
Abstract
With ever-increasing atmospheric carbon dioxide concentrations and commitments to limit global temperatures to less than 1.5 °C above pre-industrial levels, the need for versatile, low-cost carbon dioxide capture technologies is paramount. Electrochemically mediated carbon dioxide separation systems promise low energetics, modular scalability and ease of implementation, with direct integration to renewable energy for net-negative carbon dioxide operations. For these systems to be cost-competitive, key factors around their operation, stability and scaling need to be addressed. Energy penalties associated with redox-active species transport, gas transport and bubble formation limit the volumetric productivity and scaling potential due to their cost and footprint. Here we highlight the importance of engineering approaches towards enhancing the performance of redox-active electrochemically mediated carbon dioxide capture systems to enable their widespread implementation.

Similar content being viewed by others
Main
With atmospheric carbon dioxide (CO2) concentrations increasing, there is an obvious need for inexpensive, scalable and low-energy CO2 separation technologies across a range of operating conditions to mitigate the effects of climate change1. Traditional CO2 capture systems modulate temperature or pressure to swing CO2 sorption capacity2. Electrochemically mediated carbon capture (EMCC), on the other hand, with sorption capacity modulated by an applied potential, offers potentially lower energetics, direct integration with high capacity factor, low-carbon-intensity energy sources, and modular scaling and ease of implementation. EMCC can be realized in aqueous media by means of water dissociation in electrolytic cation exchange (ECC) or bipolar membrane electrodialysis (BPMED), or by the chemical looping of water oxidation and reduction products (O2 or H2; refs. 3 and 4, respectively) to drive pH changes or react with CO2 to swing CO2 capacity. Another class of EMCC introduces an additional redox-active species (in protic or aprotic media) to modulate the total CO2 sorption capacity. This Perspective focuses on the challenges faced in the development and scaled implementation of the latter class of EMCC systems using redox-active molecules. Substantial effort in the literature has focused on developing appropriate chemistries to avoid side reactions5,6,7,8,9, and little work has been directed to the engineering of such systems to address the scale of the target separations in relation to emissions associated with fossil-fuel power generation; natural gas treatment; cement, ammonia and steel production; distributed direct air capture; and, recently, marine inorganic carbon extraction.
System configurations and thermodynamics
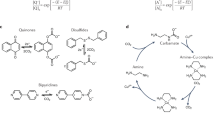
EMCC employing redox-active molecules relies on modulation of the sorption capacity for CO2 through application of an applied potential. In the simplest implementation, the applied potential acts directly on a redox-active sorbent molecule, which alters its affinity for CO2 complexation based on the oxidation state. This ‘direct’ cycle is shown in Fig. 1a for a generic redox-active sorbent, A, in aprotic media. In protic media, the reduced species, A−, can bind to protons rather than CO2 directly, resulting in a pH swing that shifts the CO2 solubility—the ‘semi-direct’ cycle shown in Fig. 1b. Alternatively, the applied potential can act on a redox-active blocker species, B, which, when activated, displaces CO2 from the electrochemically inert absorbent; this ‘indirect’ cycle is shown in Fig. 1c.

a, Direct cycle for a generic redox-active absorbent operating in aprotic media. A and A− represent the dormant and activated states, respectively. b, Semi-direct cycle operating in protic media. AH represents the activated protonated state. c, Indirect cycle: an electrochemically inert absorbent with a redox-active blocker species. B and B+ represent the dormant and activated states of the blocker, respectively. Inset graphs in a–c show the fraction of redox species active as a function of applied potential E, with the standard potential, E0, marked by a dashed gray line. d, Typical molecules employed in the direct and semi-direct cycles. e, Equilibrium binding between the activated redox sorbents in d and CO2 plotted against reduction potential (quinones, brown squares; bipyridines, red diamonds; disulfide, orange circle; transition metal complexes, purple triangles). Horizontal dashed lines indicate the minimum necessary equilibrium binding for direct air capture (blue) and flue gas (green) conditions11. Panels adapted with permission from: a,c, ref. 21, Springer Nature Ltd; e, ref. 10, Royal Society of Chemistry.
Several reviews summarize the various chemistries employed in these cycles5,6,7,8,9,10, although it is useful to briefly address one of the key challenges associated with the direct cycle. A subset of redox-active molecules used in the direct cycle are shown in Fig. 1d. The equilibrium binding of the activated form of these molecules with CO2, shown in Fig. 1e, plotted against their reduction potential, exhibits a linear trend. One key challenge arises when the region of suitable equilibrium binding constants is superimposed on that of oxygen sensitivity11,12. In the presence of oxygen, the activated redox sorbent can react with dissolved oxygen to generate superoxide, which reduces the efficiency for absorbent activation and can result in performance degradation due to deleterious side reactions. Few molecules possess the necessary traits for application in direct air capture (DAC) or flue gas capture in the direct or semi-direct cycle due to these limitations6,12. The indirect cycle offers some advantages here, although the influence of other flue-gas impurities (SOx and NOx) on cyclic stability has not been sufficiently examined and is only now just starting to be investigated in the CO2 electrolysis field13.
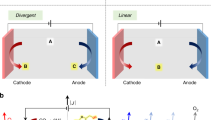
The reversible affinity of CO2 with a suitable redox-active molecule (or absorbent and blocker pair) can be exploited in various process configurations. The four-stage continuous process is the most familiar and is analogous to that of traditional thermal amine stripping, where CO2 absorption and desorption are performed in separate units. The process, shown in Fig. 2a, starts (point 1) with a CO2-saturated solution leaving the absorption unit and entering the anodic chamber of the electrochemical cell. On oxidation, the redox-active sorbent is deactivated (or blocker activated) and the sorption capacity for CO2 is reduced (point 2). The solution is directed to a separation vessel where the liberated CO2 gas disengages from the liquid sorbent, which re-enters the electrochemical cell in the cathodic chamber (point 3). The sorbent is activated (or blocker deactivated) by electrochemical reduction and returns to the absorption unit, completing the cycle.

