
Abstract
One of the paradoxes of the Archean rock record is how evidence for oxidative continental weathering is plausible when the atmosphere lacked oxygen prior to the Great Oxidation Event ca. 2.5 billion years ago. We detected reactive oxygen species (ROS) production at various silicate-water interfaces in a simulated Archean atmosphere and calculated homolysis rates of silicon–oxygen bonds in these minerals. We find that ROS can be produced while silicates were eroded and transported by streams to depositional basins. The magnitude of ROS production increases from mafic- to felsic-silicates. During early continent formation, e.g., the Kaapvaal and Pilbara basins in the Paleoarchean, the emplacement of granitoids and their subsequent exposure to physical weathering might have annually provided at least 1.73 × 108 mol O2 before the biological O2 production. Importantly, this process could have oxidatively mobilized redox-sensitive elements to seawater, increasing the availability of bioessential nutrient elements for the Archean biosphere.
Similar content being viewed by others


Introduction
The mass-independent fractionation (MIF) of sulfur isotopes resulting from photochemical reactions in the Archean atmosphere suggests free oxygen was <10–5 the present atmospheric level (PAL) before the Great Oxidation Event (GOE) ~2.5 billion years (Gyr) ago1,2. Yet paradoxically, increasing lines of geochemical evidence obtained from marine sedimentary rocks show oxidation occurred in terrestrial environments already during the Mesoarchean, and by extension the existence of O2-producing cyanobacteria. The evidence includes, the solubilization and mobilization of uranium, molybdenum, rhenium, thallium, chromium and sulfur from reduced crustal minerals to the oceans where they became incorporated into the sedimentary records3,4,5,6,7,8,9,10,11,12,13.
In order to reconcile these oxidative signals with the S-MIF record, Ono et al.14 proposed weak oxygenations of the atmosphere before the GOE. Anbar et al.4 subsequently proposed that “oxygen whiffs” provided transient pulses of oxygen to an otherwise anoxic atmosphere, although it has been argued that the whiffs were instead caused by postdepositional alteration15. Alternatively, the oxidative weathering reactions were generated within benthic microbial mats growing on sediments and soils16. Similar process of localized oxygen production in an anoxic setting (i.e., benthic oases) is observed today in Lake Fryxell, Antarctica, where benthic cyanobacteria produce O2 below the oxycline17. Photochemical oxidants formed in the atmosphere have also been proposed to have contributed to the oxidation of the Earth’s surfaces, but the extremely low production rates and the difficulty in its transportation to the ground18 imply that it was far from sufficient for the local or global oxidations observed19,20. Direct photochemical oxidation of reduced minerals in the crust is another possibility21,22,23 but whether this process causes discernable signals in S isotopic fractionations remains unknown23.
Recently, He et al.24 reported a previously unrecognized abiotic oxygen-producing pathway in the Archean. It involves the abrasion of quartz surfaces, the generation of surface-bound radicals (SBRs, e.g., ≡SiO· and ≡SiOO·) from homolytic cleavage of Si–O bonds in ≡Si–O–Si ≡ (intrinsic bonds in quartz)25 or ≡Si–O–O–Si ≡ (a peroxy linkage that is widely formed via stress-induced deformation, the condensation of two ≡Si–OH or irradiation emitted by radionuclides)26,27,28 (R1-2), and subsequently the production of reactive oxygen species (ROS, including hydroxyl radical (·OH) and hydrogen peroxide (H2O2)) on contact with water (R3-6). Specifically, those authors proposed that in turbulent subaqueous environments, such as rivers, estuaries and deltas, the process could have provided a sufficient H2O2 source that led to the generation of molecular oxygen (R7). That O2 would have then negatively impacted existing anoxygenic photosynthetic communities growing in microbial mats and/or the water column by oxidizing their electron donors, creating an evolutionary impetus for the origins of oxygenic photosynthesis. Indeed, microbial mats intercalated with sandy layers, as observed in the 3.22-billion-year-old Moodies Group in South Africa29, might have performed photosynthesis by extracting electrons from H2O2 in the weakly oxic environments.
