
Abstract
In angiogenesis with concurrent inflammation, many pathways are activated, some linked to VEGF and others largely VEGF-independent. Pathways involving inflammatory mediators, chemokines, and micro-RNAs may play important roles in maintaining a pro-angiogenic environment or mediating angiogenic regression. Here, we describe a gene expression dataset to facilitate exploration of pro-angiogenic, pro-inflammatory, and remodelling/normalization-associated genes during both an active capillary sprouting phase, and in the restoration of an avascular phenotype. The dataset was generated by microarray analysis of the whole transcriptome in a rat model of suture-induced inflammatory corneal neovascularisation. Regions of active capillary sprout growth or regression in the cornea were harvested and total RNA extracted from four biological replicates per group. High quality RNA was obtained for gene expression analysis using microarrays. Fold change of selected genes was validated by qPCR, and protein expression was evaluated by immunohistochemistry. We provide a gene expression dataset that may be re-used to investigate corneal neovascularisation, and may also have implications in other contexts of inflammation-mediated angiogenesis.
Design Type(s) | parallel group design • intervention design • in vivo design |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | DNA microarray |
Factor Type(s) | surgery |
Sample Characteristic(s) | Rattus norvegicus • cornea |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Angiogenesis drives age-related macular degeneration (AMD), retinopathy of prematurity (ROP)1, and diabetic retinopathy (DR), which can also be associated with retinal vein occlusion (RVO)2. In the cornea, corneal neovascularisation (CorNV) is driven primarily by inflammation and/or hypoxia3. These stimuli can tip the balance between antiangiogenic and proangiogenic molecules, in favour of the latter4. In recent years, anti-VEGF therapy has been used to regress immature vessels5, but despite the benefits of anti-VEGF therapy, it has been shown to regress new vessels only partially6,7, to cause neurotoxicity and in some cases to increase the risk of stroke8. In a study focused on CorNV, anti-VEGFA therapy was shown to have only a 14% efficacy in regressing neovessels during the early sprouting stages in the rat inflammatory model of angiogenesis9. On the other hand, factors other than VEGF have been identified to independently stimulate corneal angiogenesis10, while other factors may play a role in vessel maturation. For example, PDGF and Ang1 are reported to be essential for pericyte recruitment to stabilise newly formed vasculature11,12, and recently, WNT signalling enhancer R-spondin3 (RSPO3) expressed by ECs has been identified to be crucial for maintaining a remodeling vasculature13. The challenge however, is that angiogenesis in a clinical scenario such as corneal transplant rejection14, in age-related macular degeneration15, and in a tumor microenvironment16, occurs in a more complex pro-inflammatory environment, with many players involved at the gene level.
Here, we mimicked pathological angiogenesis using a novel reversible model of suture-induced inflammatory CorNV in the rat, and then studied gene expression profiles associated with angiogenic sprouting and induced blood vessel regression/remodeling. The whole transcriptome was assessed using Affymetrix Rat Gene 2.0 ST Arrays, custom designed for up to 28,407 total RefSeq transcripts coverage. Unlike traditional array designs that rely on probes designed to the first exon of the genes’ 3' end, the Affymetrix Rat Gene 2.0 ST arrays include hundreds of thousands of probes designed to every exon of every transcript represented on the array. The high transcript coverage (median of 22 probes/gene) gives a more accurate detection of genome-wide transcript expression changes compared to traditional microarrays. The whole-transcript analysis approach enables detection of multiple transcript isoforms from a given gene such as splice variants, non-polyadenylated transcripts, transcripts with alternative polyadenylation sites and truncated transcripts. The gene expression for selected genes was validated using qPCR using custom designed primers, and protein expression and localization in the tissue was evaluated by immunohistochemistry. Finally, CEL and CHIP files, and the corresponding sample metadata for each of the samples were deposited in to Gene Expression Omnibus (Data Citation 1).
