
Abstract
Background
Monogenic diabetes presents opportunities for precision medicine but is underdiagnosed. This review systematically assessed the evidence for (1) clinical criteria and (2) methods for genetic testing for monogenic diabetes, summarized resources for (3) considering a gene or (4) variant as causal for monogenic diabetes, provided expert recommendations for (5) reporting of results; and reviewed (6) next steps after monogenic diabetes diagnosis and (7) challenges in precision medicine field.
Methods
Pubmed and Embase databases were searched (1990-2022) using inclusion/exclusion criteria for studies that sequenced one or more monogenic diabetes genes in at least 100 probands (Question 1), evaluated a non-obsolete genetic testing method to diagnose monogenic diabetes (Question 2). The risk of bias was assessed using the revised QUADAS-2 tool. Existing guidelines were summarized for questions 3-5, and review of studies for questions 6-7, supplemented by expert recommendations. Results were summarized in tables and informed recommendations for clinical practice.
Results
There are 100, 32, 36, and 14 studies included for questions 1, 2, 6, and 7 respectively. On this basis, four recommendations for who to test and five on how to test for monogenic diabetes are provided. Existing guidelines for variant curation and gene-disease validity curation are summarized. Reporting by gene names is recommended as an alternative to the term MODY. Key steps after making a genetic diagnosis and major gaps in our current knowledge are highlighted.
Conclusions
We provide a synthesis of current evidence and expert opinion on how to use precision diagnostics to identify individuals with monogenic diabetes.
Plain Language Summary
Some diabetes types, called monogenic diabetes, are caused by changes in a single gene. It is important to know who has this kind of diabetes because treatment can differ from that of other types of diabetes. Some treatments also work better than others for specific types, and some people can for example change from insulin injections to tablets. In addition, relatives can be offered a test to see if they are at risk. Genetic testing is needed to diagnose monogenic diabetes but is expensive, so it’s not possible to test every person with diabetes for it. We evaluated published research on who should be tested and what test to use. Based on this, we provide recommendations for doctors and health care providers on how to implement genetic testing for monogenic diabetes.
Similar content being viewed by others

Introduction
The use of precision diabetes medicine has gained increased awareness to improve diagnosis and treatment for patients with diabetes1. While the majority of those living with diabetes globally have polygenic disorders categorized as type 1 diabetes (the predominant form in those diagnosed in childhood and early adulthood), or type 2 diabetes (the predominant form in older people), approximately 1-2% have monogenic forms of diabetes, which is most commonly found in diabetes arising in neonates through to young adulthood2. Knowledge of the exact molecular defect and mechanism of disease is crucial for precision diagnostics, which informs treatment, prognostics, and monitoring. Identification of monogenic diabetes, of which there are now over 40 different genetic subtypes, have led to improved insight into the mechanism of disease and enabled precision diabetes treatment for several of these disorders, e.g., sulfonylurea agents for the treatment of KATP neonatal diabetes3,4, HNF1A-diabetes and HNF4A-diabetes5,6,7. The genetic diagnosis of any given monogenic diabetes subtype informs precision prognostics e.g., lack of microvascular or macrovascular complications in those with a heterozygous GCK etiology and informs precision monitoring particularly in syndromic forms where the genetic diagnosis precedes the development of additional clinical features such as hepatic dysfunction and skeletal dysplasia in EIF2AK3 or hearing and vision loss in WFS18,9. Thus, diagnosing monogenic diabetes presents an opportunity to identify those who would benefit from precision medicine.
There are, however, key knowledge gaps that are obstacles for precision diagnostics in monogenic diabetes. The clinical diagnosis of diabetes is based on the measurement of a single molecule, glucose. The correct classification of diabetes relies on differentiating based on overlapping clinical features such as age, body mass index (BMI), history of diabetic ketoacidosis, glycemic response to non-insulin therapies and the selective use of C-peptide and autoantibodies10. These features are less reliable for correct diabetes classification in people of non-European ancestry, in whom the prevalence of type 2 diabetes is usually greater and often occurs from a younger age than in Europeans. The term maturity-onset diabetes of the young (MODY), frequently used to refer to common monogenic diabetes has three classical criteria: autosomal dominant inheritance pattern, onset of diabetes before 25 years, and non-insulin dependence (due to residual beta cell function)11. However, these are not specific as they overlap with the clinical features seen in type 1 and type 2 diabetes12. These classical criteria are also not sensitive, since there are spontaneous mutations occurring in individuals without a family history of diabetes, autosomal recessive cases13,14,15, later onset cases of monogenic diabetes and frequent requirement for insulin treatment. The term MODY originates from the time when the terms juvenile-onset and maturity-onset were used to distinguish between type 1 and type 2 diabetes and does not precisely distinguish the various phenotypes associated with the numerous genetic etiologies for monogenic diabetes subtypes16. Recent studies show that people with monogenic diabetes are often misdiagnosed as having type 1 diabetes or type 2 diabetes17. Given the currently prohibitive cost and low yield of universal genetic testing in the vast majority with clinically classified type 1 and type 2 diabetes12,18,19,20, there is therefore a need for more knowledge on who to test for monogenic diabetes using various clinical and biomarker-based criteria that increase the yield for this diagnosis, thereby, making such genetic testing more cost-effective.
Recent breakthroughs in sequencing technologies make it possible to sequence the entire genome of a person in less than a day21,22. Routine genome sequencing may not be appropriate for diagnosing monogenic diabetes due to costs, interpretation challenges, and ethical issues in reporting incidental findings23. Less resource-demanding technologies are exome sequencing, panel exome sequencing and next-generation sequencing (NGS) using a targeted panel where many or all monogenic diabetes genes can be investigated simultaneously24. In some instances, e.g. diagnosing a known disease-causing variant in additional family members, traditional Sanger sequencing might be preferred due to economy, speed, and reliability. The use of real-time PCR such as for detecting and quantifying mitochondrial m.3243A>G variant load, droplet digital PCR for analysis of both paternally and maternally inherited fetal alleles, copy number variant analysis for detecting gene deletions and methylation sensitive assays (e.g., for 6q24 abnormalities as a common cause of transient neonatal diabetes) are all available technologies. Thus, there are knowledge gaps regarding the choice of technology being a balance between cost, time, the degree of technical, scientific and bioinformatic expertise required, and the performance/diagnostic yield in particular diagnostic settings.
Best practices have been developed on how to report genetic findings25. The results of genetic tests may, however, be challenging to interpret26. Identifying a pathogenic variant may confirm a diagnosis of monogenic diabetes, indicate that a person is a carrier of a particular genetic variant, or identify an increased risk of developing diabetes. Although a “no pathogenic variant identified” test result often excludes a common monogenic etiology, it is quite possible for a person who lacks a known pathogenic variant to have or be at risk for alternative monogenic types of diabetes–sometimes because of limitations in technology but often due to inability to anticipate all possible genes that might be involved and limitations in our ability to interpret them depending on the technology used. In some cases, a test result might not give any useful information being uninformative, indeterminate, or inconclusive. If a genetic test finds a variant of unknown significance (VUS), it means there is not enough scientific information to confirm or refute causality of monogenic diabetes, or data are conflicting27. Two expert panels have formed to develop guidelines for reviewing evidence to determine which genes (ClinGen Monogenic Diabetes Gene Curation Expert Panel [MDEP GCEP, https://clingen.info/affiliation/40016/]) and gene variants (MDEP VCEP, https://clinicalgenome.org/affiliation/50016/) are considered causative of monogenic diabetes. However, the implementation of these guidelines by the many diagnostics laboratories around the world is likely to be variable.
The Precision Medicine in Diabetes Initiative (PMDI) was established in 2018 by the American Diabetes Association (ADA) in partnership with the European Association for the Study of Diabetes (EASD)28. The ADA/EASD PMDI includes global thought leaders in precision diabetes medicine who are working to address the burgeoning need for better diabetes prevention and care through precision medicine29. This systematic review is written on behalf of the ADA/EASD PMDI as part of a comprehensive evidence evaluation in support of the 2nd International Consensus Report on Precision Diabetes Medicine30.
To investigate the evidence for who to test for monogenic diabetes, how to test them and how to interpret a gene variant, we set out to systematically review the yield of monogenic diabetes using different criteria to select people with diabetes for genetic testing and the technologies used. In addition, we sought to develop current guidelines for genetic testing for monogenic diabetes using a systematic review and grading of the studies available. The aim for this review was to fill the knowledge gaps indicated to improve diagnostics of monogenic diabetes and hence enhance the opportunity to identify those who would benefit from precision diagnostics. The evidence underpinning the link between the genetic test result and clinical management and prognostics are covered as separate systematic reviews in this series, by other members of the Precision Medicine in Diabetes Initiative (PMDI) addressing precision treatment and prognostics for monogenic diabetes.
We provide a series of recommendations for the field informed by our systematic review, synthesizing evidence, and expert opinion. Finally, we identify major challenges for the field and highlight areas for future research and investment to support wider implementation of precision diagnostics for monogenic diabetes.
Methods
Registration
We have registered a PROSPERO (International Prospective register of Systematic Reviews) protocol (ID:CRD42021243448) at link https://www.crd.york.ac.uk/prospero/. We followed the preferred reporting items for systematic reviews and meta-analysis guidelines31.
Search strategy
We focused on seven questions for our review. For the questions of whom to test for monogenic diabetes, and which technologies should be used to test them, we searched PubMed (National Library of Medicine) and Embase.com using relevant keywords and thesaurus terms for relevant monogenic diabetes categories such as MODY, neonatal diabetes, lipodystrophy, mitochondrial, combined with key genes of interest (Supplementary Table 1). Publication date limitation was set to 1990-2022, human studies only and English as a language limitation. A first search was performed in October 2021 with an update in June 2022. For the remaining questions our search strategies were adapted to recognize guidelines already in place for these areas. Details of our PICOTS framework are provided in Supplementary Table 2.
Screening
For all questions except those relating to current guidelines, we carried out screening of papers using COVIDENCE (www.covidence.org). At least two reviewers independently screened titles and abstracts of all publications identified in the searches, blinded to each other’s decisions. Conflicts were resolved by two further reviewers. All remaining articles were retrieved and screened by at least two reviewers for eligibility, recording any reasons for exclusion. Disagreements were resolved by a third reviewer.