a–d, Simplified process configurations for four-stage (a), three-stage anodic desorption (b), three-stage cathodic absorption (c) and two-stage (d) processes. e, Deviation potential versus state of charge. Horizontal dashed lines indicate normalized CO2 partial pressure isobars; dotted lines indicate relative sorbent CO2 loading. Vertical arrows indicate the collapse from the four-stage work cycle to the two-stage cycle if processes are coupled, permitting CO2 exchange with a source or sink. Numbers correspond to those in a. f, Work of separation for two-, three- and four-stage systems at various relative Henry’s CO2 solubility, \({\widetilde{k}}\), where AD and CA refer to anodic desorption and cathodic absorption configurations, respectively.
Intensified process variations, which combine one or more of the electrochemical and gas-contacting steps, have been conceptualized14 and demonstrated in different forms. A three-stage process, where sorbent deactivation and CO2 desorption are coupled, is depicted in Fig. 2b. An alternate three-stage process, where sorbent activation is coupled with CO2 absorption, is shown in Fig. 2c. The two-stage process, which combines absorbent activation with CO2 absorption and absorbent deactivation with CO2 desorption, has been demonstrated in static (with no advection of the redox-active species), homogeneous15 and heterogeneous16,17 configurations. Figure 2d depicts a two-stage configuration with advection of a homogeneous redox-active species between gas-diffusion electrodes that provide gas exchange with the feed and sweep streams. In static heterogeneous systems (not pictured), the absorption and desorption stages are temporally separated, resulting in batch swing operation, although the introduction of gas-gating membranes offers semi-continuous operation18.
A thermodynamic comparison of these configurations can be made by tracking the open-circuit potential against the system state of charge. The use of the deviation potential (Edev), the difference between the standard redox-couple potential (E0) and the open-circuit potential, can be used in place of the open-circuit potential here, as it allows for consistent comparison across cycles19. The deviation potential, \({E}_{\rm{dev}}={\frac{{RT}}{{mF}}\mathrm{ln}(1+{K}_{{\rm{CO}}_{2}}{\widetilde{p}}_{{\rm{CO}}_{2}}^{m})}\), is a function of the normalized CO2 partial pressure, \({\widetilde{p}}_{{\rm{CO}}_{2}}\), the activated sorbent equilibrium binding constant, \({K}_{{\rm{{CO}}}_{2}}\), and number of moles of CO2 bound per mole of sorbent, m. Process configurations can then be overlaid on a plot of the deviation potential for constant normalized CO2 partial pressure and constant normalized CO2 liquid loading. The numbered points marked in Fig. 2e represent the four-stage process described previously, specifically for the up-concentration performed between 15% and 100% CO2. The work required for this separation is calculated by the path integral, \({W=\oint {E}^{\rm{dev}}{{\rm{d}}Q}}\), proportional to the contained area. The equivalent two-stage process is represented with solid lines connecting points 1 and 3. It is qualitatively clear from the contained area of each configuration that there is a substantial energy penalty associated with decoupling sorbent activation/deactivation and CO2 absorption/desorption, respectively. Figure 2f summarizes the ideal work of separation as a function of the CO2 absorbent binding equilibrium, the relative CO2 solubility and process configuration. Notably, the work of separation for two-stage systems, across all parameters examined, falls near the thermodynamic minimum.
The thermodynamic analyses present a strong motivation to pursue these configurations19,20, although the inefficiencies of real systems, including those in ancillary equipment, need to be quantified. Before discussing specific implementations, it is useful to define the key operating parameters and the derived metrics used to make comparisons. To realize changes in the state of charge of the redox-active species (or blocker), a current, I (in A), is passed through the electrochemical cell by applying a voltage, V (in V), a portion of which may be diverted to parasitic side reactions. The Faradaic efficiency, ηf, is the ratio of the time-integrated partial current, IR (that is, the current directly leading to the change in state of charge) to the time-integrated total current. High Faradaic efficiencies (>90%) are desired to maximize cyclic capacity and avoid deleterious side reactions. The current is often normalized to the electroactive surface area, yielding a current density (in A m−2), to allow for comparisons across various system sizes and geometries. Key derived system metrics are the electron utilization, ηe, defined as the ratio of the number of moles of CO2 captured/released to the number of electrons transferred (mol CO2/mol e−), and the specific energy consumption, E, defined as the energy required per mole of CO2 captured/released (kJ mol−1 CO2). The quantities, ηf, ηe and E are defined mathematically as follows:
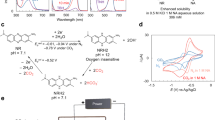
The research trajectory of electrochemically mediated amine regeneration (EMAR) presents a useful blueprint for evaluating promising chemistries, provided appropriate methods are used to determine their efficacy21. This process employs aqueous ethylenediamine as a CO2 capture sorbent and cupric ions as a blocker species. A representative EMAR process flow diagram is shown in Fig. 3a, and the accompanying chemistry cycle in Fig. 3b. Following the process flow diagram, the CO2-saturated sorbent leaving the absorption vessel (point 1) enters the anodic chamber where cupric ions are introduced, and CO2 gas is evolved. The exiting solution, now copper-rich, enters a separation vessel where CO2 gas is separated, and the liquid is returned to the cathodic chamber (point 3). The cupric ion concentration is reduced, and the solution returns to the absorption unit to complete the cycle (point 4). In this four-stage configuration, a membrane separator between the anodic and cathodic chambers is used to prevent bulk mixing of anolyte and catholyte and gas crossover, but allow for ionic conductivity. The process cycle shown in Fig. 3c is generated by combining the results of the non-ideal liquid-phase thermodynamic model and a coupled electrochemical transport model, which accounts for electrode kinetics and electrolyte advection such that the kinetic envelopes can be mapped over the thermodynamic behavior22,23,24.