Given quartz and silicate minerals have a common basic structural unit—the [SiO4] tetrahedron—breaking of Si–O bonds can also occur in a variety of silicate minerals in addition to quartz. Therefore, it is reasonable to expect that ROS could have been produced at abraded silicate-water interfaces during the Archean when exposed continental crust was eroded and the lithic debris transported downstream to depositional basins. Indeed, previous studies found high yields of ROS in the suspensions of mafic minerals that were pulverized in air with a 21% oxygen concentration30,31,32, but the role of preexisting O2 in promoting the production of SBR and ROS was neglected. In a recent work that abraded minerals in O2-defficient conditions (O2 < 0.15%), the greatest ROS production was found in felsic minerals rather than mafic minerals33. Besides the environmental O2 concentration, the temperature also affects ROS production in silicates34. It was reported that high concentrations of H2O2 could be produced via fractures of silicate rocks in the hot subsurface at temperature close to the boiling point of water35. However, evaluating the role of physical erosion and ROS production of silicate minerals under predicted Archean terrestrial surface conditions (O2 < 10–6 PAL, average surface temperature <40 °C (ref. 36)) remains poorly constrained in terms of knowing which minerals generate ROS and quantifying their O2 production potential. This clearly needs to be addressed if we are to link the redox properties of Archean sediment to the origin and evolution of cyanobacteria, or not.
Here, we conducted ROS production experiments at silicates-water interfaces, and developed a mechanochemical ROS producing model to estimate the amounts of ROS that could have been produced in the physical erosion at local and global scales. The results indicate that the physicochemical reactions on the surfaces of several silicates could have provided ~108 mol oxidants per year at the weathering interface in the Mesoarchean, and this abiotic oxidation could have supplied oxidized ions to seawater. As ROS production strongly depends on the crysto-chemistry of silicates, we argue that the mechanism was linked to felsic crust compositions and, therefore, craton formation and evolution in the Archean.
Results
We selected several major rock-forming minerals from the perspective of basic crysto-chemistry in building minerals with [SiO4] tetrahedra (Supplementary Fig. 1 and Table 1). The selected silicate minerals with varied polymerization degrees (the ratio of bridge oxygen per silicon) include quartz (Qtz), K-feldspar (Kfs), muscovite (Ms), beryl (Brl), hornblende (Hbl), spodumene (Spd), wollastonite (Wo), augite (Aug), and forsterite (Fo). Corundum (Crn, Al2O3) was further used as a proxy for minerals without Si–O bond for comparison. Ball-milling of mineral particles was performed to mimic the mechanical abrasion in an O2-free, N2 atmosphere (O2 < 0.1 ppm) reminiscent of the Archean physiochemical weathering (see ref. 24 for methods).
As ROS was previously demonstrated to form during the interactions between the abraded quartz and water24, we similarly plunged the abraded silicate minerals into deionized water without pre-existing dissolved O2 ( < 0.01 mg L–1), and the mineral-water reactions occurred in open system. ROS (e.g., ·OH and H2O2) were detected with production rates of 0.86–14.47 nmol m–2; forsterite was an exception as it did not produce ROS (Fig. 1a). Amongst these minerals, Kfs and Qtz (both tectosilicates) displayed much higher ROS production rates (13.51 and 14.47 nmol m–2) (Fig. 1a). It is noteworthy that the ROS productivity rate generally decreased in the order of tectosilicate, phyllosilicate (e.g., Ms), cyclosilicate (e.g., Brl), inosilicate (e.g., Hbl, Spd, Wo, Aug) and nesosilicate (e.g., Fo) (Fig. 1a, b), with a synchronous decrease of polymerization degree in silicates. The above-mentioned sequence of ROS production with mineral structure found in this study is generally consistent with the results of a recent work that minerals were abraded in the O2-defficient atmosphere (O2 < 0.15%)33. But it is opposite to the results of those works that performed in O2-rich atmosphere (i.e., air)31,32,37, and the reported higher ROS production of Fe-rich mafic minerals is likely attributed to the interaction between surface Fe(II) and O2 during mechanical activation, which involves the generation of surface superoxide radical (·O2–) and the subsequent production of H2O2 and ·OH in water30,38,39. Given that our experiments were conducted at room temperature (~25 °C), the ROS production is not related to thermal activation of mineral surface defects (≡SiO·). In addition, in the ROS components, the ratios of ·OH and H2O2 vary significantly amongst silicates, which is related to the specific content of dissolved ferrous iron ions released from minerals (Supplementary Table 2) because Fe(II) facilitates the decomposition of H2O2 to ·OH rapidly via the Fenton reaction30 (R8).