Inclusion/exclusion criteria
For the question of whom to test for monogenic diabetes we included original research of any study design (cohort, case-control) but not case reports, in any human population with diabetes or mild hyperglycemia in whom the yield of monogenic diabetes was provided. A minimum of 100 unrelated probands with genetic testing results using sequencing of at least one or more genes implicated in monogenic diabetes had to be provided. Studies that only tested selected variant(s) within a gene or provided an association of common variants in monogenic diabetes genes with type 2 diabetes risk were excluded. Reviews, commentaries, editorials, and conference abstracts were excluded. Other reasons for exclusion were if studies only involved animal models or in vitro data. Studies which did not provide any diabetes screening measurements or those where the outcome was not a subtype of monogenic diabetes or those focusing on treatment response or prognosis were excluded.
For the question of which technologies should be used to test for monogenic diabetes we included original research of any study design where a genetic testing methodology was employed to diagnose monogenic diabetes in any human population with diabetes, where an evaluation of a genetic testing method had been undertaken. This included mitochondrial diabetes due to the m.3243A>G variant since this has recently been shown to be a common cause of diabetes in patients referred for genetic testing for monogenic diabetes32. We excluded studies using outdated or obsolete methods very rarely used by diagnostic laboratories such as single-strand conformation polymorphism analysis. Functional studies on variants, studies detecting risk variants for polygenic forms of diabetes and linkage studies to identify candidate diabetes genes were excluded. The study had to provide a clear description of the methodology used, and studies were excluded where insufficient detail was provided.
Data extraction
From each included publication, we extracted data on the first author, publication year, and the following data: type of study, country, number of individuals genetically tested. For the question of who to test we also recorded reported race, ethnicity, ancestry or country of the study, proportion female to male, BMI, other characteristics of those who were tested such as age of diabetes diagnosis, or other clinical or biomarker criteria. Where available, the extracted data also included measures of diagnostic test accuracy including sensitivity, specificity, receiver operating characteristic curve, and the area under the curve for discriminating between those with monogenic diabetes and those with other etiologies of diabetes. For genetic testing methodology the number of genes tested and gene variant curation method. For all studies we recorded the number of individuals diagnosed with different monogenic diabetes subtypes, yield by different selection approaches or genetic testing technologies if applicable.
Data synthesis
For the question of whom to test for monogenic diabetes, we summarized the total number of monogenic diabetes studies concerning neonatal diabetes, gestational diabetes, and other atypical presentations of diabetes. For each of these presentations of diabetes we grouped them according to whether they were tested for a single gene, small (2–5 genes) or a large gene panel \(\le\) 6 genes. We also summarized the studies where possible by whether they included international cohorts or those that includes individuals of predominantly European or non-European descent. If self-reported race, ethnicity or ancestry information of the population was not provided, those studies conducted in countries with predominantly non-European populations were allocated to the latter group.
Critical appraisal and grading the certainty of evidence
A ten-item checklist for diagnostic test accuracy studies33 was used to assess the methodological quality of each study by two critical appraisers, and any conflicts were resolved by a third reviewer for Questions 1 and 2. This tool is designed to evaluate the risk of bias relating to diagnostic accuracy studies using three items regarding patient selection and seven items regarding the index test. Patient selection items included whether there was a consecutive or random sample of patients enrolled (Item 1). This was interpreted as yes if the cohort described consecutive enrollment or a random sample from any given collection of individuals. For items 4-8, the index test was defined as the clinical features or biomarkers used to select individuals for genetic testing. The genetic test was considered the reference test, of which the current reference standard was decided to be at least a six-gene panel, including the genes most associated with the phenotype category. This for the neonatal diabetes phenotype category was considered to include ABCC8, KCNJ11, INS, GCK, EIF2AK3, PTF1A, and for non-neonatal beta-cell monogenic diabetes category was considered to include GCK, HNF1A, HNF4A, HNF1B, ABCC8, KCNJ11, INS and m.3243A>G. The reference standard genetic test for diabetes associated with a lipodystrophy phenotype category was considered to include at least PPARG and LMNA. Item eight, regarding an appropriate interval between the index test and the reference test to ensure that the status of the individual could not have meaningfully changed, was deemed not applicable to monogenic diabetes as the genetic test result remains stable throughout the person’s lifetime, hence a total of 9 items of this checklist were scored for each paper. We then synthesized the data from tabulated summaries and assessed the certainty of evidence by using the GRADE approach34.
The GRADE approach for diagnostic tests and test strategies was applied to answer the clinical question of who with diabetes should be offered the reference genetic test if we could not afford to provide this to everyone. The aim of the test (i.e., the clinical features and/or biomarkers) was to perform a triage function for selecting those with diabetes who had a greater likelihood of having a monogenic diabetes etiology, which when correctly diagnosed would enhance their clinical management. In assigning levels of evidence to the included studies considering various triage tests, 5 criteria were used as per the Canadian guidelines for grading evidence for diabetes studies35. Firstly, independent interpretation of the triage test results, without knowledge of the diagnostic standard (reference genetic test result) which was item 4 of the risk of bias tool. This was considered to always be the case, given that clinical features and laboratory biomarkers (triage tests) were assessed independently of the genetic testing and variant curation (reference test). Secondly, item 7 of the risk of bias tool, independent interpretation of the diagnostic standard (the reference genetic test result) without knowledge of the triage test result, was also considered to always be the case because those instances in which interpretation of the genetic test was done with more detailed knowledge of the clinical features, could not be gleaned from the papers. Whilst gene variant curation often relies on knowledge of the clinical features and laboratory biomarkers, this criterion was not deemed sufficiently informative for decisions about grading the evidence for the question of whom to offer genetic testing for monogenic diabetes. Thirdly, the selection of people suspected (but not known) to have the disorder was considered for the summary of the evidence and related to item two of the bias tool of avoiding a case-control design. Fourthly, a reproducible description of the test and diagnostic standard was considered. Finally, at least 50 patients with and 50 patients without monogenic diabetes was a key criterion that was considered. This criterion was incorporated into the inclusion criteria for studies considered relevant for the question of whom to test, by having a minimum of 100 unrelated probands with genetic testing results. However, depending on the selection criteria used, the genetic etiologies tested for and the size of the study, the number of patients who were confirmed as having monogenic diabetes did not always exceed 50 patients. To derive the overall level of evidence for the published studies for any group of triage tests, all five criteria had to be present for level 1, four criteria for level 2, three criteria for level 3 and one or two criteria for level 4 evidence. We developed guideline recommendations for whom to test for monogenic diabetes by assigning grade A for those criteria that were supported by best evidence at level 1, grade B for those that were supported by best evidence at level 2, grade C for those that were supported by best evidence at level 3 and Grade D for those that were supported by level 4 or consensus. Details of our pipeline for assessing the level of evidence and grade are outlined in Supplementary Figure 2.
Answering Questions 3 (On what basis is a gene considered a cause of monogenic diabetes), 4 (On what basis is a variant considered a cause of monogenic diabetes), and 5 (How should a gene variant causing monogenic diabetes be reported) are central to putting knowledge about monogenic diabetes etiology into practice. Currently, individual laboratories select the genes to include on NGS panels, interpret variants according to internal guidelines, and create reports based on internal procedures. Recognizing the need for clarity and consistency in these areas, several national and international guidelines have been developed and refined. It was recognized that several general resources exist for assessing whether a gene is implicated in a disease, including the crowd-sourced UKPanelApp36 and the ClinGen evidence-based Gene-Disease Validity framework37. It was also noted that the ClinGen MDEP GCEP (https://clinicalgenome.org/affiliation/40016/) has convened to apply the ClinGen evidence-based framework to monogenic diabetes. Therefore, a de novo systematic evidence review for this question was not considered necessary or useful for this document, but rather a description of these existing resources and how they can be accessed. Similar to question 3, for question 4, it was recognized that consensus guidelines for assessing the role of specific genetic variants in disease were issued jointly by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) in 201538 and the Association for Clinical Genomic Science in 2020. The ACMG/AMP guidelines have been expanded and refined by ClinGen38,39,40,41,42, and the ClinGen MDEP VCEP (https://clinicalgenome.org/affiliation/50016/) has convened to develop gene-specific rules for applying the guidelines to monogenic diabetes. For reporting genetic testing results (Question 5), there are some general published consensus guidelines38,43,44, and a limited emerging literature reporting studies evaluating report utility45 that was deemed not sufficient for a systematic evidence review. In this document, these are summarized and recommendations specific to monogenic diabetes are proposed based on existing practice.
For our evaluation of the next steps after a diagnosis of monogenic diabetes (Question 6), we excluded articles that either did not answer the question or only included a cursory general mention of the value of genetic testing for management. We reviewed the remaining 36 publications, consisting of specific case studies, cohorts, and review articles. Twelve papers discussed monogenic diabetes testing and/or treatment in adults and children. Seven articles described strategies for testing and/or management of monogenic diabetes during pregnancy. Three articles focused on maternally inherited diabetes and deafness (MIDD), five centered on neonatal diabetes, and nine covered syndromic forms of monogenic diabetes, including Wolcott-Rallison, Alström, and Wolfram syndromes. We then reviewed the literature for additional published studies relating to the steps after monogenic diabetes diagnosis. Information from publications was combined with expert advice from genetic counselors and physicians who specialize in monogenic diabetes clinical care. This section includes recommendations for results disclosure, cascade testing and addressing non-medical issues that may arise. We direct the reader to other systematic reviews in this series for prognostics and treatment recommendations. To evaluate the challenges for diagnostic testing for monogenic diabetes (Question 7) we screened 455 abstracts for challenges for the field of monogenic diabetes diagnosis of which 41 were screened as full text articles and 14 taken forward for full text extraction.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Question 1—Who to test for monogenic diabetes?
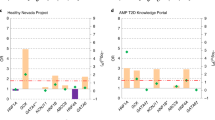
For the question of who to test for monogenic diabetes, a total of 12,896 records were retrieved. In Covidence, 2,430 duplicates were identified. We included 100 publications from 10,469 publications screened (Supplementary Fig. 1A). The key data from each of the 100 studies were included in Supplementary Data 1 and the 10-item checklist assessments for these papers were summarized in Fig. 1A. The summary of evidence from the included studies is detailed in Tables 1–2. We also provide a list of recommendations based on this evidence in Table 3.