The dataset described and presented here offers a means to identify and understand transcripts and gene interactions in angiogenesis driven primarily by inflammation. In addition to the coding transcripts, the microarray chips used in this study also capture information about miRNAs, which are reported to be key regulators for biological processes such as the modulation of tip cell behaviour in zebrafish17. Also, using the rat alkali burn model, miR-446 among others was suggested to have a role in protecting the transplanted cornea by suppressing its target Prox1 (ref. 18). The provided dataset therefore also provides a means to investigate miRNAs associated with angiogenic sprouting and regression in the cornea. In addition, the data files described here were previously analyzed in-depth, and it was shown that suture removal induced blood vessel remodelling, and that many genes classified as either pro-angiogenic/pro-inflammatory or anti-angiogenic/pro-maturation were identified. At the phenotypic level, blood vessel remodelling was characterized by an increase in macrophages, and a reduction of granulocytes in the cornea19.
Methods
All methods described here are either an extract or an expansion of those recently published, GeneChip WT PLUS Reagent Kit, P/N 703174 Rev. 2 (Affymetrix Inc), and RMA method19,20.
Rats
Ethical application, strain and housing conditions of the animals used for this study were previously detailed19. Using 12–16 weeks old rats, the suture model of inflammatory corneal neovascularisation was used as previously described21.
Study design and sample collection
Two 10-0 nylon sutures were placed in the temporal side of the cornea of the right eye of the rat at 1.5 mm from the cornea-scleral border (limbus), to induce inflammation leading to limbal capillary sprouting and CorNV. After 4–5 days following suturing (0 h time point, when new sprouts had invaded the clear cornea one-half to two-thirds of the distance to the suture), rats were divided into two groups, called suture IN and suture OUT. In the suture IN group, sutures were left in place for an additional 24 h before harvesting cornea tissue for gene expression analysis. In the suture OUT group, both sutures were removed at 0 h and corneas were harvested 24 h later for gene expression analysis. The control group consisted of rats whose corneas were not sutured. In both suture IN and suture OUT arms, only the vascularised area of the tissue was excised and immediately immersed in RNA later and stored at −20 °C. For each of the four groups (i.e., Control, 0 h, 24 h suture IN, 24 h suture OUT), n=4 rats were used, and the identical phenotype was confirmed by in vivo slit lamp and IVCM imaging at 0 h. A detailed graphical illustration of the experimental design we followed was previously published19.
Cornea neovascularisation phenotype
Prior to gene expression analysis, morphological data was collected using a clinical slit lamp camera and an in vivo confocal microscope (IVCM). The morphological data was analysed for changes in neovessels (for example vessel density and splitting), and for the dynamics of the infiltrating inflammatory cells. From this data, we confirmed the induction of a remodelling phenotype following the removal of the angiogenic stimulus19. These findings were of importance as they served as a foundation for the design and planning of the microarray experiments that followed, and whose dataset we now describe here. In Fig. 1a are the slit lamp images of corneas in suture IN and suture OUT groups, indicating new capillary sprouting from the limbus into the cornea. Figure 1b is an example of IVCM images showing two types of inflammatory cells known to infiltrate the sutured cornea, which in this model were affected by removal of the angiogenic stimulus.

In a,b are slit lamp images of a neovascularised cornea in the suture IN and suture OUT groups respectively. In c,d are IVCM images of granulocyte and macrophage cell types infiltrating into the neovascularised cornea, respectively.
RNA isolation, quantitation and quality verification
A single cornea per sample (non-pooled) was used for total RNA extraction. Tissues were preserved in RNA later immediately after harvest and stored at −20 °C. Cornea tissue was disrupted using a hand-held tissueRuptor and disposable Probe (Qiagen, Hilden, Germany), in a 2 ml micro centrifuge tube, on ice. Total RNA was then extracted from the lysate (Qiagen, Hilden, Germany), without DNase treatment. RNA concentration was then determined using a Nanodrop spectrophotometer 2000 (Thermo scientific), and the RNA quality verified using an Agilent 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit Quick Start guide, Agilent Technologies). RNA Integrity Number (RIN)≥7 was set as the cut off for selecting RNA samples for downstream analysis. Samples with RIN values less than the set cut-off were excluded from the analysis process, and were replaced with fresh sample that met the inclusion criteria. Speed vac was used to concentrate samples whenever needed. RNA samples were temporarily stored at −20 °C following extraction and for both microarray and qPCR analysis, the RNA was used for analysis not longer than 7 days’ post extraction. See Fig. 2a for an illustration of the workflow that was followed during sample processing and qPCR analysis.