a ROS production of the abraded silicates. The error bars present standard deviations of three independent replicates. b Schematic diagram of [SiO4] polymerization in silicates in the order of the number of bridge oxygens (BO) shared between [SiO4] (the yellow polyhedron) or [AlO4] (the pink polyhedron) in minerals (modified from Hurowitz et al.31). c EPR spectra of the abraded silicates. Three SBR species were identified, i.e., peroxy radical (≡SiOO·, gzz = 2.064; gyy = 2.007; gxx = 2.002), surface-bound superoxide ion (≡Si+O2·–, gzz = 2.044; gyy = 2.010; gxx = 2.002), and E’ center (Si·, g// = 2.001; g┴ = 2.000).
We note that the variation in ROS productivity in silicates shows strong correlation to the density of SBR in the abraded minerals, which were measured by electron paramagnetic resonance (EPR) spectroscopy (Fig. 1c). As indicated by the intensities of the EPR signals, there were remarkable differences in SBR density amongst the different silicates. The SBR densities of the abraded tectosilicates (e.g., Qtz and Kfs) were expectedly higher than other silicates, in concert with their high ROS productivities. By contrast, due to a great incorporation of metals (e.g., Fe and Mg) into mineral structures, the linkage styles of [SiO4] tetrahedra become isolated, and the ratio of bridge oxygens decreases and the SBR density in abraded minerals reduces. The SBR signals in abraded Fe-poor minerals with low polymerization degree of [SiO4] (Spd and Brl, wt (FeO) <0.5%) are much weaker than that of the abraded tectosilicates, while the SBR signals in other abraded mafic minerals (e.g., Fo, Aug, and Hbl) are readily shielded by the paramagnetic Fe (wt (FeO) >8%) (Supplementary Table 1).
Variations in ROS productivity and SBR density are intrinsically related to the crystallographic structure of silicates. SBRs are generated by the probabilistic homolysis of Si–O bonds, which form more readily when there is a higher covalent proportion in the bonds25,30,40. For instance, the calculated percentage of covalent component for the Si–O bonds in Qtz (~42%) is higher than that in Fo (~37%)41, which is in good accordance with the in our experimental results. Thus, variations in the covalent component for the Si–O bonds amongst different silicates would influence their homolysis rates and SBR densities.
To evaluate the homolysis rates of Si–O bonds in silicates, an energetic statistical approach—the Boltzmann Distribution—was employed to quantify the mechanochemical potentials of Si–O bonds in silicates (Methods). The estimated values of homolysis rates of Si–O bonds display a large variation amongst the different silicates (Table 1). As the content of SiO2 increases from mafic to felsic minerals, the polymerization degree of [SiO4] increases from ino- to tectosilicate42, correspondently leading to the formation of more bridge oxygens that connect [SiO4] tetrahedra (Fig. 1b). Notably, felsic silicates (e.g., Qtz and Kfs), with a maximum degree of polymerization of [SiO4], show relatively high homolysis rates (e.g., 1.00 for quartz), which are 7~8 orders of magnitude higher than that of the mafic minerals such as wollastonite and augite (Table 1). Our calculations demonstrate that the homolysis rates of Si–O bonds in silicates are generally consistent with the measured SBR density and ROS production results (Fig. 1a), while some other structural subfactors (e.g., cleavage in Brl, see Supplementary Discussion) also affect the ultimate SBR density and subsequent ROS yielding.