A Results for papers from question 1. B Results for papers from question 2. The horizontal axis for the heatmap refer to questions 1–10 of the 10-item JBI checklist. Green is Yes, Red is No, Gray is unclear. Non-applicable answers were left blank. A completes list of papers in (A, B) can be found in Supplementary Data 1 and 2.
In neonatal diabetes there were a total of 13 studies, of which three included those diagnosed with diabetes within 24 months of age, three within 12 months of age and the rest within six months of age (Table 1). There was only one study which used the reference standard large gene panel for neonatal monogenic diabetes diagnosis, while the rest did not. The highest yield of 82% was obtained in a single international cohort study of 1,020 patients diagnosed with diabetes within six months of age using a large 23-gene panel46. Of these, 46% had KCNJ11 or ABCC8 followed by INS as the next common etiology.
For neonatal diabetes diagnosed between 6–12 months the yield was 0-28% derived from six studies containing sample sizes of 18 to 145 individuals tested using only KCNJ11, ABCC8, INS genes. No cases of monogenic diabetes were found in the small subpopulations tested with diabetes diagnosed 12-24 months in three studies sequencing KCNJ11 and INS genes only (n = 58–70). The risk of bias criterion for patient selection was high for one study in which a random sample of patients had not been enrolled47. Two studies were deemed to be at risk of bias due to not all receiving the reference test48,49 (Fig. 1A).
The selection by age of diabetes diagnosis between six and 12 months for neonatal diabetes genetic testing was supported by level 1 evidence from 1 large study and thereby supports this being a Grade A recommendation (Table 3). Selection by age of diabetes diagnosis between 6 and 12 months was supported by a yield of 6% to 28% by level 2 evidence from six studies, although these were limited by only testing for INS or KCNJ11 and ABCC8. Selection by age of diabetes diagnosis beyond 12 months for monogenic diabetes testing was not supported by three studies examining those diagnosed with diabetes up to 24 months. These failed to find any cases of monogenic diabetes although these were limited by only testing for KCNJ11 or INS etiologies in small cohorts with diabetes diagnosed between 12-24 months (level 2 evidence).
In gestational diabetes mellitus (GDM), the recommendation that all women without obesity should be tested for GCK etiology (Table 3) were derived from a total of four studies which examined GCK diagnosis only, of which three were in predominantly European women50,51,52, and one study was in Chinese women53 (Table 1). The yield for GCK etiology ranged from 1–6% in otherwise unselected women with GDM, however, increased to 22% when only non-obese women were selected for GCK testing52. The use of fasting glucose of 5.5 mmol/L or higher was demonstrated to have the highest yield (3/118) for GCK, with only 1/129 women with fasting glucose below 5.5 mmol/L found to have GCK etiology and 0/109 women with fasting glucose below 5.1 mmol/L50. In this 2014 study the trade off between specificity and sensitivity of detection for GCK among the women with GDM (and fasting glucose above 5.5 mmol/L), by various BMI thresholds is shown (Table 1). Given none of these studies included over 50 women with GCK who had been suspected but not known to have the disorder in the study, the highest level of evidence was graded at level 2. Other than only testing for the single gene GCK, there were no other concerns about bias in these studies.
For GCK testing in those without GDM, the recommendation to provide this for those with persisting, mild hyperglycemia at any age, in the absence of obesity (Table 3) is based on a total of 13 studies of which 11 were in predominantly European populations (Table 1). Overall, there was frequent assessment of bias in patient selection criteria used in all but one study. There were 5 studies with either unclear or no defined thresholds provided for persistency or stability of mild hyperglycemia using various fasting glucose and or HbA1C thresholds. The yield for GCK etiology ranged from 0% in unselected cases of hyperglycemia and increased to 30-74% in those with persistent, stable, mild hyperglycemia (Table 1). There was only one Italian study of 100 individuals that compared two testing strategies54. This study demonstrated that the yield for GCK increased in those with impaired fasting glucose and without diabetes autoantibodies from 32% when one MODY criteria was added compared to 88% when non-obese and lack of diabetes medications were added. However, this study characteristics provided level 3 evidence. The yield for GCK-etiology in a Chinese study which used mild, fasting hyperglycemia and low triglycerides was relatively low (2% vs 0.5% in discovery and replication datasets of n = 545 and n = 207 respectively)55. However, in a mixed ethnicity population in the USA, selection of those with persistent, mild fasting hyperglycemia plus either family history or BMI below 30 kg/m2 or diabetes diagnosis age below 30 years produced a yield of 55%56. Overall, four studies supported level 1 evidence for selecting those with persisting, mild, fasting hyperglycemia for GCK testing.
The recommendation to provide monogenic diabetes testing to people without obesity under the age of 30 years who are either autoantibody negative and/or have retained C-peptide levels was derived from a total of 60 studies. These examined the yield of monogenic diabetes beyond the neonatal period, of which 43 were in predominantly European populations. Of these, 25 studies utilized the reference standard of the large-gene panel (8 in non-European populations). The yield varied by the triage test strategy utilized to select individuals for genetic testing and those receiving the large-gene panel had a greater yield than smaller or single-gene approaches. Younger age of diagnosis of diabetes (thresholds included below 15, below 18, below 25, below 35 and below 40 years) and negative diabetes autoantibodies were the most common triage test strategy. Excluding those with type 1 diabetes using either negative diabetes autoantibodies or presence of C-peptide or both was frequently employed. With the large-gene panel approach, the yield for a monogenic etiology ranged from 0.7% to 34%. There was low yield of 18/2670 (0.7%) in those with negative antibodies who had diabetes diagnosed above the age of 40 years57. In suspected MODY cohorts, the yield was 16% to 23% (Table 1). Most of such studies were assessed as having bias in patient selection and many did not have a clear description of “suspected MODY” (Fig. 1B). One French study of 1564 individuals provided the comparative yield for (a) 3 clinical criteria of diabetes diagnosis age of 15–40 years, BMI below 30 kg/m2, and family history of diabetes which was 20% vs (b) for any 2 of these clinical criteria the yield was 16% vs (c) diabetes diagnosis of 15–40 years and BMI below 25 kg/m2 the yield was 34%58. In a Turkish cohort of children with diabetes (diabetes diagnosis age IQR 5–12 years), with either low Type 1 diabetes genetic risk score (T1GRS) or moderate T1GRS and negative diabetes autoantibodies the yield was yield of 34/236 (14%). This included 14/34 autosomal recessive cases, with approximately 20% prevalence of consanguinity in the tested population59. While there was considerable heterogeneity in selection criteria used, the best evidence was at level 1 for selecting those diagnosed with diabetes below the age of 30 years who are either autoantibody negative/and or have retained C-peptide (for lowering probability of type 1 diabetes) and those without obesity (for lowering probability of type 2 diabetes), for testing for monogenic diabetes using the reference large gene panel.
In people with diabetes, there were 2 studies which evaluated the yield for PPARG or LMNA etiology. There was low yield of 0.6% for PPARG etiology in a Chinese study of people diagnosed with diabetes between 18–40 years who were antibody negative and C-peptide positive60. However, in those selected on the basis of lipodystrophy and/or severe insulin resistance, the yield was 9.7% for LMNA etiology in a French study61.
For HNF1B yield in populations selected for diabetes, there were 4 studies (Supplementary Data 2, Fig. 1B). A yield of 2.4% and 2.9% for HNF1B etiology for diabetes was found in a UK and Chinese study respectively. In the UK study, those referred for monogenic diabetes who had no other common etiology detected received HNF1B sequencing and doseage analysis. In the Chinese study, older adults with diabetes who were antibody negative and had either renal structural abnormalities or impaired renal function, were tested.
Besides the yield of monogenic diabetes using simple clinical criteria, the sensitivity and specificity of various clinical risk scores and biomarkers are summarized in Table 2. The clinical risk scores have been derived for either a single genetic etiology (eg: HNF1B risk score)62,63 or a group of genetic etiologies collectively (eg: “MODY risk calculator” for subtypes GCK, HNF1A, HNF4A)63. The utility of routine biochemical biomarkers such as hsCRP or antibodies have been investigated for distinguishing several genetic etiologies (HNF1A, HNF4A, HNF1B) from Type 1 or Type 2 diabetes, or for distinguishing HNF1A specifically from Type 2 diabetes using HDL-cholesterol or hsCRP64,65,66,67,68,69. Low T1GRS has been used as a triage test in addition to negative diabetes antibodies for selecting individuals for broad monogenic diabetes panel testing59,70. Other non-routine biomarkers such as lipid fractions and glycan moieties regulated by HNF1A have been explored for distinguishing HNF1A from other diabetes subtypes, but these have added complexity and cost above clinical features71,72, without better informing whom to test for the greater yield of monogenic diabetes provided from large-gene panel testing.
The use of 8 simple clinical features in the logistic regression model based MODY risk calculator distinguished the 3 common monogenic diabetes subtypes GCK, HNF1A, HNF4A collectively from Type 1 diabetes or Type 2 diabetes with a c-statistic of 0.98 and 0.95 respectively. This indicates excellent discrimination between Type 1 diabetes or Type 2 diabetes and the 3 common monogenic diabetes subtypes GCK, HNF1A, HNF4A collectively, as well as when comparing Type 1 and Type 2 diabetes with HNF1A/4A and GCK etiologies separately. However, limitations of this calculator include lack of validation for non-European poluations, those diagnosed with diabetes above 35 years, those with other forms of monogenic diabetes, and weaker performance in insulin-treated patients where the probability of type 1 diabetes is high. The use of negative antibodies and detectable C-peptide biomarkers to exclude Type 1 diabetes and select individuals for monogenic diabetes testing had higher yield than the use of MODY probability calculator pre-test probability of >25%. The latter had a high PPV (40%), but missed more cases (55%) compared with the antibody/C-peptide biomarker pathway (PPV 20%). Traditional MODY criteria (age at diabetes diagnosis less than 25 years, non-insulin requiring and a parent affected with diabetes) had a PPV of 58%, yet missed even more cases (63%) compared with the biomarker pathway.
Question 2—How to test for monogenic diabetes?