In both cases, cornea tissue previously preserved in RNA later was recovered by thawing at room temperature. The tissue was then disrupted and total RNA extracted from the lysate. The RNA was quantified, purity verified and integrity checked prior to the selected analysis. Separate groups of rats were used for qPCR and microarray analysis. At least three biological replicates were used for each case (i.e., qPCR and microarray analysis). The number of replicates was chosen to increase the statistical reliability of the generated dataset (R2>0.96 for samples within a specific sample group).
Microarray target preparation, hybridisation and data acquisition
All procedures for microarray analysis were performed at the core facility, cell biology laboratory at Linköping University. Gene expression analysis was performed using the whole-transcriptome Rat Gene 2.0 ST microarrays (Affymetrix Inc). An input of 100 ng of total RNA was mixed with the Poly-A RNA Spike-in controls and used for microarray target preparation following the manufacturer’s instructions (GeneChip WT PLUS Reagent Kit, P/N 703174 Rev. 2, Affymetrix Inc). Fifteen Rat Gene 2.0 ST arrays were used to correspond to 4 microarray chips per experimental time point (except for the 0 h, where 3 microarrays were used as one as one sample was lost (spillage) during target processing). In summary; single-stranded cDNA with T7 promoter sequence at the 5' end was synthesised from total RNA using primers containing a T7 promoter sequence. Following DNA polymerase and RNase H, double-stranded cDNA was synthesised from the single-stranded cDNA. After this, cRNA was synthesized and amplified by in vitro transcription (IVT) of the second-stranded cDNA template using T7 RNA polymerase. The cRNA was purified and the yield verified using Nanodrop 2000. Reverse transcription of cRNA using 2nd cycle primers synthesised 2nd-Cycle Single-Stranded cDNA. In order to receive single-stranded cDNA, the 2nd-Cycle Single-Stranded cDNA was treated with RNase H to hydrolyse cRNA template, which was purified and quantified as already described above. Following fragmentation of the Single-stranded cDNA using uracil-DNA glycosylase (UDG) and apurinic/apyrimidinic endonuclease 1 (APE 1) at the unnatural dUTP residues, the fragmented cDNA was labeled by terminal deoxynucleotidyl transferase (TdT) using the Affymetrix proprietary DNA Labeling Reagent that was covalently linked to biotin. The labeled cDNA was then hybridised to the Affymetrix Rat Gene 2.0 ST arrays in a 645 hybridisation oven and washed in Fluidics Station 450, according to the User's Guide (AGCC, P/N 08–0295, Affymetrix Inc). The chips were then scanned according to the user Manual for Cartridge Arrays (PN 702731, Affymetrix Inc). The acquired image files were examined for hybridisation of the internal Poly-A RNA Spike-in controls. DAT and CEL files were then generated from the probe intensity files using GeneChip Command Console Software (AGCC). See Fig. 2b for an illustration of the workflow that was followed during sample and microarray preparation and analysis.
Analysis of microarray files
Raw CEL files were converted into expression measures, background-corrected, and data-normalized using Affymetrix Expression Console software and Affymetrix transcription console (TCA) v. 3.0 tool.
The Poly-A RNA Spike-in controls that were previously mixed with the pure and intact RNA sample Figure 3a,b, were successfully detected on the microarray chips Fig. 3c. Pearson’s Correlations showed high sample correlation with sample groups, with a higher correlation found between the control samples Fig. 3d. Also, the control samples are separate from the experimental samples. Robust Multi-array Average (RMA) for background correction, normalisation and computation of probe-set level expression of the samples was successful Fig. 3e,f.

All CHIPs passed the initial quality checks. (a) RNA purity as determined by Nanodrop 2000, (b) is the gel size separation of the 18S and 28S subunits shown as distinct bands, and S, integration number. Results in b were obtained using RNA 6000 pico kit following the eukaryote total RNA Pico assay. (c) Boxplot of four of Affymetrix Spike-In controls, lys-M, phe-M, thr-M and dap-M illustrating the range of signal (log2) over the experiment as well as quality control of the arrays technical run. (d) Illustrates the Pearsson Correlation for all samples after DABG correction and RMA normalization (figure constructed using R/Bioconducter and package pheatmap). (e,f) Boxplot showing raw log2 Probe Cell Intensities, before (e) and after the DABG correction and RMA normalized log2 Expression Signal (f) for all samples.