Discussion
Our experimental results show that during the physical weathering of Earth’s surface—in the absence of atmospheric O2—SBRs generated by the homolysis of Si–O bonds in silicate minerals split water molecules and release oxidants (ROS and O2) as well as H2 (the gas product in the reaction between ≡Si· and H2O, refs. 24,34). The significance of these observations is that physical weathering of silicates not only provides a source of O2 for oxidative weathering at rock-water interfaces, but also allows H2 to escape to the atmosphere which further contributed to the irreversible oxidation of Earth’s surface (as per ref. 43). Crucially, the production of mechanochemical oxidants likely increased concomitant with the evolution of mineral composition and physical weathering rates of the upper continental crust (UCC).
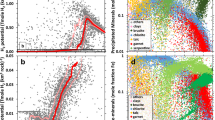
To elucidate the link between the flux of mechanochemical oxidants and the weathering of the UCC, we calculated the potential ROS productions by various silicate rocks based on our experimentally measured ROS production of minerals (Methods). The total number of electrons, e(T), that can be stripped from reductants by ·OH and H2O2, was used to characterize the contribution of certain silicate rocks in oxidative weathering. As Fig. 2a shows, both e(T) and the ROS production of silicate rocks increase along with the increment in SiO2 content, indicating that the felsic rocks have greater potential in producing ROS than mafic rocks when being mechanically eroded. This feature is correlated to the proportion of three-dimensional structural units with different polymerization degrees in the petrogenetic melts, which increases as the magma evolves to more felsic, i.e., granitic magmas42.

a The correlation between estimated ROS production and SiO2 in crustal rocks based on a compilation of 2632 reported magmatic rocks (see Supplementary Dataset). Data from GEOROC (ref. 93). The triangle, circle and star are ·OH production, H2O2 production and e(T), respectively, while each color corresponds to a certain silicate rock. b Historical changes in e(T) during physical erosion and weathering of silicate rocks in juvenile crust (the change of SiO2 content was obtained from Dhuime et al.44). The e(T) in physical weathering of silicate rocks in juvenile crust is calculated by bringing the model SiO2 into the equation of e(T) in Fig. 2a.
Because the SiO2 content in the Earth’s crust changed with time44,45, the ROS-producing capacity of continent weathering must also have changed with time (Fig. 2b). Indeed, there is a great change in mineral composition of the continental crust in the Archean due to the formation of tonalite-trondhjemite-granodiorite (TTG) via partial melting of hydrous basalt. Although the composition of the juvenile continental crust was predominantly mafic before 3.0 Gyr (SiO2 ∼48–50%)44,46, felsic rocks already occurred in the terrestrial crust by the Paleoarchean47,48,49. The Paleoarchean east Pilbara terrane of the Pilbara Craton, western Australia, and the Barberton granitoid-greenstone terrane of the eastern Kaapvaal Craton in southern Africa contain Earth’s oldest (3.65–3.22 Gyr), best-preserved continental nuclei50. During the growth of these crusts, thick intervals of felsic volcanics covered the basal units (3.49–3.46 Gyr), then voluminous TTG were emplaced (3.49–3.42 Gyr) and granites intruded widely, followed by a pulse of K-granite magmatism at ∼3.1 Gyr51,52. Similarly, voluminous granitoid magmatism also occurred during the growth of Singhbum Craton (India) that produced a polyphase suite of ∼3.36- to 3.24-Gyr granitoids and felsic orthogneiss (∼3.53 to 3.36 Gyr) and potassic granites (∼3.13 to 3.08 Gyr)53. With the emplacement of volumetric granitoids in these early cratons, a favorable provenance for Si–O bond homolysis and ROS production would have been formed (Figs. 2–3).

During early crustal thickening of cratons, formation and emplacement of voluminous felsic rocks provided large positive buoyancy, resulting in elevated topography and high denudation rates. This geodynamic process runs like a giant crushing machine, and crust uplifting fed the felsic rocks to tectonic denudation and physical weathering, in which ROS are generated during transport from land to the oceans. Those regions with intensive physical weathering may have acted as an effective “oxygen-producing factory” on the Archean terrestrial surface. A possible side-effect was localized oxidative weathering and supply of dissolved oxidized species (e.g., sulfate, molybdate) to the oceans.