a, Simplified process flow diagram of the EMAR process, indicating three desorbers operating in series, each segment at a different operating potential. PC, partial condenser. b, EMAR chemistry cycle: CO2 is chemically bound to aqueous ethylenediamine, forming a carbamate, and cupric ions (blocker species) are introduced in the anode chamber, forming the amine–Cu complex, which releases CO2. c, Energy mapping of a six-module electrochemcal desorption unit, shown as a three-stage anodic desorption configuration where CO2 desorption occurs at ambient pressure as a function of relative copper loading (in weight fraction, xCu = 2cCu/cEDA, where c is concentration and EDA is ethylenediamine). The green area marks the thermodynamic minimum. The expanded region represents the necessary overpotentials to drive the given current for the required desorption rate. The red lines mark the electrode potentials, where all anodes are grounded to 0 V. The dashed gray line indicates the potential of the cathode if a single module were to be used for desorption. Panel a adapted with permission from ref. 22, Elsevier. Panels reproduced with permission from: b, ref. 27, Elsevier; c, ref. 23, American Chemical Society.
Starting with the development of chemistry-specific thermodynamic models, the deviation from the ideal thermodynamic case, presented above, can be established. The selection or development of an appropriate liquid-phase model depends on the availability of relevant chemical data. For instance, many chemistries are evaluated in organic solvents or ionic liquids and show substantial changes based on electrolyte composition or the presence of hydrogen-bonding compounds12,25,26. The effect of ion migration can be determined22, and direct the selection and evaluation of appropriate separators or ion-exchange membranes. It is necessary to determine the activation kinetics of the redox species such that coupled transport models can be used to characterize the system overpotentials required to sustain reasonable currents and transport rates, as well as to identify areas for improvement. Redox-active species transport in the four-stage EMAR continuous-flow process, for example, presented quite reasonable overpotentials, aided by the use of turbulence promoters to improve the power density and to reduce system energetics, footprint and cost. Further analysis indicated that substantial energy was required when operating with a single electrode pair, where inefficiencies arise due to performing the entire state of charge swing with that one electrode set22. Figure 3c shows the various operating potentials (solid red lines) of six sequential modules to achieve the desired state of charge swing, and the gray dashed line marks the operating potential if a single module were used instead. These studies inform the electrochemical reactor design, operating window and scaling potential of these systems to industrially relevant applications. Analyses of this nature for generic and specific two-stage systems, in which the coupling of electrochemical kinetics and interfacial transport contributes additional complexity, have not yet been performed.
Transport considerations in EMCC
The four-stage systems are the simplest and most widely used configurations for evaluating EMCC performance (for example, EMAR27, liquid quinone28, MnO229,30, amino-pyridine31 and phenazines32,33,34). The transport within the electrochemical reactor, for a homogeneous redox sorbent flowing between parallel plates, is shown in Fig. 4a. To achieve the necessary swing in the state of charge of a redox-active sorbent (or blocker), the electrochemical reactor must be sized with the appropriate residence time for the achievable current density. The current density, a function of electrode kinetics, mass-transport limitations and ohmic losses, dictates the volumetric productivity of sorbent regeneration and ultimately CO2 removal. The electrode and electrolyte design can be tuned to improve the electrochemical kinetics, although mass-transfer limitations will always be present. Increasing the superficial velocity, incorporating turbulence promoters35 or using flow through porous electrodes offer means to reduce these limitations, although they have consequences in terms of reduced sorbent swing and elevated pressure drop. Inspiration from the adjacent redox flow battery36 and CO2 electrolysis37 fields can help in this regard, where the internal design of EMCC cells has not yet been a major focus. Alternate methods to reduce mass-transfer limitations include operating with pulsed flow38 or potential39,40,41, or providing external stimuli to the electrochemical cell, such as sonication42. Any improvements in volumetric productivity need to be weighed against energy consumption, cell assembly size and complexity, and reliability.
Transport considerations in two-stage systems are more complex and have fewer demonstrations. One embodiment, shown in Fig. 4b, employs redox-active species immobilized on a conductive substrate, which is in contact with a porous conductive layer that facilitates in-plane gas transport and through-plane electron transport. Absorption and desorption are temporally separated in this static heterogeneous configuration—Fig. 4b depicts the absorption stage, where CO2 breakthrough behavior is a function of the ratio of advective and diffusional transport, CO2 reaction kinetics and charging rate. To maximize sorbent utilization and yield favorable breakthrough behavior, the charging rate must be greater than the rate of CO2 absorption. Such configurations have employed ionic liquid electrolytes due to their favorable properties (low volatility, high stability window, non-flammability, selectivity for CO2 (Henry’s Law)), but the low CO2 diffusion coefficients (<10−10 m2 s−1) limit absorption rates even with thin electrodes (hundreds of micrometers). The development of microstructured porous materials that house the redox-active sorbent, while permitting advective gas transport, could substantially improve absorption rates by reducing the average CO2 diffusion lengths in electrolyte volumes to charged active sites. Three-dimensional printing of conductive polymers43, engineering of porous layered materials, or employing functionalized hollow fibers, recently demonstrated for CO2 capture and release via joule heating44,45, could be employed to reduce mass-transfer resistances. System energy penalties associated with lower ionic and electronic conductivities. as well as pressure drops, must be weighed against improvements in absorption rate and scaling potential. Parametric studies of coupled electrochemical transport models can inform the design space and trade-offs in such new geometries.