Processing and normalization of the raw-files, and CEL files was done using the Affymetrix Expression Console built in algorithm called ‘Detection above background’ (DABG) and the RMA method, which uses a multi-array approach in its normalization method. The DABG algorithm is intended to serve as a confidence score in lieu of Affymetrix previous Detection call (‘Present’ or ‘Absent’). In short, DABG is a detection metric generated by comparing ‘Perfect Match’ probes to a distribution of background probes. This comparison yields a P-value which is then combined into a probe set level P-value using the Fischer equation. More information regarding the DABG algorithm is found at: http://media.affymetrix.com/support/technical/whitepapers/exon_background_correction_whitepaper.pdf. The RMA package includes background correction, normalization and computation of probe-set level expression. The data was then log2 transformed for further biological downstream analysis. Comprehensive literature about the RMA method is available elsewhere20.
Verification of fold change expression by quantitative real-time PCR (qPCR)
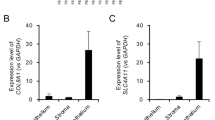
A separate group of rats was used for this purpose. Total RNA was extracted as illustrated in Fig. 2a above. From the RNA, cDNA was synthesised at 42 °C for 2 h with a heated lid ON (Invitrogen). Custom Taqman primers were obtained from TaqMan, Applied Biosystems for Timp1, Serpinb2 and Reg3g. The primers were used with TaqMan Fast Advanced Master Mix (Applied Biosystems). Three biological, and 2 technical replicates were used per sample and the analysis was performed using CFX 96 Touch Real-Time PCR detection system (Bio-Rad). Threshold cycle (Ct) values were normalised to GAPDH, and gene expression was measured by the relative quantitation method (2−ΔΔCt). The fold change expression profile between suture IN and suture OUT as determined by qPCR in general was in agreement with the expression profile determined by the microarray analysis Fig. 4a,b, with further qPCR results available19.

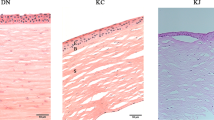
(a) is the microarray log2 fold change expression, while (b) is the corresponding qPCR log2 fold change expression. n=3 and 4, for (a,b) respectively. For comparison of the log2 fold change between suture IN and suture OUT, an unpaired two-tailed t-test was performed to compare the means between suture IN and suture OUT. No multiple test correction was performed for the two-group comparisons. The t test was performed in both the microarray and qPCR datasets independently. In both (a,b), asterisks indicate significance for P<0.05 level, error bars represent standard deviation. Data was analysed using GraphPad Prism 5 (GraphPad Software, Inc. CA 92037 USA). In (c) are immunofluorescent images from cornea sections indicating the corresponding protein expression across treatments. DAPI labels cell nuclei in blue, while the target proteins are indicated in green color.
Protein expression and localisation by immunohistochemistry
Following qPCR confirmation of the fold change, protein expression and localisation in cornea tissue cross-sections was investigated. A separate set of three rats per group was used for this purpose. Immunohistochemistry was performed to assess the expression and localisation for Timp1, Serpinb2 and Reg3g, using primary polyclonal antibodies; orb195994 and orb25969 from Biorbyt for Timp1 and Reg3g respectively, and ABIN1860122 from antibodiesonline.com, for Serpinb2. For detection, Alexa 488 secondary fluorescent antibody was used. From the analysis, Timp1 was expressed and localised within the stroma and endothelium, Reg3g localised to the stroma and Serpinb2 to the epithelium, stroma and endothelium Fig. 4. The protein expression of several further genes in the corneal tissue has been provided elsewhere19.
Data Records
CEL and CHIP files associated with the samples analysed in this study are deposited at gene expression omnibus with the accession number GSE81418. The detailed information regarding the samples is tabulated in Table 1.
Technical Validation
A number of precautions were taken into consideration during the experimental procedure. Rats were randomly assigned to experimental groups (suture IN and suture OUT) at 0 h. The rat ID assigned to the rats at the beginning of each experiment was maintained throughout the entire experimental process, and this included both microarray and qPCR validation of gene expression data. Following rat sacrifice, cornea tissue was harvested from the eye within one minute, and immediately immersed in RNA later and stored at −20 °C. This practice was instated to minimize alterations in gene expression that could arise from cold ischemia22. Surgical equipment were cleaned between tissue extraction from different animals, and after each experimental day. At each time point, four biological replicates were used. RNA was extracted from individual rats without pooling of any samples. Only high quality RNA (refer to the RIN values) was used for both microarray and qPCR analysis.