At the same time, the progressive crustal maturation and thickening driven by voluminous granitoid magmatism additionally led to buoyant rise of continental crust, followed by continental exposure and subaerial weathering51,54,55,56. When cratons were in isostatic uplift, felsic rocks became subject to physical erosion, with the denudation rate increasing greatly as the craton elevation increased from seafloor to ~3 km in elevation during the Mesoarchean (Fig. 3)51,53,57,58. Specifically, the Kaapvaal-and-Pilbara cratons accreted at 3.3–3.1 Gyr, which also triggered regional-scale compressional deformation59. Such tectonic events greatly contributed to reworking of the preexisting crust, which enhanced excavation of silicate rocks, weathering and erosion, transport of detrital material from land to the oceans, and ultimately deposition of coarse clastic sequences51,59,60. It is during the transport from fluvial environments to coastal environments that we argue quartz- and feldspar-rich rocks would have undergone abrasion to continuously produce SBRs and ROS (Fig. 3). Importantly, the mechanochemical oxidation of crustal minerals was independent of atmospheric composition.
To provide some perspective on the potential magnitude of this process, for instance at 3.2 Gyr ago, we took the Himalayan mountains as an example to estimate the ROS production that might have resulted from the potentially intense mountain denudation in the Kaapvaal-and-Pilbara continents (Methods). Our calculations suggest that weathering of the Himalayan mountains could theoretically provide ROS at the flux of 1.79 × 108 mol y–1. We then extrapolated the mechanochemical ROS producing model to continental weathering in the Mesoarchean, and calculated the global ROS production based on possible Archean parameters, including the mechanical denudation rate53 and the land area that was proposed in continental growth models (~4% (ref. 61) and ~7% (ref. 62)) (Methods). The results show that about 3.92 × 108~6.71 × 108 mol of ROS was produced per year in Mesoarchean continental weathering. According to the capability of extracting electron from reductants, the equivalent amount of O2 is 1.73 × 108~2.97 × 108 mol y–1 (the converting factors for H2O2 and ·OH to O2 is 0.5 and 0.25, respectively, see Methods, Supplementary Tables 3–5). And if the continental surface area at 3.2 Gyr is close to the present-day level (27.5%) (refs. 63,64), the estimated ROS production is 2.63 × 109 mol y–1, equivalent to a O2 amount of 1.17 × 109 mol y–1. These estimated mechanochemical ROS productions are comparable to the production of sulfate by photochemical pyrite weathering in the Mesoarchean (about 109 mol y–1)23. We further propose that in the late Archean, the ROS production should have increased with enlarged land area to physical weathering (Supplementary Fig. 2), along with the transition from mafic to felsic continental crust (Fig. 2). Besides, the river length also increased with the growth of continental crust, and the silicate debris were finely pulverized in longer fluvial transport, leading to increased production of SBRs and ROS. Although the pH of world-average river water increased in the late Archean65, it had little effect on the ROS flux because the ROS production of H2O2 and ·OH changes insignificantly in the range of pH (6.0 ~ 7.4).
Specifically, uplift of Archean mountains enhanced physical weathering, erosion, and transport of lithic fragments via streams, while river incision and groundwater weathered underlying bedrock66. In the absence of plant root systems, the Archean terrains would have been subject to rapid migration of riverbeds and a predisposition for wide, braided streams to transport and disperse large volumes of supermature quartz-rich sands weathered from the source terrains25,67,68. Remarkably, there are modern examples; measurements of ROS have been reported in groundwater along the Himalayan frontal thrust that flows through crushed quartzite bedrock69. The concentration of H2O2 from the Sohna thermal spring water falling along the active Sohna fault and geothermal field is in a range of 0.53–0.56 mg L–1 or 15.59–16.47 μM, while the background concentration is 0.14 mg L–1 or 4.12 μM. Because the presence of H2O2 is largely dependent on the circulation of groundwater through the crushed quartzite (≤77 °C) at a depth of ~915 m, H2O2 is inferred to form in reactions between anoxic groundwater and mineral defects (e.g., peroxy radicals), while H2 gas is generated and released to the fracture networks69,70. The natural occurrence in the Himalayas is consistent with laboratory simulations that H2O2 formed at stressed rock-water boundaries27, and provides support for our hypothesis that considerable ROS production took place in Archean mountainous terrains.
