads of those corresponding pools could be retrieved. Assembled Illumina contig data is available at NCBI assembly under accession numbers GCA_001618795 (ZS97, Data Citation 6) and GCA_001618785 (MH63, Data Citation 7).
The final genome pseudomolecules (Version 1) for each reference genome were deposited in NCBI assembly under accession numbers GCA_001623345 (ZS97RS1, Data Citation 8) and GCA_001623365 (MH63RS1, Data Citation 9).
Technical Validation
Essentially, each genome equivalent BAC library was freshly grown in copied sets of 384-well plates and three dimensional pooling was performed on the bacterial cells followed by pool growth and plasmid DNA extraction using in-house alkaline lysis chemistry. DNA pools were digested by restriction enzymes (EcoRI/MseI) followed by ligation of pool-dimension oligomers that were designed to specifically locate BAC clone addresses and associate to sequences. After mixed molecule amplification, Illumina sequencing was performed and the resulting data was parsed for 50-bp sequence tag identification to each specific BAC clone address, and for producing bands files as input into FPC. FPC was run under high stringency (HS) settings: first with a ‘tolerance=0 [fixed], Cutoff=1e-15’, then the DQ-option (in 3 steps: Cutoff=1e-18, 1e-21, 1e-24) was employed to split problematic contigs. After the resulting HS PMs were generated, we performed an Ends-to-Ends merge step (Cutoff=1e-9) and the incorporation of remarked singletons to contigs (Cutoff=1e-12) to produce reduced stringency (RS) maps. The WGP RS maps were manually edited by integration with our previous low coverage PMs11.
For plasmid DNA extraction, MTP clone plates were thawed and inoculated into deep well blocks containing 1.2 ml of 2XYT+12.5 μgml−1 chloramphenicol and grown with shaking for exactly 18 h at 37 °C. The pooled BAC cells were collected, washed with water, and individually prepped to isolate plasmid DNA using standard alkaline lysis with Qiagen P1, P2 and P3 buffers. Following DNA quantification of each pooled sample (i.e., 12–16 ug of DNA/pool) a 20-kb PacBio library was constructed following manufacturer’s protocols, that included BluePippin (Sage Science) size selection of templates, and sequenced in a SMRT cell with P5/C3 chemistry for three hours on a PacBio RS II instrument. Once the sequence was generated, an HGAP (v2 or v3)15 assembly run was performed under default settings (minimum polymerase read length=100 bp, minimum polymerase read quality=80, and minimum subread length=500 bp), except ‘Minimum Seed Read Length’ as mean length determined by reads of insert from each SMRT cell and ‘Genome Size’ as estimated lengths of total BACs in a pool. Detailed settings for each run can be extracted from the HGAP job archives at the iPlant Datastore. During ‘postHGAP’ processing, we used fixed parameters for sequence circularization (minOverlap=500 bp, overlapIdentity=95%) and BAC Id assignments (minCloneTagNumber=5, tagMatchIdentity=100%, tagMatchPercent=80%; besMatchIdentity=98% if no WGP tag(s) is available).
In the GPM ‘assemblyRun’ step for building BAC-based sequence contigs, the default parameters for merging two BAC sequences were ‘minOverlapSeqToSeq=1000 bp’ and ‘identitySeqToSeq=99%’, plus the overlaps were required to be at the ends of both sequences. We used the Nipponbare RefSeq17 as a reference to assign chromosome numbers to assembled contigs, as well as to order and orientate them. Additionally, only one copy of redundant overlapping sequence was retained in an assembled contig, with no preference on determining which BAC sequence piece was kept. However, non-gapped sequences had higher priority over gapped ones. All contigs were manually checked and edited as needed through the GPM16 assembly viewer. When using assembled Illumina contigs to fill gaps between two BAC-based contigs, we only selected the Illumina contigs that could fully connect two neighboring BAC-based contigs, and importantly, such overlaps (‘minOverlapSeqToSeq=1000 bp’ and ‘identitySeqToSeq=99%’) were required to occur at the ends of both contigs. When redundancies were found in these regions, the BAC-based sequence pieces were always retained in the final genome assemblies.
This paper is the first release of the raw data for the assembly of the ZS97 and MH63 indica rice genomes, and also provides the first versions of two sets of high quality pseudomolecules to the scientific community. DNA sequencing technologies and sequence assembly programs change rapidly, and the datasets presented here contain multiple types of sequencing reads which can be used to develop new methodologies and software tools as test inputs.
Additional information
How to cite this article: Zhang, J. et al. Building two indica rice reference genomes with PacBio long-read and Illumina paired-end sequencing data. Sci. Data 3:160076 doi: 10.1038/sdata.2016.76 (2016).
References
References
IRRI. World Rice Statistics 1990 (International Rice Research Institute, 1991).
Huang, X. et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 42, 961–967 (2010).
Xie, W. et al. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc. Natl. Acad. Sci. USA 112, E5411–E5419 (2015).
Yu, S. B. et al. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 94, 9226–9231 (1997).