For the question of which technologies should be used to test for monogenic diabetes, we included 32 studies from 2,102 publications screened (Supplementary Fig. 1B). A total of 32 studies which accessed 76 different genes were analyzed (Supplementary Data 2, Table 4) and assessed for methodological quality (Fig. 1B). NGS was the most used technique, with 16/22 NGS studies using a targeted panel. Where NGS was employed, the monogenic diabetes diagnostic yield increased by around 30% compared to Sanger sequencing of GCK, HNF1A and HNF4A alone, and resulted in the (often unexpected) diagnosis of rare syndromic forms of diabetes, most commonly m.3243A>G. NGS technologies also enabled the diagnosis of multiple monogenic subtypes in the same patient, and diagnosed patients who were missed by previous Sanger sequencing due to allelic drop-out. Gene agnostic exome and genome strategies were rarely used and did not increase diagnostic yield. Copy-number variant (CNV) analysis (by Multiplex-Ligation-Dependent Probe Amplification [MLPA] or NGS) increased diagnostic yield mostly by detecting HNF1B deletions. Non-coding variants were rare but important findings and required genome sequencing or specific targeting of non-coding mutation loci. A high diagnostic yield (74%) was reported when performing Sanger sequencing of GCK in patients with a clinical suspicion of GCK due to persistent, mild, fasting hyperglycemia. Similarly, variants in KCNJ11, ABCC8 and INS accounted for 50% of neonatal diabetes mellitus (NDM) cases and were sequenced by Sanger first in some studies. 6q24 abnormalities were also a common cause of NDM and required a specific methylation-sensitive assay to detect them. Recessively inherited and syndromic forms of monogenic diabetes were predominant in countries with high rates of consanguinity. Real-time PCR and pyrosequencing were highly sensitive and specific techniques for detecting m.3243A>G and quantifying heteroplasmy, and ddPCR successfully determined all fetal genotypes in a cell-free fetal DNA prenatal testing study of 33 pregnancies. Based on our systematic review of the literature we can make several recommendations (Table 5).
Question —What is the basis for considering a gene as a cause of monogenic diabetes?
A general evidence-based framework for evaluating gene disease validity has been developed by the ClinGen and published by an inter-institutional group of clinical and molecular genetics and genomics experts37. This framework involves evaluating case level, segregation, and functional data for previously reported variants and functional data for the gene itself to classify gene-disease validity relationships into Definitive, Strong, Moderate, Limited, Disputed, or Refuted categories based on a point system combined with expert consensus for the final assignment. Tools for implementing this are available at the ClinGen website. The international MDEP GCEP has convened with the goal of curating gene-disease validity for monogenic diabetes genes and has completed the common genes (https://www.clinicalgenome.org/affiliation/40016/) and is working on expanding beyond these genes. Other general repositories for gene-disease validity curation include the crowd-sourced Genomics England PanelApp36. For monogenic diabetes, a curated list of monogenic diabetes genes is available at the website for the University of Exeter, where most of the research and clinical monogenic diabetes testing for the UK is conducted (https://www.diabetesgenes.org/).
Over recent years the increased availability of high throughput sequencing has led to a substantial increase in the number of genes reported to cause monogenic diabetes. The evidence that supports these gene-disease relationships does, however, vary widely. Whilst there is overwhelming genetic evidence that established the etiological role of genes such as HNF1A, HNF4A and GCK, recent studies that have investigated variation in genes such as BLK, KLF11 and PAX4 in large population datasets have not supported their role in causing monogenic diabetes73, and these genes were recently refuted as monogenic diabetes genes by the MDEP GCEP.
The consensus opinion of the writing group was that a gene should only be considered causative of monogenic diabetes if it meets the criteria set out in expertly curated guidelines that have been developed to validate gene-disease relationships. These guidelines have already been applied to many of the monogenic diabetes genes by the ClinGen MDEP GCEP. We recommend continued efforts to curate new and updated existing monogenic diabetes genes for gene-disease validity be centralized with the MDEP GCEP. Those interested in contributing to this effort should engage with the MDEP GCEP to ensure that genes used in monogenic diabetes have been curated for gene disease validity in a process that is evidence based and updated on a standard schedule as directed by ClinGen.
Question 4—On what basis should a variant be considered a cause of monogenic diabetes?
In 2015, the ACMG and AMP developed general guidelines for the interpretation of sequence variants38. The ClinGen Sequence Variant Interpretation (SVI) Working Group has published multiple updates to these original guidelines39,40,41,42. The Association for Clinical Genomic Science (ACGS) voted to adopt these guidelines74. These guidelines have undergone several updates. ClinGen’s MDEP VCEP has modified these general guidelines for three common monogenic diabetes genes (HNF1A, HNF4A and GCK); these guidelines account for many issues inherent in the difficulty in interpreting monogenic diabetes variants and can be used as a framework for interpreting variants in genes for which rules have not yet been established.
The ACMG/AMP guidelines were developed through an evidence-based process involving the sharing, developing, and validating of variant classification protocols among over 45 laboratories in North America. They incorporate various types of evidence to determine if a variant is pathogenic, likely pathogenic, of uncertain significance (VUS), likely benign, or benign. Examples of the types of evidence include: frequency in public databases such as gnomAD; the segregation of a variant with a disease phenotype; results of computational (in silico) prediction programs; de novo status; functional studies; frequency of variant in cases vs. controls; the presence of other pathogenic variants at the same nucleotide or within the same codon; the location of a variant (i.e., if it is within a well-established functional domain or mutational hotspot); and whether a variant has been found in a patient with a phenotype consistent with the disease. MDEP gene-specific rules incorporate experts’ unpublished case data and knowledge of monogenic diabetes phenotype and prevalence in recommending the evidence and thresholds to apply.
Continued work by MDEP VCEP is needed to develop applications of the guidelines tailored to additional monogenic diabetes types and genes. Improvement in de-identified case-sharing platforms is needed to promote maximizing the ability to gather the evidence needed to evaluate pathogenicity.
Question 5—How should a variant in a monogenic diabetes gene be reported?
Well written general guidelines for the reporting of genetic test results are available43,44,75,76,77,78,79 and this review will therefore summarize the basic requirements and focus on reporting monogenic diabetes tests.
We summarize the recommendations for reporting results for a range of different testing scenarios and methodologies (Table 4). A single page report with appendices is preferred. The report should restate the reason for testing, including the clinical characteristics/phenotype of the patient. The report must include a headline result or summary that clearly states the outcome of the test for the patient – this may be stating whether a diagnosis of monogenic diabetes has or has not been made, or whether a patient is or is not genetically predisposed to monogenic diabetes. Patients with specific subtypes may respond well to certain therapies and this should be noted in the report. Testing should be offered to at-risk family members, which may be diagnostic, predictive or carrier testing. Special care should be taken when reporting variants in syndromic diabetes genes in patients with isolated diabetes. The risk to future offspring should be stated according to mode of inheritance. The report should not use terms positive or negative for describing test results. Variants should be reported in a table that includes the HUGO gene name, zygosity of the variant, both nucleotide and protein level descriptions using HGVS nomenclature, genomic coordinates and the classification of the variant based on the ACMG/AMP 5 level classification system38. Benign and likely benign variants should not be reported. Class 3 (VUS or VOUS) variants should be reported based on professional judgment, the level of supporting evidence and on whether additional investigations can be undertaken to change the classification such as testing of other affected relatives, further biochemical testing, or additional functional laboratory investigations. Evidence used to classify the variant should be clearly outlined. Technical information should be provided in a section separate from results and interpretation and will include details of the methodology and gene or genes tested. If the testing performed does not cover all known genes and possible mutations, then this should be stated as a limitation with recommendations for further genetic testing (e.g., NGS or MLPA analysis). Laboratory reports should avoid the terminology “MODY” given its lack of precision in sensitivity or specificity for any given genetic etiology of monogenic diabetes. Instead the use of the gene name hyphenated with hyperglycemia, diabetes severe insulin resistance, lipodystrophy or syndrome as appropriate is recommended. Examples are GCK-related hyperglycemia, HNF1Adiabetes, INSR-severe insulin resistance, PPARG-lipodystrophy, mt.3243 A > G syndrome. Alternatively, the term “monogenic diabetes” followed by “subtype gene name” eg: monogenic diabetes subtype HNF4A may be used.
The structure, format, and content of monogenic diabetes testing reports will vary widely between laboratories across the world. Standardization is difficult due to variability in mandatory report content, such as legal disclaimers, and the ability to include clinical recommendation. But there are essential reporting best practices that should be adopted by all laboratories irrespective of local reporting policies. We recommend that laboratories performing monogenic diabetes testing participate in the EMQN’s monogenic diabetes EQA scheme (www.emqn.org) which aims to educate and improve quality of diagnostic testing and reporting for this condition. Future research is advised to engage patients, providers, and other stakeholders in the design and evaluation of readability, comprehension, and application of information contained in genetic testing reports for monogenic diabetes.
Question 6—Research Question: What are the next steps after a diagnosis of monogenic diabetes?
A systematic, comprehensive, and collaborative approach is required after making a monogenic diabetes diagnosis after conducting genetic testing. Our guidance for the next steps after diagnosis of monogenic diabetes focuses on the following: (1) practical recommendations for providing the diagnosis results and clinical follow-up, (2) reviewing genetic testing reports, (3) family testing for adults and children, (4) legal considerations for this diagnosis, (5) considering psychological impact of diagnosis, and (6) recommendations for addressing VUS results and negative monogenic diabetes testing despite atypical features to a patient’s diabetes presentation. In the following paragraphs, the term “clinician” can refer to a physician or genetic counselor. Genetic counselors are specially trained to communicate complex genetic information, facilitate family testing, and address psychosocial issues that may arise with a new diagnosis; thus, we recommend having a genetic counselor as part of the care team if possible. Upon receipt of a genetic test result diagnosing monogenic diabetes (i.e., pathogenic, or likely pathogenic variant identified), the clinician should schedule a 30–60 min in-person or telehealth appointment with the patient/family80. We do not recommend that results be disclosed via an electronic health record (EHR) portal or by non-clinical staff.