a, Electrochemical cell used in a four-stage continuous-flow process. b, Two-stage static heterogeneous electrochemical cell, shown during the absorption stage. c, Two-stage static homogeneous cell assembly. d, Two-stage homogeneous continuous-flow cell. Blue wavy lines indicate CO2 diffusive flux, im indicates migration of ions, straight blue lines behind a wavy front indicate in-plane plug flow advective CO2 transport through porous media with axial dispersion, and straight black lines indicate laminar advective redox-active species transport. Vertical and horizontal gray lines in a–d between the anodic and cathodic chambers (or electrodes in b) indicate a separator or ion-selective membrane.
An alternate two-stage configuration, in which all processes occur simultaneously in the same assembly, is shown in Fig. 4c. In this static-homogeneous configuration, the redox-active sorbent is ‘shuttled’ between electrodes. At the cathode, the sorbent is activated and binds with CO2 introduced through the gas-diffusion cathode. At the anode, the sorbent-CO2 adduct is deactivated and releases CO2 through the gas-diffusion anode. Initially modeled14,46 and demonstrated in the flat cell geometry shown, the device was refined to utilize hollow fibers as gas-diffusion electrodes to increase the current density15. This cell architecture inherently includes an inefficiency associated with back-diffusion of CO2 between the pure CO2 sweep stream and the dilute absorption stream (15% CO2). A minimum operating cell current is therefore required to overcome CO2 back-diffusion (influenced by the CO2 diffusion coefficient and inter-electrode spacing), which ultimately reduces electron utilization for the target separation. Liquid-phase mass transfer limits the achievable cell current, as net-zero pulsed flow, or convective mixing (and other active methods mentioned previously), can reduce the concentration boundary layers, although increased CO2 back-diffusion will also result. Again, additional energy requirements and the integrity of the gas-diffusion electrodes should be considered.
Incorporating fluid circulation to reduce mass-transfer resistances results in the configuration Fig. 4d, where a membrane prevents convective mixing of the bulk electrolyte. Although the presence of the membrane limits CO2 back-diffusion (depending on the chemistry, electrolyte system, membrane perm-selectivity and so on), there is still a minimum necessary current at which the cell must be operated to avoid underutilization of the redox-active sorbent. For the example separation between 15% CO2 and a 100% pure CO2 sweep stream, the anolyte enters (ideally) equilibrated with 15% CO2 and is exposed to the 100% CO2 sweep stream via a gas-diffusion layer. The cell operating current must be sufficient to raise the partial pressure of CO2 in the electrolyte to greater than that in the sweep stream. Below this level, CO2 will be absorbed from the sweep stream. This represents a similar inefficiency to the membrane-less cell, where electron utilization is compromised by all processes occurring in a single assembly. Conversely, if the volumetric production of CO2 from the oxidation reaction exceeds the transport capability of the gas-diffusion layer (or otherwise) to remove such volumes, bubble formation, and the consequences thereof, may plague operation. Additionally, the energy balances of the cell assemblies must be considered to determine the relative effect of sorption/desorption and sorbent activation/deactivation reaction enthalpies and joule heating on thermal operation. Failure to do so may conflate temperature- and electrochemically driven CO2 capture and release. The development of coupled transport, electrochemical and chemical kinetic models is necessary to guide appropriate cell assembly design and operation, as well as quantify the realized benefit of coupling all processes.
Although two-stage devices potentially offer lower energy requirements and high volumetric productivity, coupled electrochemical and transport models are necessary to guide the design and operating limits, and also quantify trade-offs. The relative simplicity of four-stage configurations and potential applications in retrofitting existing CO2 capture infrastructure warrant further investigation, where the most important chemistry-agnostic developments can be made to avoid the detrimental effects of bubble formation on cell performance.
Bubble management
Bubbles present great challenges for electrochemical processes, substantially limiting the current density by actively blocking electrode area and increasing ohmic losses47,48. The turbulence produced by bubbles may also contribute to electrode degradation and cause entrapment of gas pockets in continuous systems. The impact of CO2 bubbles on EMCC processes has only recently been highlighted, but it is an area that requires additional focus in future designs. Figure 5a presents an image of the anode outlet of an operating EMCC unit and shows the issue—the volume ratio of CO2 gas generated relative to the circulating electrolyte28. Bubble formation has been studied extensively in the context of water electrolysis and fuel cells47. Recent efforts have begun to investigate other non-aqueous electrochemical systems that involve gas49, but few have investigated EMCC systems, although similar strategies can be employed. Relevant examples of bubble remediation include passive (surface modification50, electrolyte modification51) or active (sonication, applied magnetic fields) techniques or cell design (gas-diffusion electrodes52, incorporation of hollow fibers15, or staged desorption), as shown in Fig. 5b–d. Our group has demonstrated the use of surfactants to minimize the impact of bubbles on the electrochemically mediated amine regeneration process51, as well as the use of hollow fibers to more rapidly remove CO2 from the anolyte15,53, and we have analyzed the influence of operating pressure on system energetics24. These strategies have focused on either limiting the initial formation of bubbles by removing CO2 before reaching the saturation limit, increasing the saturation limit by operating the electrochemical cell at pressure before flashing the stream to a lower pressure to remove CO2, or limiting the size of bubbles by solution engineering. This range of strategies highlights the applicability of methods investigated for bubble remediation in other electrochemical technologies for EMCC. All these strategies require additional evaluation of their practicality at scale, as active techniques bring concerns over energetic requirements, and passive techniques may substantially increase cell or system costs. The case of operation at pressure may be more challenging to investigate at the laboratory scale and require substantially more complex bench set-ups, but is quite common for larger industrial processes.