In the oxidative weathering on Archean continents, this oxygen-producing pathway could also have facilitated the release of trace metals associated with reduced minerals, such as sulfides. With intense rock fracturing processes in the Archean mountains, the waterflow was oxygenated and the H2O2 concentration might be comparable to that of the Himalayas (the average H2O2 concentration from above (16.03 μM) in Sohna spring water). The micromolar level H2O2 can efficiently oxidize dissolved reductants, such as the rapid oxidation of Fe2+ in Fenton reaction (R8) at the rate of 4.0 × 108 M–1 s–1, and even small organics71. Given the wide occurrence of sulfide minerals in detrital sediments72, we used the oxidation rate laws of pyrite and molybdenite73 to further evaluate the feasibility of oxidative weathering by mechanochemical oxidants. Assuming all H2O2 dissociate to dissolved O2 (R7) at the absence of dissolved reductants, the dissolved O2 concentration of waterflows is ~8.02 μM. The calculated results suggest that it is sufficient to oxidize pyrite and molybdenite at relatively rapid rates of 7.45 × 10–11 and 1.42 × 10–11 mol m–2 s–1 (25 °C, pH = 5), respectively, which are faster than that of silicates, such as forsterite and albite (1.2 × 10–12 mol m–2 s–1 and 1.19 × 10–11 mol m–2 s–1, 25 °C, pH = 5)74.
At a broader scale, besides promoting chemical dissolution via exposing fresh rock surface to water, physical weathering of silicates directly promoted the transport of those mobilized metals (such as Cu, Zn, Mo and Co) via rivers to the oceans. Those metals, in turn, were either assimilated by marine benthic and/or planktonic microorganisms, or adsorbed onto suspended sediment (e.g., clay minerals) or chemical precipitates (e.g., iron oxyhydroxides) and deposited to the seafloor where they potentially become incorporated into the sedimentary rock record75. In the case of the former, transport of metals required in enzymes (e.g., Mo in nitrogenase) would facilitate enhanced primary productivity, and ultimately help drive the microbial diversification via the ‘bioinorganic bridge’76 or through providing a greater flux of electron acceptors (e.g., sulfate) that could alter the biogeochemical cycling of carbon77. In sum, the low but ubiquitous abiotic oxygen-producing process served as a link between the coevolution of the early Archean lithosphere and biosphere.
Methods
Sample preparation and storage
Quartz was purchased from Richjoint (Shanghai, China), while other silicate minerals (K-feldspar, muscovite, beryl, hornblende, spodumene, wollastonite, augite, forsterite and corundum) were collected from the depository of the Key Laboratory of Mineralogy and Metallogeny, Chinese Academy of Sciences, Guangzhou, China.
The silicate minerals were crushed and mesh-sieved. The 0.85–2.0 mm particles were chosen and washed with deionized water, dried in oven at 110 °C for 24 h and stored in a Mikrouna glovebox (N2 > 99.999%, H2O < 0.1 ppm, O2 < 0.1 ppm) for further experiments. In the glovebox, 50 g of particles was loaded into a zirconia ceramic jar with 150 g balls. The jar was filled with the same atmosphere as in the glovebox (i.e., ultrapure nitrogen), sealed, moved out of the glovebox and transferred to a planetary ball mill (Fritsch, Pulverisette 6) for grinding at 350 revolutions per minute for 5 h. After grinding, the jar was moved back to the glovebox, and the abraded minerals were collected and sealed in glass bottles, stored in the glovebox to protect the surface reactive sites.