After a very brief reminder of what the genetic test analyzed, we recommend the clinician describe the identified variant in patient-friendly language (e.g., a single spelling error in the genetic code) and review how disease-causing variants in the gene impair glucose metabolism. The clinician can explain the evidence used to classify the variant as disease-causing which is often included in the genetic testing report, e.g., if the variant was previously identified in patients with monogenic diabetes or experimental evidence demonstrated loss of function. The clinician should describe the general features of the type of monogenic diabetes indicated by the genetic change, including the inheritance pattern of the disorder, specifying those features that are consistent with the patient’s clinical picture. If the type of monogenic diabetes is characterized by variable expressivity and/or reduced penetrance, these concepts should be introduced to the patient/family, providing specific examples from the disorder at hand. HNF1B syndrome is a prime example of variable expressivity, as the renal and extra-renal phenotypes (diabetes, genital malformations, pancreatic hypoplasia, abnormal liver function) vary among affected individuals, even within the same family81,82. The patient/family should be provided a copy of the report for their records. Additionally, a document describing the variant identified and avenues for variant-specific testing can be provided to the patient to distribute to family members if family testing is being pursued. Upon reflecting on the diagnosis, patients may feel relief at a genetic etiology for their symptoms, while others may feel angry or annoyed if they were initially misdiagnosed and prescribed suboptimal treatment83,84,85,86,87,88. Feelings of frustration should be validated. Some patients may find solace in hearing that knowledge and testing of monogenic diabetes have both evolved greatly over time and we hope more diagnoses will be made moving forward. Patients may also be helped by speaking to other patients with monogenic diabetes. At this time, formal support groups are limited for monogenic diabetes, but the provider can consider connecting patients with monogenic diabetes given mutual consent. Patients, providers, and researchers are in the process of creating a consortium for communication and support regarding monogenic diabetes called the Monogenic Diabetes Research and Advocacy Consortium (MDRAC, mdrac.org). Yearly follow-up can be suggested to continue to provide updates on the monogenic diabetes diagnosis, prognosis, and treatment in addition to any new information on the gene and genetic variant identified.
Results of genetic testing should be discussed in context of the family history. The most common forms of monogenic diabetes, HNF1A, HNF4A, and GCK etiologies, are dominantly inherited, and the vertical transmission of diabetes or hyperglycemia is often evident in the pedigree8,89,90,91. If a disease-causing variant in one of these conditions is identified in a parent of an affected individual, there is a 50% chance that siblings and children of the proband will inherit the variant. The absence of a family history of diabetes may suggest that a variant associated with a dominant condition is de novo in the proband. If parents test negative and maternity and paternity are confirmed, the recurrence risk in siblings is approximately 1%, which accounts for the possibility of gonadal mosaicism92. De novo disease-causing variants have been reported and are especially common in HNF1B81,93,94. With HNF1B etiology, the family history may also include genital tract malformations, renal cysts, or pancreatic hypoplasia81. Recurrence risk of recessive forms of monogenic diabetes, such as Wolcott-Rallison syndrome (due to EIF2AK3 mutations) or Thymine Responsive Megaloblastic Anemia (TRMA) syndrome, is 25% in offspring when both the proband and their partner are carriers89. Recurrence risk of monogenic diabetes caused by the mitochondrial DNA MIDD (maternally inherited diabetes and deafness) variant (m.3243A>G) is essentially zero when the sperm-producing parent has the variant, as mitochondria are passed down through the oocyte. All offspring and maternal relatives of the egg-producing parent will inherit the variant, albeit at varying heteroplasmy95.
Affected family members of individuals with molecular confirmation of monogenic diabetes should be offered variant-specific testing of the familial variant, a process known as cascade testing8,80. For probands with GCK- related hyperglycemia, it is important to also discuss cascade testing of family members with gestational diabetes and pre-diabetes, since this is characterized by stable, mild fasting hyperglycemia that is clinically asymptomatic and can also impact pregnancy management in a gestational parent with apparent GDM. Aparently unaffected or undiagnosed first-degree adult relatives of probands with GCK etiology should be counseled that if they wish to undergo a fasting glucose test; if normal, a diagnosis of GCK- hyperglycemia is highly unlikely and genetic testing is unnecessary8,96,97. If they have an elevated fasting glucose test, then they should undergo a cascade genetic test to clarify whether GCK is the etiology.
The risks and benefits of testing, and possible results of testing, should be reviewed in all cases to allow the family to make autonomous testing decisions consistent with their goals and values. Possible benefits of genetic testing include the ability to obtain or advocate for more appropriate treatment, reduced anxiety, and uncertainty, decreased stigma, knowledge of recurrence risk, and the ability to plan for the future80,83,87,98. Risks may include increased anxiety, trouble adjusting to a new diagnosis, or learning unexpected information85. The risk of insurance discrimination may also need to be reviewed, as different countries have instituted varying rules regulating the use of genetic information in insurance underwriting.
Additional ethical and psychosocial issues surrounding a genetic diagnosis should also be discussed when considering predictive testing in a minor. The clinical relevance of an HNF1A or HNF4A positive genetic test would likely have minimal clinical relevance prior to adolescence, and thus we generally discourage genetic testing in young children. Indeed, adolescents in families with HNF1A diabetes preferred testing in adolescence when parents and their children can engage in joint decision-making regarding genetic testing84. We do not recommend testing asymptomatic children for GCK, given this is a benign condition and there are potential adverse psychosocial effects of being labeled as “sick”98. Also, the “GCK related hyperglycemia” diagnosis can lead to problems achieving life, long-term care, or disability insurances since this may be classified as a monogenic form of diabetes. However, in practice this is a benign condition that often fulfills the glucose criteria for prediabetes rather than diabetes and does not impact any increased cardiovascular risk or progression to type 2 diabetes that is frequently seen in people with prediabetes. If a child in a family with a GCK etiology is incidentally found to have hyperglycemia, their pediatrician should be informed of the familial variant and familial variant testing can proceed to avoid unnecessary treatment.
These strategies also respect the autonomy of the child to make an informed decision about testing when they are able. However, the significant fear of uncertainty that some parents of at-risk children feel should not be dismissed. Genetic testing may decrease anxiety in parents, allow them to gradually introduce the disorder to their child in a developmentally appropriate manner, and empower them to prepare for the future83,98.
A positive result of genetic testing would replace the prior diagnosis (of type 1 or type 2 diabetes) with a diagnosis of monogenic diabetes. The clinician should review the prognosis of the condition and potential changes in medical management (e.g., no treatment in GCK- hyperglycemia and sulfonylurea treatment with HNF1A and HNF4A monogenic diabetes8,97. We refer the reader to the recommendations generated by the Monogenic Diabetes Precision Prognostics and Therapeutics groups for additional information which we also provide a high-level summary of in Fig. 2.

Examples of genetic forms of diabetes identified through precision diagnostics and how these lead to precision treatment and prognostics. Current gaps and challenges identified through the systematic review are highlighted.
There may be instances when an asymptomatic family member has a positive result on genetic testing. In this case, the high risk of developing diabetes or hyperglycemia should be emphasized and a plan for monitoring blood glucose should be developed if appropriate. Of course, an asymptomatic family member with negative variant-specific testing may still be at risk of developing more common forms of diabetes80. A notable exception to this is that asymptomatic family members, with negative genetic testing of probands with MIDD, are still at risk for diabetes, hearing loss, and potentially other symptoms of mitochondrial disease given the variability in heteroplasmy of the m.3243A>G variant among different body tissues95. A negative result of variant-specific testing in a family member with diabetes would indicate another etiology for the diabetes diagnosis, such as type 1 or type 2 diabetes.
Question 7—What are the current challenges for the field in precision diagnostics for monogenic diabetes?
We reviewed the abstracts of 455 abstracts and the full text of 42 papers before extracting data from 14 articles meeting our criteria. Several key themes emerged which present on-going challenges for the field of precision diagnostics for monogenic diabetes. With the generation of exome and now genome sequencing data from both larger clinical cohorts and biobanks we have new insights into variant frequency and penetrance. For some previously reported pathogenic variants in monogenic diabetes genes there is now evidence for reduced penetrance in unselected populations and that individuals carrying the variants do not necessarily display the hallmark characteristics (e.g BMI < 30 kg/m2, age of diagnosis <35 years) of monogenic diabetes99,100,101,102. The interpretation of novel rare variants in monogenic diabetes genes is challenging, functional studies can assist but multiple assays are required in concert with frequency and clinical data103,104. Functional studies are slow, lack standardization and are usually retrospectively performed after variant discovery. Efforts to generate variant maps in genes of interest are a potential route forward but will require coordination and implementation of standards105. The perpetuation of errors in the literature remains a concern with ongoing reporting of novel variants in genes which are not considered by experts in the field to be causal for monogenic diabetes106. Whilst the reporting of potential novel genes can be misleading as they do not necessarily meet the criteria for classification as a novel genetic cause of diabetes107. There remain inequalities in sequencing data across diverse ancestries and populations even when there are examples of the importance of rare variation in monogenic diabetes genes108,109,110.
Finally, barriers to genetic testing remain including limited provider awareness of monogenic diabetes. It is important that all clinicians treating diabetes patients are considering monogenic etiologies as a potential diagnosis, especially when diagnosis can occur in adults that have had diabetes since youth111. Future research should focus on increasing representation of sequence data in monogenic diabetes genes in diverse populations, generating variant maps of clinically actionable diabetes genes and continued efforts to share knowledge and expertise of monogenic diabetes in underserved communities and populations.
Discussion
To diagnose monogenic diabetes offers an opportunity to find those who can benefit from precision medicine89,112. This systematic review has summarized and quantified data on the yield of monogenic diabetes detection using various selection criteria in different populations with diabetes. The greatest yield for monogenic diabetes is in those diagnosed with diabetes in the first 6 months of age, when the background prevalence of type 1 diabetes is low. There is progressively lower additional yield in those diagnosed between 6–9 months, or 6–12 months or above 12 months. Dominantly inherited subtypes such as KCNJ11, ABCC8 and INS account for the majority of neonatal diabetes in non-consanguineous families, while in consanguineous populations recessively inherited subtypes such as EIF2AK3 are more common. The ongoing detection of the commonest subtype of neonatal diabetes due to KCNJ11/ABCC8 mutations who can be transferred to inexpensive sulfonylurea treatment makes testing all those below 12 months still cost-saving, so long as 3% of those screened have a monogenic diabetes diagnosis that is treatment-changing8. Further large-scale studies need to be performed using large-gene panels in those with diabetes diagnosed between 12 and 24 months to ascertain further triaging features that could improve the yield of diagnosis of monogenic diabetes cases amongst the increasing majority who would have type 1 diabetes during early childhood. The prevalence of type 2 diabetes is generally only seen with severe obesity in early childhood with onset generally well above 24 months of age, so excluding obesity in those selected for genetic testing is less of a consideration in this very low age group. With increasing age of diabetes diagnosis, there are additional criteria required to lower the probability of type 1 diabetes (such as autoantibody negative and or retained C-peptide), and type 2 diabetes (such as the absence of obesity).