a, CO2 gas evolution from the anode of an EMCC unit during operation. b–d, Bubble management methods: active ultrasonication (b), with ‘off’ (left) and ‘on’ (right) showing cavitation and the collapse of larger bubbles due to pressure fluctuations; passive electrode surface chemistry or structure modification (c), showing a standard planar electrode with large-surface-area bubble wetting (top) and a textured electrode minimizing surface occlusion by the presence of bubbles (bottom); cell modification (d) with gas-diffusion electrodes or modified hollow-fiber electrodes for gas removal. e,f, Scaled reactor configurations: four-stage configuration with multiple electrochemical cells to meet capacity, shown with EMAR chemistry (e), and two-stage configuration at scale employing cross-flow layered hollow-fiber electrodes. The configuration in f is chemistry-agnostic. Panels reproduced with permission from: a, ref. 28, Elsevier; e, ref. 24, American Chemical Society; f, ref. 15, American Chemical Society. Panel d adapted with permission from ref. 15, American Chemical Society.
Scaling and economic considerations
The challenge of designing scaled EMCC systems is compounded by the relatively little attention given to developing system targets and metrics. To appropriately design large-scale capture systems, it is important to consider relevant metrics at target applications such that system optimization and research at earlier stages can be most relevant and ultimately useful. Important metrics to consider with given applications include the volume of gas to be processed, the desired capture fraction of CO2 in the treated gas, the availability of waste heat or cooling water, and the availability of renewable-energy sources at common target locations. Generalized techno-economic models incorporating these factors and considering electrochemical processes have not been developed but would be valuable to the field.
The discussion so far has been largely chemistry-agnostic, although when considering scaling and process economics, chemistry specifics become important. The performance of the full EMCC cycle, as well as capital and operating costs (for example, the cost, toxicity, and lifetime of the redox-active species, electrolyte components, and membrane separator; the cost and replacement time of the electrodes and ancillary equipment), should be factors guiding development even at the laboratory scale, where performance or cost trade-offs can be identified. For example, improvements in the cell energetics and stability in EMAR provided by surfactant additives may negatively impact the absorption performance due to electrolyte foaming in traditional gas–liquid contacting equipment51. Meanwhile, sorbent modification that improves cell energetics in EMAR54, and additives that avoid oxygen sensitivity with redox-active organic molecules12, increase the pumping requirements due to elevated viscosity and require solution make-up due to volatility, respectively, each representing increases in system operating expenses.
There are few studies that have considered the economics of full processes incorporating EMCC. The most advanced example available was developed for the EMAR process, for application at a coal-fired power plant24. This model incorporated a full thermodynamic and kinetic model of the electrochemical cell, requiring substantial investigation of these parameters over several previous publications22,55,56, as well as the design and optimization of the process model and separation scheme. This model highlighted several critical parameters for process cost that may be relevant to other flowing electrochemical systems, including the high cost of ionomer-based membrane separators employed in many electrochemical cells, the electrode and cell cost, and the potential for cost savings with operation at elevated temperature. The importance of membrane cost was also noted in a recent analysis of a bipolar membrane electrodialysis process57. The economics of the EMAR process were also strongly dependent on metrics common to other CO2 capture technologies, such as the swing in sorbent capacity between the loaded and unloaded state. For the electrochemical process, this swing in sorbent capacity introduced an added complexity due to the relationship between sorbent conversion and electrochemical potential and, in particular, the shift in the electrochemical potential as a function of position within the cell. This prompted the consideration of segmented electrodes or series reactors operating at potentials tailored for the sorbent conversion in that area, as depicted in Fig. 3c, and the consideration of electrochemical cell trains, like those illustrated in Fig. 5e ref. 24. This type of new research direction prompted by advanced cell modeling and process analysis is an example of the importance of such efforts.
The complexity of developing a model like that for EMAR also illustrates the importance of identifying simplified, generalized parameters that may be targeted by researchers at earlier stages. Such models and targets exist for other technologies aimed at electrochemical applications58. In the CO2 reduction literature, a commonly cited target is a current density of 100 mA cm−2 (refs. 59,60,61). This target was developed based on a number of generalized process analyses and techno-economic modeling of CO2 reduction products and cell costs58,62. We suggest that similar targets may be possible within the field of EMCC, although these will probably be dependent on system format. It is useful to determine the limiting current density in example EMCC systems as a proxy for scaling potential. This is proportional to the product of the mass-transfer coefficient and redox-active species concentration, when the redox-active species concentration at the electrode surface is maintained at zero and mass transfer is maximized. The limited solubility of some promising homogeneous sorbents32 (<100 mM) hinders their application at scale, and solubilizing agents to increase active-component concentrations can present additional diffusional limitations. For comparison, traditional amine scrubbers operate with 30 wt% sorbent to minimize the required circulation rate and associated operating expenses63,64.