Hua, J. et al. Genetic dissection of an elite rice hybrid revealed that heterozygotes are not always advantageous for performance. Genetics 162, 1885–1895 (2002).
Hua, J. et al. Single-locus heterotic effects and dominance by dominance interactions can adequately explain the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 100, 2574–2579 (2003).
Huang, Y. et al. Heterosis and polymorphisms of gene expression in an elite rice hybrid as revealed by a microarray analysis of 9198 unique ESTs. Plant Mol. Biol. 62, 579–591 (2006).
Zhou, G. et al. Genetic composition of yield heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 109, 15847–15852 (2012).
Huang, X. et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun 6, 6258 (2015).
Zhang, J. et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 10.1073/pnas.1611012113 (2016).
Wang, X. et al. Global genomic diversity of Oryza sativa varieties revealed by comparative physical mapping. Genetics 196, 937–949 (2014).
van Oeveren, J. et al. Sequence-based physical mapping of complex genomes by whole genome profiling. Genome Res. 21, 618–625 (2011).
Nelson, W. & Soderlund, C. Integrating sequence with FPC fingerprint maps. Nucleic Acids Res. 37, e36 (2009).
Kim, H. et al. Comparative physical mapping between Oryza sativa (AA genome type) and O. punctata (BB genome type). Genetics 176, 379–390 (2007).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563–569 (2013).
Zhang, J. et al. Genome puzzle master (GPM): an integrated pipeline for building and editing pseudomolecules from fragmented sequences. Bioinformatics 10.1093/bioinformatics/btw370 (2016).
International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature 436, 793–800 (2005).
Schneeberger, K. et al. Reference-guided assembly of four diverse Arabidopsis thaliana genomes. Proc Natl Acad Sci USA 108, 10249–10254 (2011).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Kelley, D. R., Schatz, M. C. & Salzberg, S. L. Quake: quality-aware detection and correction of sequencing errors. Genome Biol 11, R116 (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Kim, J. et al. Reference-assisted chromosome assembly. Proc Natl Acad Sci USA 110, 1785–1790 (2013).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Pop, M., Phillippy, A., Delcher, A. L. & Salzberg, S. L. Comparative genome assembly. Brief Bioinform 5, 237–248 (2004).
Yu, J. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 96, 79–92 (2002).
Delcher, A. L., Phillippy, A., Carlton, J. & Salzberg, S. L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30, 2478–2483 (2002).
Huang, X. CAP3: A DNA Sequence Assembly Program. Genome Res 9, 868–877 (1999).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Data Citations
GenBank KG737749–KG771717 (2013)
GenBank KG702200–KG737748 (2013)
NCBI Sequence Read Archive SRP071597 (2016)
NCBI Sequence Read Archive SRP071598 (2016)
NCBI Sequence Read Archive SRP071944 (2016)
NCBI Assembly GCA_001618795 (2016)
NCBI Assembly GCA_001618785 (2016)
NCBI Assembly GCA_001623345 (2016)
NCBI Assembly GCA_001623365 (2016)
Acknowledgements
This work was supported by grants from the National Natural Science Foundation (No. 31330039), National 863 Project (No. 2014AA10A600) and the 111 Project of China to Q.Z., the Start-up Fund of the National Key Laboratory of Crop Genetic Improvement to J.Z., and the Bud Antle Endowed Chair of Excellence in Agriculture and Life Sciences, and the AXA Chair in Genome Biology and Evolutionary Genomics to R.A.W.
Author information
Authors and Affiliations
Contributions
J.Z., L.L.C., R.A.W. and Q.Z. designed and conceived research; M.L. built the BAC libraries; J.Z., T.M., M.L. and J.L.G. constructed the WGP physical maps; D.K., S.L., J.T. and B.E.P.H. prepared DNA for PacBio sequencing; D.K., D.C., S.L., J.T., C.E.M., Y.Y. and A.D. performed PacBio sequencing; J.Z., L.L.C., S.S., D.C., W.L., T.M., W.B.J. F.X., J.M.S., B.D., W.X. and Y.L. performed sequence assembly and analysis; J.Z., L.L.C., W.X., L.X., C.W., Y.X., D.Z., S.Y., Y.Z., G.W., R.A.W. and Q.Z. contributed new reagents/analytic tools; J.Z., L.L.C., R.A.W. and Q.Z. wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Zhang, J., Chen, LL., Sun, S. et al. Building two indica rice reference genomes with PacBio long-read and Illumina paired-end sequencing data. Sci Data 3, 160076 (2016). https://doi.org/10.1038/sdata.2016.76
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2016.76
This article is cited by
-
Development of Whole-Genome Agarose-Resolvable LInDel Markers in Rice
Rice (2020)
-
A benchmark of transposon insertion detection tools using real data
Mobile DNA (2019)
-
The rice genome revolution: from an ancient grain to Green Super Rice
Nature Reviews Genetics (2018)
-
Assembly of an early-matured japonica (Geng) rice genome, Suijing18, based on PacBio and Illumina sequencing
Scientific Data (2017)