The recommendations for restricting testing to the selected groups of individuals with diabetes provided in this systematic review, have considered the highest diagnostic yields reported in the literature, balancing high specificity of clinical and/or biochemical features with high sensitivity for monogenic diabetes. As sensitivity for identifying monogenic diabetes subtypes increases with the use of less stringent clinical characteristics, the number needed to test to find one positive case increases, so choosing absolute thresholds of key characteristics is a matter of balancing the costs of the genetic test, against the benefits of correctly identifying individuals with monogenic diabetes. Different health systems should consider adapting these recommended thresholds for their population contexts to allow for systematic genetic testing in those with diabetes whose probability of having a monogenic diabetes etiology meets their cost-effectiveness threshold for implementing through clinical care pathways for diabetes.
Currently, we cannot afford to offer a genetic test to everyone with diabetes. Such an “all testing” approach would find every case of monogenic diabetes but would have a low diagnostic yield. In a USA study, monogenic diabetes genetic screening approach was modeled as being cost-effective at 6% yield and cost-saving at 30% yield for GCK, HNF1A, HNF4A, with an estimated combined prevalence of 2%, implicating the most clinically actionable changes to therapy113. In a UK study, selecting individuals for genetic testing among those diagnosed below 30 years, using either a clinical prediction model or a biomarker strategy (with negative antibodies and retained C-peptide) was deemed cost-saving, assuming cost-benefit from stopping insulin treatment for misdiagnosed type 1 diabetes in this age group114. This model assumed a prevalence of the most cost-saving monogenic diabetes types as 2.4% (GCK 0.7%, HNF1A 1.5% and HNF4A 0.2%). Clearly, once monogenic diabetes has been identified in a proband, cascade screening of family members maximizes cost-savings, given the yield is at least 50% in autosomal dominant subtypes of monogenic diabetes.
The use of NGS targeted large-gene panel is the recommended testing technology with variant curation developed by ClinGen. However, the high diagnostic yield of GCK etiology in patients with persistently mild fasting hyperglycemia means Sanger sequencing of this gene alone may be offered for such individuals. Laboratory reports should replace the imprecise term MODY with monogenic diabetes and the gene name.
In this article, we provide recommendations on practical steps for communicating a diagnosis of monogenic diabetes to patients, methods for family testing, and considering the psychological impact of diagnosis (Table 3 & 5; Fig. 2). The practice of communicating genetic testing results for monogenic diabetes to patients with a genetic diagnosis is evolving as monogenic diabetes testing becomes more prevalent. Although communicating genetic testing results for disease-causing variants is more straightforward, it remains challenging in communicating results of a VUS or a no genetic diagnosis resulting in a patient with a clearly atypical presentation of diabetes. Fortunately, collaborative efforts in variant curation and precision medicine research will continue to reduce the ambiguity in VUS or no diagnosis results and improve our ability to effectively provide recommendations for diagnosis, treatment and family testing for people who undergo monogenic diabetes testing.
The major strength with our study is that to the best of our knowledge it is the first comprehensive overview of all available evidence on diagnostics of monogenic diabetes based on screening more than 12,500 peer reviewed articles published during the last 32 years extracting data from >100 studies that met the predefined criteria. This makes it easier for healthcare professionals to make evidence-based decisions. We used rigorous and transparent methods, leading to a higher quality of the evidence for how to use precision diagnostics in monogenic diabetes than other types of studies. Moreover, we aimed to reduce bias in the selection of studies, data extraction, and analysis making our findings more reliable and credible. Finally, our systematic review is an efficient way to identify knowledge gaps and prioritize future research, as it avoids duplication of efforts and resources.
Our study also has several limitations. For the question of who to test, the index/triage test of clinical or laboratory biomarkers used to select people for monogenic diabetes testing would ideally be compared with the reference standard of genetically testing all individuals with diabetes (without any such selection) however, such studies were rare. Most were cohort or cross-sectional studies in patients with diabetes diagnostic uncertainty, that only genetically tested a smaller sample by certain criteria, so it was not possible to discern the number of cases missed (false negatives) with this approach. Only a few studies directly compared two or more approaches in the same study population, so most recommendations were based on comparative yields in different populations. Whilst syndromic forms of monogenic diabetes (such as mitochondrial diabetes and deafness, severe insulin resistance, lipodystrophy) were included in the search strategy, there were not sufficient papers that included at least 100 genetically tested individuals to permit graded recommendations on whom to select for genetic testing when these additional features were present.
Our review was limited by the availability of relevant studies, and sometimes there were not enough high-quality studies to draw meaningful conclusions. Hence, we were not able to address Questions 3-7 initially or ultimately (for Questions 6-7) by a systematic review using the method offered by Covidence. However, the co-authors have been working on diagnostics of monogenic diabetes for 10-30 years and are experts in the field. We therefore used expert opinion for the Questions 3–7. Another weakness is that only papers in the English language were included in the analysis. Thus, non-English papers potentially offering useful information on Questions 1-2 were missed. It is, however, not likely since we defined a cut-off of 100 study individuals undergoing genetic testing to ensure a high scientific quality. Conducting the systematic review was a time-consuming process searching for and evaluating many studies. It was also resource-extensive necessitating a team of trained researchers and specialized software. And we cannot completely exclude publication bias, where only studies with significant results are published, and non-significant results are not reported. Despite some limitations, we believe our systematic review will prove a valuable tool in precision diagnostics of monogenic diabetes providing high-quality evidence to inform clinical decision-making.
What is needed next? Our systematic review reveals that improved access to genetic testing for monogenic diabetes to prevent health disparities is important. There are issues regarding equity and utility in non-European countries where background prevalence of type 2 diabetes is higher. Ethnicity dependent thresholds for overweight or obese categories need to be considered when implementing the recommendations of whom to test in different geographical regions with distinct ethnic groups. Cultural factors may also influence the acceptability of screening for monogenic diabetes which needs further study and education. Moreover, type 1 diabetes genetic risk score (a tool for using common susceptibility variants for type 1 diabetes to pre-assess the likelihood of having type 1 vs. other types of diabetes) data has not been well characterized in these countries. Another step is generation of and access to systematic measurements of autoantibodies and C-peptide for people diagnosed with diabetes under the age of 45 years with the addition of validated ancestry-appropriate type 1 diabetes genetic risk score data. This information would be advantageous to better discriminate monogenic diabetes from type 1 diabetes. What is more, improvement in de-identified case-sharing platforms is needed to promote maximizing the ability to gather the evidence needed to evaluate pathogenicity. As such, continued work by expert panels such as the MDEP VCEP is warranted to develop guidelines for which gene variants should be considered causative of monogenic diabetes as well as applications of the guidelines tailored to additional monogenic diabetes types and genes. One relevant instrument is generation of deep mutational scanning maps of monogenic diabetes genes to aid variant classification. It is also important to remember that the genetic and genomic testing landscape is ever evolving, with a strong possibility of universal genome sequencing in the future, which would reduce concerns on whom to test but place an even higher burden on having adequate tools, expertise, and workforce for interpretation. Finally, with the increased numbers of people being given a genetic diagnosis of monogenic diabetes, further studies to evaluate whether this actually results in improved management due to capacity of medical services are needed. Further clinical guidance is needed for steps following monogenic diabetes testing which includes genetic counseling, subsequent referrals, and family testing in addition to research on the outcomes of implementation.
References
Gloyn, A. L. & Drucker, D. J. Precision medicine in the management of type 2 diabetes. Lancet Diabetes Endocrinol. 6, 891–900 (2018).
International Diabetes Federation IDF Diabetes eAtlas (International Diabetes Federation, 2022).
Bowman, P. et al. Effectiveness and safety of long-term treatment with sulfonylureas in patients with neonatal diabetes due to KCNJ11 mutations: an international cohort study. Lancet Diabetes Endocrinol. 6, 637–646 (2018).
Pearson, E. R. et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med. 355, 467–477 (2006).
Pearson, E. R. et al. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362, 1275–1281 (2003).
Christensen, A. S. et al. Efficacy and safety of glimepiride with or without linagliptin treatment in patients With HNF1A diabetes (maturity-onset diabetes of the young type 3): a randomized, double-blinded, placebo-controlled, crossover trial (GLIMLINA). Diabetes Care 43, 2025–2033 (2020).
Tuomi, T., Honkanen, E. H., Isomaa, B., Sarelin, L. & Groop, L. C. Improved prandial glucose control with lower risk of hypoglycemia with nateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care 29, 189–194 (2006).
Greeley, S. A. W. et al. ISPAD Clinical Practice Consensus Guidelines 2022: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 23, 1188–1211 (2022).
Stone, S. I., Abreu, D., McGill, J. B. & Urano, F. Monogenic and syndromic diabetes due to endoplasmic reticulum stress. J Diabetes Complications 35, 107618 (2021).
Shields, B. M. et al. Can clinical features be used to differentiate type 1 from type 2 diabetes? A systematic review of the literature. BMJ Open 5, e009088 (2015).
Fajans, S. S., Bell, G. I. & Polonsky, K. S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med. 345, 971–980 (2001).
Flannick, J., Johansson, S. & Njolstad, P. R. Common and rare forms of diabetes mellitus: towards a continuum of diabetes subtypes. Nat. Rev. Endocrinol. 12, 394–406 (2016).
Misra, S. et al. Homozygous hypomorphic HNF1A alleles are a novel cause of young-onset diabetes and result in sulfonylurea-sensitive diabetes. Diabetes Care 43, 909–912 (2020).
Gloyn, A. L. et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 350, 1838–1849 (2004).