For two- and three-stage capture processes where CO2 capture occurs coincidentally with sorbent activation, the electrode area and the area for CO2 uptake from the gas are coupled. This means that CO2 flux from the gas is directly related to the current density at the electrode surface. Thus, process targeting may be able to provide very direct target metrics for electrode current densities in two- and three-stage EMCC processes. As an example, to fully remove CO2 from the exhaust of a 600 MWe coal power plant at ~500 m3 s−1 and 13% CO2 with a cell area comparable to that required for traditional membrane separation techniques (1 to 10 million m2)65, a current density of ~3–30 mA cm−2 would be required. Electrochemical cell area and traditional membranes require substantially different assumptions as to cost, as the cell requires two electrodes and a separator, while the membrane requires a pressure differential and typically advanced membrane materials. Furthermore, the calculation for membranes referenced here assumes a CO2 purity near 50%, whereas electrochemical technologies may produce streams of pure CO2 (ref. 15). We encourage contributions examining process and system costs for related electrochemical technologies, such that these comparisons can be rigorously examined. We recently examined experimentally the impact of alternate geometries to produce large cell areas in tubular geometries, and similar advances have been examined in related redox flow battery systems as well15,66. The new geometries may offer greater volumetric productivity and favorable module scaling, as depicted in Fig. 5f.
Three- and four-stage electrochemical systems that decouple the electrode area from gas contacting and mass transfer require substantially different assumptions to produce targets for electrochemical system design. As discussed, the swing in sorbent capacity between loaded and unloaded states is a key design metric for such systems, and this swing dictates the necessary conversion of the electrochemical species within a single pass of an electrochemical cell28,56. Conversion of species within a flowing electrochemical system is dependent on flow rates and current, and the capture rate in steady-state operation is only dependent on the absolute current value. Targets for current density in practical systems must thus be related to flow rate or system size to be meaningful.
The differences in scaling between two- and four-stage systems should also be noted. In either technology the electrochemical cell area would scale approximately linearly with system size, meaning that the cost of the cell and related components would also vary near linearly (depending on cost reductions due to bulk modular production), because additional current can only be produced from additional cell area. That additional cell area would only operate at the same current density as the existing cell, producing a linear relationship. Systems with decoupled gas contacting stages may, however, scale differently due to the economies of scale produced with absorption columns and related separations devices that have been produced for many years. These differences between electrochemical technologies and traditional separations may enable EMCC in smaller applications that would otherwise be infeasible for large absorption columns, but may preclude them from large applications where substantial savings may be realized for traditional separations, unless alternate factors such as space constraints play an important role in design decisions. It should be noted, however, that innovations in separations system design may be inspired by the new chemistries available within EMCC, such as the large-scale hollow-fiber contactor that we recently considered67.
Outlook
Electrochemically mediated carbon capture has several promising characteristics: modularity, low energy consumption and integration with high-capacity-factor, low-carbon-intensity energy sources. With an appropriate redox-active chemistry, several system configurations are plausible, with various trade-offs. The electrochemical cell in four-stage configurations offers a simpler design that can be readily scaled to match the required CO2 flow rate in existing carbon capture infrastructure, or in the development of distributed ambient-air capture67. Two- and three-stage configurations offer potentially smaller footprints with lower energy consumption, with possible compromises in sorbent utilization. These trade-offs need to be better characterized with generalized transport models that inform scaling and economic analyses. Further work on minimizing mass-transfer resistances to improve cell current density and reducing the detrimental effects of bubble formation on these systems will enable their consideration for scaled implementation. Lastly, more general costing and scoping of EMCC systems can inform key areas of improvement and allow more direct comparisons to existing separation methods.
References
IPCC Climate Change 2021: The Physical Science Basis (eds Masson-Delmotte, V. et al.) (Cambridge Univ. Press, 2021).
Halliday, C. & Hatton, T. A. Sorbents for the capture of CO2 and other acid gases: a review. Ind. Eng. Chem. Res. 60, 9313–9346 (2021).
Zhu, P. et al. Continuous carbon capture in an electrochemical solid-electrolyte reactor - Supporting Information. Nature 618, 959–966 (2023).
Jiang, W. et al. Electrochemically regenerated amine for CO2 capture driven by a proton-coupled electron transfer reaction. Ind. Eng. Chem. Res. 61, 13578–13588 (2022).
Liu, Y., Lucas, É., Sullivan, I., Li, X. & Xiang, C. Challenges and opportunities in continuous flow processes for electrochemically mediated carbon capture. iScience 25, 105153 (2022).
Renfrew, S. E., Starr, D. E. & Strasser, P. Electrochemical approaches toward CO2 capture and concentration. ACS Catal. 10, 13058–13074 (2020).
Kang, J. S., Kim, S. & Hatton, T. A. Redox-responsive sorbents and mediators for electrochemically based CO2 capture. Curr. Opin. Green Sustain. Chem. 31, 100504 (2021).
Gurkan, B. et al. Perspective and challenges in electrochemical approaches for reactive CO2 separations. iScience 24, 103422 (2021).
Zito, A. M. et al. Electrochemical carbon dioxide capture and concentration. Chem. Rev. 123, 8069–8098 (2023).
Barlow, J. M. et al. Molecular design of redox carriers for electrochemical CO2 capture and concentration. Chem. Soc. Rev. 51, 8415–8433 (2022).
Sullivan, B. P., Krist. K. & Guard, H. E. (eds) Electrochemical and Electrocatalytic Reactions of Carbon Dioxide (Elsevier, 1993).
Barlow, J. M. & Yang, J. Y. Oxygen-stable electrochemical CO2 capture and concentration with quinones using alcohol additives. J. Am. Chem. Soc. 144, 14161–14169 (2022).