Usage Notes
Microarrays have been used previously with great success as a tool for an unbiased identification of novel pathways and regulatory molecules in the pathology of different diseases23,24. These datasets therefore provide a solid basis for the investigation of specific research questions of interest e.g. angiogenic sprouting, remodeling of capillaries, relationship between inflammatory cells and capillaries, and hypoxia among others. The microarray chips used in this study also provide information regarding microRNAs that are key regulators of gene expression. Using this dataset, the role of these molecules can be explored to address questions regarding the functionality of both known and novel gene targets that are key in inflammation-driven angiogenesis in the cornea. To explore such possibilities, different Gene Set Expression Analysis (GSEA) software can be used. These are available both as public tools such as STRING and Cytoscape with for instance the Bingo or ClueGO, and also as commercially available software such as QIAGEN's Ingenuity Pathway Analysis tool. Information pertaining miRNAs can also be exploited by using specific kits designed for miRNA extraction and for qPCR validation of gene expression. These kits are commercial and readily available.
Probe IDs with missing gene symbols can be updated by performing a Batch Query at NetAffx Analysis Center tool-Affymetrix. Gene fold change expression can be obtained by comparing each treatment group to the non-sutured control sample group. The corresponding p and q values can also be calculated since the study has enough degrees of freedom in its statistical design. For the adjusted P-value calculation (False Discovery Rate (FDR) or q value) researchers commonly use the Benjamini-Hochberg statistical method. However, other methods exist and are used. The method of choice is dependent on the study design.
Additional Information
How to cite this article: Mukwaya, A. et al. A microarray whole-genome gene expression dataset in a rat model of inflammatory corneal angiogenesis. Sci. Data 3:160103 doi: 10.1038/sdata.2016.103 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Cao, R., Jensen, L. D., Soll, I., Hauptmann, G. & Cao, Y. Hypoxia-induced retinal angiogenesis in zebrafish as a model to study retinopathy. PLoS ONE 3, e2748 (2008).
Cugati, S., Wang, J. J., Rochtchina, E. & Mitchell, P. Ten-year incidence of retinal vein occlusion in an older population: the Blue Mountains Eye Study. Archives of ophthalmology 124, 726–732 (2006).
Safvati, A., Cole, N., Hume, E. & Willcox, M. Mediators of neovascularization and the hypoxic cornea. Current eye research 34, 501–514 (2009).
Azar, D. T. Corneal angiogenic privilege: angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing (an American Ophthalmological Society thesis). Trans Am Ophthalmol Soc 104, 264–302 (2006).
Korn, C. & Augustin, H. G. Mechanisms of vessel pruning and regression. Developmental cell 34, 5–17 (2015).
Chang, J.-H. et al. Corneal neovascularization: an anti-VEGF therapy review. Survey of ophthalmology 57, 415–429 (2012).
Dicato, M. A . Side effects of medical cancer therapy: prevention and treatment (Springer Science & Business Media, 2012).
Schmid, M. K. et al. Efficacy and adverse events of aflibercept, ranibizumab and bevacizumab in age-related macular degeneration: a trade-off analysis. British Journal of Ophthalmology, 99, 141–146 (2015).
Mirabelli, P., Peebo, B. B., Xeroudaki, M., Koulikovska, M. & Lagali, N. Early effects of dexamethasone and anti-VEGF therapy in an inflammatory corneal neovascularization model. Experimental eye research 125, 118–127 (2014).
Cao, R. et al. Mouse corneal lymphangiogenesis model. Nature protocols 6, 817–826 (2011).
Armulik, A., Abramsson, A. & Betsholtz, C. Endothelial/pericyte interactions. Circulation research 97, 512–523 (2005).
Aguilera, K. Y. & Brekken, R. A. Recruitment and retention: factors that affect pericyte migration. Cellular and Molecular Life Sciences 71, 299–309 (2014).