Raimondo, A. et al. Phenotypic severity of homozygous GCK mutations causing neonatal or childhood-onset diabetes is primarily mediated through effects on protein stability. Hum. Mol. Genet. 23, 6432–6440 (2014).
Murphy, R., Ellard, S. & Hattersley, A. T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 4, 200–213 (2008).
Pihoker, C. et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J. Clin. Endocrinol. Metab. 98, 4055–4062 (2013).
Johansson, B. B. et al. Targeted next-generation sequencing reveals MODY in up to 6.5% of antibody-negative diabetes cases listed in the Norwegian Childhood Diabetes Registry. Diabetologia 60, 625–635 (2017).
Shepherd, M. et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the u.k. pediatric diabetes population with monogenic diabetes. Diabetes Care 39, 1879–1888 (2016).
Katashima, R., Matsumoto, M., Watanabe, Y., Moritani, M. & Yokota, I. Identification of novel GCK and HNF4alpha gene variants in Japanese pediatric patients with onset of diabetes before 17 years of age. J. Diabetes Res. 2021, 7216339 (2021).
Ewans, L. J. et al. Whole exome and genome sequencing in mendelian disorders: a diagnostic and health economic analysis. Eur. J. Hum. Genet. 30, 1121–1131 (2022).
Gorzynski, J. E. et al. Ultrarapid nanopore genome sequencing in a critical care setting. N. Engl. J. Med. 386, 700–702 (2022).
Clayton, E. W. Ethical, legal, and social implications of genomic medicine. N. Engl. J. Med. 349, 562–569 (2003).
Hu, T., Chitnis, N., Monos, D. & Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 82, 801–811 (2021).
Austin-Tse, C. A. et al. Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genom. Med. 7, 27 (2022).
Misra, S. & Owen, K. R. Genetics of monogenic diabetes: present clinical challenges. Curr. Diab. Rep. 18, 141 (2018).
Burke, W., Parens, E., Chung, W. K., Berger, S. M. & Appelbaum, P. S. The challenge of genetic variants of uncertain clinical significance: a narrative review. Ann Intern Med 175, 994–1000 (2022).
Chung, W. K. et al. Precision medicine in diabetes: a consensus report from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 43, 1617–1635 (2020).
Nolan, J. J. et al. ADA/EASD precision medicine in diabetes initiative: an international perspective and future vision for precision medicine in diabetes. Diabetes Care 45, 261–266 (2022).
Tobias, D. et al. Second International Consensus Report on Gaps and Opportunities for the Clinical Translation of Precision Diabetes Medicine. Nature Med. (in press). https://doi.org/10.1038/s41591-023-02502-5.
Page, M. J. et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ 372, n71 (2021).
Colclough, K., Ellard, S., Hattersley, A. & Patel, K. Syndromic monogenic diabetes genes should be tested in patients with a clinical suspicion of maturity-onset diabetes of the young. Diabetes 71, 530–537 (2022).
Whiting, P. F. et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann. Intern. Med. 155, 529–536 (2011).
Diabetes Canada Clinical Practice Guidelines Expert, C. Methods. Can. J. Diabetes 42, S6–S9 (2018). https://doi.org/10.1016/j.jcjd.2017.10.002.
Sherifali, D. et al. The diabetes health coaching randomized controlled trial: rationale, design and baseline characteristics of adults living with type 2 diabetes. Can. J. Diabetes 43, 477–482 (2019).
Martin, A. R. et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 51, 1560–1565 (2019).
Strande, N. T. et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am J Hum Genet 100, 895–906 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Abou Tayoun, A. N. et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 39, 1517–1524 (2018).
Brnich, S. E. et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 12, 3 (2019).
Tavtigian, S. V., Harrison, S. M., Boucher, K. M. & Biesecker, L. G. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat 41, 1734–1737 (2020).
Tavtigian, S. V. et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 20, 1054–1060 (2018).
Deans, Z. C. et al. Recommendations for reporting results of diagnostic genomic testing. Eur. J. Hum. Genet. 30, 1011–1016 (2022).
Farmer, G. D., Gray, H., Chandratillake, G., Raymond, F. L. & Freeman, A. L. J. Recommendations for designing genetic test reports to be understood by patients and non-specialists. Eur. J. Hum. Genet. 28, 885–895 (2020).
Haga, S. B. et al. Developing patient-friendly genetic and genomic test reports: formats to promote patient engagement and understanding. Genome Med. 6, 58 (2014).
De Franco, E. et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet 386, 957–963 (2015).
Edghill, E. L. et al. HLA genotyping supports a nonautoimmune etiology in patients diagnosed with diabetes under the age of 6 months. Diabetes 55, 1895–1898 (2006).
Rubio-Cabezas, O., Flanagan, S. E., Damhuis, A., Hattersley, A. T. & Ellard, S. KATP channel mutations in infants with permanent diabetes diagnosed after 6 months of life. Pediatr. Diabetes 13, 322–325 (2012).
De Franco, E. et al. Biallelic PDX1 (insulin promoter factor 1) mutations causing neonatal diabetes without exocrine pancreatic insufficiency. Diabet. Med. 30, e197–e200 (2013).
Chakera, A. J. et al. The 0.1% of the population with glucokinase monogenic diabetes can be recognized by clinical characteristics in pregnancy: the Atlantic Diabetes in Pregnancy cohort. Diabetes Care 37, 1230–1236 (2014).
Gjesing, A. P. et al. High prevalence of diabetes-predisposing variants in MODY genes among danishwomen with gestational diabetes mellitus. J. Endocrine Soc. 1, 681–690 (2017).
Zubkova, N. et al. High frequency of pathogenic and rare sequence variants in diabetes-related genes among Russian patients with diabetes in pregnancy. Acta Diabetol. 56, 413–420 (2019).
Jiang, Y. et al. Identification and management of GCK-MODY complicating pregnancy in Chinese patients with gestational diabetes. Mol. Cell Biochem. 477, 1629–1643 (2022).
Aloi, C. et al. Glucokinase mutations in pediatric patients with impaired fasting glucose. Acta Diabetol. 54, 913–923 (2017).
Ma, Y. et al. A new clinical screening strategy and prevalence estimation for glucokinase variant-induced diabetes in an adult Chinese population. Genet. Med. 21, 939–947 (2019).
Carmody, D. et al. GCK-MODY in the US National Monogenic Diabetes Registry: frequently misdiagnosed and unnecessarily treated. Acta Diabetol. 53, 703–708 (2016).
Bansal, V. et al. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Med. 15, 213 (2017).
Donath, X. et al. Next-generation sequencing identifies monogenic diabetes in 16% of patients with late adolescence/adult-onset diabetes selected on a clinical basis: a cross-sectional analysis. BMC Med. 17, 132 (2019).
Patel, K. A. et al. Systematic genetic testing for recessively inherited monogenic diabetes: a cross-sectional study in paediatric diabetes clinics. Diabetologia 65, 336–342 (2022).
Gong, S. et al. Genetics and clinical characteristics of PPARγ variant-induced diabetes in a Chinese Han population. Front. Endocrinol. (Lausanne) 12, 677130 (2021).
Decaudain, A. et al. New metabolic phenotypes in laminopathies: LMNA mutations in patients with severe metabolic syndrome. J. Clin. Endocrinol. Metab. 92, 4835–4844 (2007).
Clissold, R., Shields, B., Ellard, S., Hattersley, A. & Bingham, C. Assessment of the HNF1B score as a tool to select patients for HNF1B genetic testing. Nephron 130, 134–140 (2015).
Shields, B. M. et al. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia 55, 1265–1272 (2012).
Juszczak, A. et al. Plasma fucosylated glycans and c-reactive protein as biomarkers of HNF1A-MODY in young adult-onset nonautoimmune diabetes. Diabetes Care 42, 17–26 (2019).
McDonald, T. J. et al. Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from Type 1 diabetes. Diabet. Med. 28, 1028–1033 (2011).
McDonald, T. J. et al. Lipoprotein composition in HNF1A-MODY: differentiating between HNF1A-MODY and type 2 diabetes. Clin. Chim. Acta 413, 927–932 (2012).
McDonald, T. J. et al. High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care 34, 1860–1862 (2011).
Owen, K. R. et al. Assessment of high-sensitivity C-reactive protein levels as diagnostic discriminator of maturity-onset diabetes of the young due to HNF1A mutations. Diabetes Care 33, 1919–1924 (2010).
Thanabalasingham, G. et al. A large multi-centre European study validates high-sensitivity C-reactive protein (hsCRP) as a clinical biomarker for the diagnosis of diabetes subtypes. Diabetologia 54, 2801–2810 (2011).
Yaghootkar, H. et al. Type 1 diabetes genetic risk score discriminates between monogenic and Type 1 diabetes in children diagnosed at the age of <5 years in the Iranian population. Diabet. Med. 36, 1694–1702 (2019).
Mughal, S. A. et al. Apolipoprotein M can discriminate HNF1A-MODY from Type 1 diabetes. Diabet. Med. 30, 246–250 (2013).
Thanabalasingham, G. et al. Mutations in HNF1A result in marked alterations of plasma glycan profile. Diabetes 62, 1329–1337 (2013).
Laver, T. W. et al. Evaluation of evidence for pathogenicity demonstrates That BLK, KLF11, and PAX4 should not be included in diagnostic testing for MODY. Diabetes 71, 1128–1136 (2022).
Ellard, S. et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. https://www.acgs.uk.Com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf (Association for Clinical Genomics Science, 2020).
Vears, D. F. et al. Points to consider for laboratories reporting results from diagnostic genomic sequencing. Eur. J. Hum. Genet. 26, 36–43 (2018).
Vears, D. F., Senecal, K. & Borry, P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur. J. Med. Genet. 60, 553–558 (2017).
Davis, K. W., Hamby Erby, L., Fiallos, K., Martin, M. & Wassman, E. R. A comparison of genomic laboratory reports and observations that may enhance their clinical utility for providers and patients. Mol. Genet. Genomic Med. 7, e00551 (2019).
Cresswell L. et al. General Genetic Laboratory Reporting Recommendations. https://www.acgs.uk.com/quality/best-practice-guidelines/#GeneralGuidelines (Association for Clinical Genomics Science, 2023).