Van Daele, S. et al. How flue gas impurities affect the electrochemical reduction of CO2 to CO and formate. Appl. Catal. B Environ. 341, 123345 (2024).
Gurkan, B., Simeon, F. & Hatton, T. A. Quinone reduction in ionic liquids for electrochemical CO2 separation. ACS Sustainable Chem. Eng. 3, 1394–1405 (2015).
Diederichsen, K. M., DeWitt, S. J. A. & Hatton, T. A. Electrochemically facilitated transport of CO2 between gas diffusion electrodes in flat and hollow fiber geometries. ACS ES&T Eng. 3, 1001–1012 (2023).
Voskian, S. & Hatton, T. A. Faradaic electro-swing reactive adsorption for CO2 capture. Energy Environ. Sci. 12, 3530–3547 (2019).
Hemmatifar, A., Kang, J. S., Ozbek, N., Tan, K. J. & Hatton, T. A. Electrochemically mediated direct CO2 capture by a stackable bipolar cell. ChemSusChem 15, e202102533 (2022).
Liu, Y. et al. Electrochemically mediated gating membrane with dynamically controllable gas transport. Sci. Adv. 6, 22–24 (2020).
Shaw, R. A. & Hatton, T. A. Electrochemical CO2 capture thermodynamics. Int. J. Greenh. Gas Control 95, 102878 (2020).
Clarke, L. E., Leonard, M. E., Hatton, T. A. & Brushett, F. R. Thermodynamic modeling of CO2 separation systems with soluble, redox-active capture species. Ind. Eng. Chem. Res. 61, 10531–10546 (2022).
Diederichsen, K. M. et al. Electrochemical methods for carbon dioxide separations. Nat. Rev. Methods Prim. 2, 2354–2374 (2022).
Wang, M., Hariharan, S., Shaw, R. A. & Hatton, T. A. Energetics of electrochemically mediated amine regeneration process for flue gas CO2 capture. Int. J. Greenh. Gas Control 82, 48–58 (2019).
Wang, M. & Hatton, T. A. Flue gas CO2 capture via electrochemically mediated amine regeneration: desorption unit design and analysis. Ind. Eng. Chem. Res. 59, 10120–10129 (2020).
Wang, M., Shaw, R., Gencer, E. & Hatton, T. A. Technoeconomic analysis of the electrochemically mediated amine regeneration CO2 capture process. Ind. Eng. Chem. Res. 59, 14085–14095 (2020).
Xu, Y. et al. Assessing the kinetics of quinone-CO2 adduct formation for electrochemically mediated carbon capture. ACS Sustain. Chem. Eng. 11, 11333–11341 (2023).
Gurkan, B., Simeon, F. & Hatton, T. A. Quinone reduction in ionic liquids for electrochemical CO2 separation - supporting information. ACS Sustain. Chem. Eng. 3, 1394–1405 (2015).
Wang, M. et al. Flue gas CO2 capture via electrochemically mediated amine regeneration: system design and performance. Appl. Energy 255, 113879 (2019).
Diederichsen, K. M., Liu, Y., Ozbek, N., Seo, H. & Hatton, T. A. Toward solvent-free continuous-flow electrochemically mediated carbon capture with high-concentration liquid quinone chemistry. Joule 6, 221–239 (2022).
Rahimi, M. et al. Carbon dioxide capture using an electrochemically driven proton concentration process. Cell Rep. Phys. Sci. 1, 100033 (2020).
Rahimi, M., Catalini, G., Puccini, M. & Hatton, T. A. Bench-scale demonstration of CO2 capture with an electrochemically driven proton concentration process. RSC Adv. 10, 16832–16843 (2020).
Seo, H., Rahimi, M. & Hatton, T. A. Electrochemical carbon dioxide capture and release with a redox-active amine. J. Am. Chem. Soc. 144, 2164–2170 (2022).
Seo, H. & Hatton, T. A. Electrochemical direct air capture of CO2 using neutral red as reversible redox-active material. Nat. Commun. 14, 313 (2023).
Jin, S., Wu, M., Gordon, R. G., Aziz, M. J. & Kwabi, D. G. pH swing cycle for CO2 capture electrochemically driven through proton-coupled electron transfer. Energy Environ. Sci. 13, 3706–3722 (2020).
Jin, S., Wu, M., Jing, Y., Gordon, R. G. & Aziz, M. J. Low energy carbon capture via electrochemically induced pH swing with electrochemical rebalancing. Nat. Commun. 13, 2140 (2022).
Leitz, F. B. & Marinčić, L. Enhanced mass transfer in electrochemical cells using turbulence promoters. J. Appl. Electrochem. 7, 473–484 (1977).
Ke, X. et al. Rechargeable redox flow batteries: flow fields, stacks and design considerations. Chem. Soc. Rev. 47, 8721–8743 (2018).
Quentmeier, M., Schmid, B., Tempel, H., Kungl, H. & Eichel, R. A. Toward a stackable CO2-to-CO electrolyzer cell design─impact of media flow optimization. ACS Sustain. Chem. Eng. 11, 679–688 (2023).
Pérez-Gallent, E. et al. Overcoming mass transport limitations in electrochemical reactors with a pulsating flow electrolyzer. Ind. Eng. Chem. Res. 59, 5648–5656 (2020).
Pei, S., You, S. & Zhang, J. Application of pulsed electrochemistry to enhanced water decontamination. ACS ES&T Eng. 1, 1502–1508 (2021).
Xu, Y. et al. Self-cleaning CO2 reduction systems: unsteady electrochemical forcing enables stability. ACS Energy Lett. 6, 809–815 (2021).