Scholz, B. et al. Endothelial RSPO3 Controls Vascular Stability and Pruning through Non-canonical WNT/Ca 2+/NFAT Signaling. Developmental cell 36, 79–93 (2016).
Maguire, M. G. et al. Risk factors for corneal graft failure and rejection in the collaborative corneal transplantation studies. Ophthalmology 101, 1536–1547 (1994).
Stanton, C. M. & Wright, A. F. in Retinal Degenerative Diseases 251–257 (Springer, 2014).
Coussens, L. M. & Werb, Z. Inflammation and cancer. Nature 420, 860–867 (2002).
Nicoli, S., Knyphausen, C.-P., Zhu, L. J., Lakshmanan, A. & Lawson, N. D. miR-221 is required for endothelial tip cell behaviors during vascular development. Developmental cell 22, 418–429 (2012).
Seo, M., Choi, J.-S., Rho, C. R., Joo, C.-K. & Lee, S. K. MicroRNA miR-466 inhibits Lymphangiogenesis by targeting prospero-related homeobox 1 in the alkali burn corneal injury model. Journal of biomedical science 22, 3 (2015).
Mukwaya, A. et al. Factors regulating capillary remodeling in a reversible model of inflammatory corneal angiogenesis. Scientific Reports 6, 32137 (2016).
Irizarry, R. A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Peebo, B. B., Fagerholm, P., Traneus-Röckert, C. & Lagali, N. Cellular level characterization of capillary regression in inflammatory angiogenesis using an in vivo corneal model. Angiogenesis 14, 393–405 (2011).
David, K. et al. Surgical procedures and postsurgical tissue processing significantly affect expression of genes and EGFR-pathway proteins in colorectal cancer tissue. Oncotarget 5, 11017–11028 (2014).
Tarca, A. L., Romero, R. & Draghici, S. Analysis of microarray experiments of gene expression profiling. American journal of obstetrics and gynecology 195, 373–388 (2006).
Nai, W. et al. Identification of novel genes and pathways in carotid atheroma using integrated bioinformatic methods. Scientific reports 6, 18764 (2016).
Data Citations
Mukwaya, A. NCBI Gene Expression Omnibus GSE81418 (2016)
Acknowledgements
The authors wish to acknowledge the technical support offered by Åsa Schippert and Annette Molbaek at the Linköping University core facility laboratory during microarray processing and data acquisition. The authors also like to acknowledge financial contribution to this study in the form of a project grant from the Swedish Research Council (Grant No. 2012–2472), and an unrestricted basic research grant from Bayer HealthCare AB, Solna, Sweden. The funders did not participate in the design or conduct of the study, or interpretation of results. The authors like to acknowledge the Bioinformatics Infrastructure for Life Sciences (BILS) Sweden, for their financial contribution.
Author information
Authors and Affiliations
Contributions
A.M., B.P. and N.L. developed the concept of the study. A.M., B.P., A.L., L.D.E.J. and N.L. designed the experiments. A.M., B.P., M.X., A.Z., A.L. and N.L. performed in vivo experiments. A.M. processed microarrays and qPCR. A.M. and J.M.L. performed data quality control. J.M.L. performed data normalisation. A.M., J.M.L. and N.L. wrote the manuscript and all authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Mukwaya, A., Lindvall, J., Xeroudaki, M. et al. A microarray whole-genome gene expression dataset in a rat model of inflammatory corneal angiogenesis. Sci Data 3, 160103 (2016). https://doi.org/10.1038/sdata.2016.103
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2016.103
This article is cited by
-
Discovery of novel L-type voltage-gated calcium channel blockers and application for the prevention of inflammation and angiogenesis
Journal of Neuroinflammation (2020)
-
Microarray profiling of miRNA and mRNA expression in Rag2 knockout and wild-type mouse spleens
Scientific Data (2018)
-
Selective IKK2 inhibitor IMD0354 disrupts NF-κB signaling to suppress corneal inflammation and angiogenesis
Angiogenesis (2018)
-
Time-dependent LXR/RXR pathway modulation characterizes capillary remodeling in inflammatory corneal neovascularization
Angiogenesis (2018)
-
Genome-wide expression datasets of anti-VEGF and dexamethasone treatment of angiogenesis in the rat cornea
Scientific Data (2017)