ACMG. The American College of Medical Genetics Technical Standards for Clinical Genetics Laboratories https://www.acmg.net/ACMG/Medical-Genetics-Practice-Resources/Genetics_Lab_Standards/ACMG/Medical-Genetics-Practice-Resources/Genetics_Lab_Standards.aspx?hkey=0e473683-3910-420c-9efb-958707c59589 (2023).
Liljestrom, B. et al. Genetic testing for maturity onset diabetes of the young: uptake, attitudes and comparison with hereditary non-polyposis colorectal cancer. Diabetologia 48, 242–250 (2005).
Clissold, R. L., Hamilton, A. J., Hattersley, A. T., Ellard, S. & Bingham, C. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nat. Rev. Nephrol. 11, 102–112 (2015).
Wentworth, J. M. et al. Maturity-onset diabetes of the young type 5 in a family with diabetes and mild kidney disease diagnosed by whole exome sequencing. Intern. Med. J. 44, 1137–1140 (2014).
Shepherd, M. et al. Predictive genetic testing in maturity-onset diabetes of the young (MODY). Diabet. Med. 18, 417–421 (2001).
Liljestrom, B. et al. Adolescents at risk for MODY3 diabetes prefer genetic testing before adulthood. Diabetes Care 30, 1571–1573 (2007).
Shepherd, M. & Hattersley, A. T. I don’t feel like a diabetic any more’: the impact of stopping insulin in patients with maturity onset diabetes of the young following genetic testing. Clin. Med. (Lond.) 4, 144–147 (2004).
Guan, Y., Maloney, K. A. & Pollin, T. I. Patient perspectives on the diagnostic journey to a monogenic diabetes diagnosis: barriers and facilitators. J. Genet. Couns. 29, 1106–1113 (2020).
Bosma, A. R., Rigter, T., Weinreich, S. S., Cornel, M. C. & Henneman, L. A genetic diagnosis of maturity-onset diabetes of the young (MODY): experiences of patients and family members. Diabet. Med. 32, 1385–1392 (2015).
Lindner, T. H. et al. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum. Mol. Genet. 8, 2001–2008 (1999).
Zhang, H., Colclough, K., Gloyn, A. L. & Pollin, T. I. Monogenic diabetes: a gateway to precision medicine in diabetes. J. Clin. Invest. 131. https://doi.org/10.1172/JCI142244 (2021).
Stein, S. A., Maloney, K. L. & Pollin, T. I. Genetic counseling for diabetes mellitus. Curr. Genet. Med. Rep. 2, 56–67 (2014).
American Diabetes, A. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 44, S15–S33 (2021).
Harper, P. S. Practical Genetic Counseling 6th edn (Hodder Education Publishers, 2004).
Edghill, E. L. et al. HNF1B deletions in patients with young-onset diabetes but no known renal disease. Diabet. Med. 30, 114–117 (2013).
Bellanne-Chantelot, C. et al. Clinical spectrum associated with hepatocyte nuclear factor-1beta mutations. Ann. Intern. Med. 140, 510–517 (2004).
Murphy, R., Turnbull, D. M., Walker, M. & Hattersley, A. T. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet. Med. 25, 383–399 (2008).
Slingerland, A. S. Monogenic diabetes in children and young adults: challenges for researcher, clinician and patient. Rev. Endocr. Metab. Disord. 7, 171–185 (2006).
Chakera, A. J. et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care 38, 1383–1392 (2015).
Shepherd, M., Hattersley, A. T. & Sparkes, A. C. Predictive genetic testing in diabetes: a case study of multiple perspectives. Qual. Health Res. 10, 242–259 (2000).
Bansal, V., Boehm, B. O. & Darvasi, A. Identification of a missense variant in the WFS1 gene that causes a mild form of Wolfram syndrome and is associated with risk for type 2 diabetes in Ashkenazi Jewish individuals. Diabetologia 61, 2180–2188 (2018).
Goodrich, J. K. et al. Determinants of penetrance and variable expressivity in monogenic metabolic conditions across 77,184 exomes. Nat. Commun. 12, 3505 (2021).
Laver, T. W. et al. The Common p.R114W HNF4A mutation causes a distinct clinical subtype of monogenic diabetes. Diabetes 65, 3212–3217 (2016).
Wright, C. F. et al. Assessing the pathogenicity, penetrance, and expressivity of putative disease-causing variants in a population setting. Am. J. Hum. Genet. 104, 275–286 (2019).
Althari, S. et al. Unsupervised clustering of missense variants in HNF1A using multidimensional functional data aids clinical interpretation. Am. J. Hum. Genet. 107, 670–682 (2020).
Mazzaccara, C. et al. Mitochondrial diabetes in children: seek and you will find it. PLoS ONE 7, e34956 (2012).
Majithia, A. R. et al. Prospective functional classification of all possible missense variants in PPARG. Nat. Genet. 48, 1570–1575 (2016).
Ushijima, K. et al. KLF11 variant in a family clinically diagnosed with early childhood-onset type 1B diabetes. Pediatr. Diabetes 20, 712–719 (2019).
Graff, S. M. et al. A KCNK16 mutation causing TALK-1 gain of function is associated with maturity-onset diabetes of the young. JCI Insight 6 (2021). https://doi.org/10.1172/jci.insight.138057 (2021).
Consortium, S. T. D. et al. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311, 2305–2314 (2014).
Hegele, R. A., Cao, H., Harris, S. B., Hanley, A. J. & Zinman, B. Hepatocyte nuclear factor-1 alpha G319S. A private mutation in Oji-Cree associated with type 2 diabetes. Diabetes Care 22, 524 (1999).
Hegele, R. A., Hanley, A. J., Zinman, B., Harris, S. B. & Anderson, C. M. Youth-onset type 2 diabetes (Y2DM) associated with HNF1A S319 in aboriginal Canadians. Diabetes Care 22, 2095–2096 (1999).
Kleinberger, J. W. & Pollin, T. I. Undiagnosed MODY: Time for Action. Curr. Diab. Rep. 15, 110 (2015).
Bonnefond, A. et al. Monogenic diabetes. Nat. Rev. Dis. Primers 9, 12 (2023).
Naylor, R. N. et al. Cost-effectiveness of MODY genetic testing: translating genomic advances into practical health applications. Diabetes Care 37, 202–209 (2014).
Peters, J. L., Anderson, R. & Hyde, C. Development of an economic evaluation of diagnostic strategies: the case of monogenic diabetes. BMJ Open 3. https://doi.org/10.1136/bmjopen-2013-002905 (2013).
Faguer, S. et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int. 86, 1007–1015 (2014).
Shields, B. M. et al. Population-based assessment of a biomarker-based screening pathway to aid diagnosis of monogenic diabetes in young-onset patients. Diabetes Care 40, 1017–1025 (2017).
Ma, Y. et al. New clinical screening strategy to distinguish HNF1A variant-induced diabetes from young early-onset type 2 diabetes in a Chinese population. BMJ Open Diabetes Res. Care 8. https://doi.org/10.1136/bmjdrc-2019-000745 (2020).
Bellanné-Chantelot, C. et al. High-sensitivity C-reactive protein does not improve the differential diagnosis of HNF1A-MODY and familial young-onset type 2 diabetes: a grey zone analysis. Diabetes Metab. 42, 33–37 (2016).
Acknowledgements
The ADA/EASD Precision Diabetes Medicine Initiative (PMDI), within which this work was conducted, has received the following support: The Covidence license was funded by Lund University (Sweden) for which technical support was provided by Maria Björklund and Krister Aronsson (Faculty of Medicine Library, Lund University, Sweden). Administrative support was provided by Lund University (Malmö, Sweden), University of Chicago (IL, USA), and the American Diabetes Association (Washington D.C., USA). The Novo Nordisk Foundation (Hellerup, Denmark) provided grant support for in-person writing group meetings (PI: L Phillipson, University of Chicago, IL).
S.M. has a personal award from Wellcome Trust Career Development scheme (223024/Z/21/Z) and holds Institutional funds from the NIHR Biomedical Research Centre Funding Scheme. P.R.N. was supported by grants from the European Research Council (AdG #293574), Stiftelsen Trond Mohn Foundation (Mohn Center of Diabetes Precision Medicine), the University of Bergen, Haukeland University Hospital, the Research Council of Norway (FRIPRO grant #240413), the Western Norway Regional Health Authority (Strategic Fund “Personalised Medicine for Children and Adults”), and the Novo Nordisk Foundation (grant #54741). J.M.I. was supported by a grant from the National Institute of Health (K08DK133676). S.E.F. is supported by a Wellcome Trust Senior Research Fellowship (Grant Number 223187/Z/21/Z). ALG is a Wellcome Trust Senior Fellow (200837/Z/16/Z) and is also supported by NIDDK (UM-1DK126185). EDF is a Diabetes UK RD Lawrence Fellow (19/005971). KRO is supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Consortia
Contributions
Review design: R.M., K.C., T.I.P., P.R.N., L.K.B., K.R.O., A.L.G. Systematic review implementation: R.M., K.C., T.I.P., J.I.M., P.S., K.A.M., C.S., J.M., S.M., I.A., E.dF., S.E.F., P.R.N., L.K.B., K.R.O., A.L.G. Data extraction manuscripts: R.M., K.C., T.I.P., J.M.I., P.S., L.K.B., K.R.O., A.L.G. Manuscript writing: R.M., K.C., T.I.P., L.K.B., K.A.M., P.R.N., K.R.O., A.L.G. Project Management: R.M., L.K.B., K.R.O., A.L.G.
Corresponding authors
Ethics declarations
Competing interests
R.M. has received honoraria for consulting or educational activities for Eli Lilly, Novo Nordisk and Boeringer Ingelheim. S.M. has Investigator initiated funding from DexCom and serves on the Board of Trustees for the Diabetes Research & Wellness Foundation (UK). LK Billings: Received honoraria for consulting activities for Novo Nordisk, Bayer, Lilly, Xeris, and Sanofi which are unrelated to this present work. ALG’s spouse holds stock options in Roche and is an employee of Genentech. No other authors have competing interests.
Peer review
Peer review information
Communications Medicine thanks Fumihiko Urano and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Murphy, R., Colclough, K., Pollin, T.I. et al. The use of precision diagnostics for monogenic diabetes: a systematic review and expert opinion. Commun Med 3, 136 (2023). https://doi.org/10.1038/s43856-023-00369-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43856-023-00369-8