Jeon, H. S. et al. Selectivity control of Cu nanocrystals in a gas-fed flow cell through CO2 pulsed electroreduction. J. Am. Chem. Soc. 143, 7578–7587 (2021).
Compton, R. G., Eklund, J. C., Page, S. D., Mason, T. J. & Walton, D. J. Voltammetry in the presence of ultrasound: mass transport effects. J. Appl. Electrochem. 26, 775–784 (1996).
Xie, X. et al. Liquid-in-liquid printing of 3D and mechanically tunable conductive hydrogels. Nat. Commun. 14, 4289 (2023).
Lee, Y. H. et al. Controlled synthesis of metal-organic frameworks in scalable open-porous contactor for maximizing carbon capture efficiency. JACS Au 1, 1198–1207 (2021).
Lee, W. H. et al. Sorbent-coated carbon fibers for direct air capture using electrically driven temperature swing adsorption. Joule 7, 1241–1259 (2023).
Singh, S., Stechel, E. B. & Buttry, D. A. Transient modeling of electrochemically assisted CO2 capture and release. J. Electroanal. Chem. 799, 156–166 (2017).
Angulo, A., van der Linde, P., Gardeniers, H., Modestino, M. & Fernández Rivas, D. Influence of bubbles on the energy conversion efficiency of electrochemical reactors. Joule 4, 555–579 (2020).
He, Y. et al. Strategies for bubble removal in electrochemical systems. Energy Rev. 2, 100015 (2023).
Leonard, M. E. et al. Editors’ Choice—Flooded by success: on the role of electrode wettability in CO2 electrolyzers that generate liquid products. J. Electrochem. Soc. 167, 124521 (2020).
Lake, J. R., Soto, Á. M. & Varanasi, K. K. Impact of bubbles on electrochemically active surface area of microtextured gas-evolving electrodes. Langmuir 38, 3276–3283 (2022).
Rahimi, M., Zucchelli, F., Puccini, M. & Hatton, T. A. Improved CO2 capture performance of electrochemically mediated amine regeneration processes with ionic surfactant additives. ACS Appl. Energy Mater. 3, 10823–10830 (2020).
Gendel, Y., Roth, H., Rommerskirchen, A., David, O. & Wessling, M. A microtubular all CNT gas diffusion electrode. Electrochem. Commun. 46, 44–47 (2014).
Hatton, T. A., Shaw, R. A., Wang, M. & Voskian, S. Methods and systems for removing CO2 from a feed gas. US patent US11446604B2 (2019).
Rahimi, M. et al. An electrochemically mediated amine regeneration process with a mixed absorbent for postcombustion CO2 capture. Environ. Sci. Technol. 54, 8999–9007 (2020).
Wang, M., Herzog, H. J. & Hatton, T. A. CO2 capture using electrochemically mediated amine regeneration. Ind. Eng. Chem. Res. 59, 7087–7096 (2020).
Stern, M. C. & Hatton, T. A. Bench-scale demonstration of CO2 capture with electrochemically-mediated amine regeneration. RSC Adv. 4, 5906–5914 (2014).
Sabatino, F. et al. Evaluation of a direct air capture process combining wet scrubbing and bipolar membrane electrodialysis. Ind. Eng. Chem. Res. 59, 7007–7020 (2020).
Orella, M. J., Brown, S. M., Leonard, M. E., Román-Leshkov, Y. & Brushett, F. R. A general technoeconomic model for evaluating emerging electrolytic processes. Energy Technol. 8, 1900994 (2020).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
Reyes, A. et al. Managing hydration at the cathode enables efficient CO2 electrolysis at commercially relevant current densities. ACS Energy Lett. 5, 1612–1618 (2020).
Verma, S., Kim, B., Jhong, H. R. M., Ma, S. & Kenis, P. J. A. A gross-margin model for defining technoeconomic benchmarks in the electroreduction of CO2. ChemSusChem 9, 1972–1979 (2016).
Kohl, A. L. & Nielsen, R. B. Gas Purification (Gulf Publishing Company, 1997).
Rochelle, G. T. Amine scrubbing for CO2 capture. Science 325, 1652–1655 (2009).
Merkel, T. C., Lin, H., Wei, X. & Baker, R. Power plant post-combustion carbon dioxide capture: an opportunity for membranes. J. Memb. Sci. 359, 126–139 (2010).
Wu, Y. et al. A submillimeter bundled microtubular flow battery cell with ultrahigh volumetric power density. Proc. Natl Acad. Sci. USA 120, e2213528120 (2023).
Diederichsen, K. M. & Hatton, T. A. Nondimensional analysis of a hollow fiber membrane contactor for direct air capture. Ind. Eng. Chem. Res. 61, 11964–11976 (2022).
Author information
Authors and Affiliations
Contributions
M.M.-H., K.M.D. and T.A.H. contributed to conceptualization, writing of the original draft, and review and editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
T.A.H. is a co-founder and Scientific Advisory Board member of Verdox, Inc.
Peer review
Peer review information
Nature Chemical Engineering thanks Klaus Lackner and Chang-Ha Lee for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Massen-Hane, M., Diederichsen, K.M. & Hatton, T.A. Engineering redox-active electrochemically mediated carbon dioxide capture systems. Nat Chem Eng 1, 35–44 (2024). https://doi.org/10.1038/s44286-023-00003-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44286-023-00003-3
This article is cited by
-
Redox-mediated electrochemical liquid–liquid extraction for selective metal recovery
Nature Chemical Engineering (2024)
-
Water management and heat integration in direct air capture systems
Nature Chemical Engineering (2024)