To check if the atmospheric O2 leaked into the jar during ball milling, we performed blank runs using the same ball milling procedures mentioned above without the amendment of minerals. The O2 concentration in the headspace of the ball-milling jar was measured at the beginning and the end of the ball milling (T = 0 h and 5 h). In the measurements, any potential contamination from atmospheric O2 should be avoided. A 10 mL gastight syringe was vacuumed and filled with high-purity nitrogen for 5 times, and 2 mL of gas in the headspace of the jar was sampled with the syringe in the glovebox (O2 < 0.1 ppm), during which the syringe and the jar reached equilibrium by pumping the syringe 6 times. The syringe was pushed into the GC inlet when the vacuum valve is open, and the needle was evacuated for 3 min to exclude the atmospheric O2 during the transport in air. The vacuumed valve was closed and the syringe valve was open to inject gas sample. The volume of the zirconia balls used for grinding is 25.64 cm3, which was obtained by dividing the weight (150 g) by its density (5.85 g cm–3). The volume of the headspace for the 500 cm3 jar was 474.36 cm3 (=500 − 25.64) after adding the zirconia balls. Because the working gas pressure of the glove box was ~0.1 MPa and the temperature was 25 °C, the amount of O2 was calculated (the volume of 1 mol gas is 22.5 L under this condition). At the beginning of the ball milling, the measured O2 concentration in the jar was ~4 ppm (0.0004 ± 0.0001 v/v%), the volume of the O2 was ~0.0017 cm3 and its amount was ~7.73 × 10–8 mol; at the end of the 5-hour ball milling, the concentration slightly increased to ~11 ppm (0.0011 ± 0.0002 v/v%), the volume of the O2 was ~0.0051 cm3 and its amount was ~2.25 × 10–7 mol. If 2.25 × 10–7 mol of O2 totally bond to the abraded surface of silicate minerals and then involve into ROS production, it is normalized to the weight of silicate minerals (50 g) to give 4.5 × 10–9 mol O2 g–1. This conservative O2 concentration was about 3- to 8-fold lower than the ROS production in felsic minerals (Fig. 1a, Supplementary Table 3). In addition, given the negligible ROS production of forsterite, we think that the trace O2 in the headspace has little effect on the experimental results of the mafic minerals.
Experimental procedures
Experiments on ROS production for all silicate minerals are conducted in the glovebox (N2 > 99.999%, H2O < 0.1 ppm, O2 < 0.1 ppm). For measuring the production of ROS, suspensions were prepared by submerging the washed particles or abraded powders of silicate minerals (0.50 g) into the deionized water (5.00 mL, 18.2 MΩ cm−1, DO < 0.01 mg L–1) with magnetic stirring. The deionized water was set as blank controls. The filtered liquids were collected for measuring the concentrations of hydroxyl radical and hydrogen peroxide.
Measurements of reactive oxygen species (·OH and H2O2)
The production of hydroxyl radical (·OH) was measured by determining the concentrations of the oxidative products (p-Hydroxybenzoic acid, p-HBA) of benzoic acid (BA)78. For the measurement of p-HBA, the filtered liquid sample was rapidly mixed with methanol to quench further oxidation of p-HBA and the p-HBA concentration was measured by a high-performance liquid chromatography. Detail steps see ref. 24, in which the detection limit of p-HBA was optimized to 0.01 μM, equivalent to 0.059 μM ·OH.
The hydrogen peroxide (H2O2) concentration was determined by using Leuco Crystal Violet (LCV) technique79. With the catalysis of horseradish peroxidase, H2O2 can dissociate into ·OH, and LCV can be oxidized to crystal violet cation (CV+) by the ·OH. The concentration of CV+ could be measured by a UV-Vis spectrophotometer at the wave length of 590 nm. The detailed H2O2 detection procedures followed those described in ref. 24.
Measurements of headspace gas
The headspace gas of the sealed jar was determined by gas chromatography (GC, Agilent Technologies 7890-0322). Each gas sample was injected into the GC inlet connected with a vacuum glass system. The carrying gas was Ar. The gas analyzer included two thermal conductivity detectors for the analysis of permanent gases and a flame ionization detector for the analysis of hydrocarbon gases, as well as five rotary valves and seven columns. This enabled the analysis of all gaseous components with a single injection.
Measurements of surface-bound radicals
Surface-bound radicals created via grinding were measured by electron paramagnetic resonance (EPR) on a Bruker A300-10-12 spectrometer. The settings for the EPR measurements were as follows: center field, 3320 Gauss; sweep width, 500 Gauss; microwave frequency, 9.297 GHz; modulation frequency, 100 kHz; power, 2.28 mW; and temperature, 77 K.
Characterization of the abraded minerals
X-ray diffraction (XRD) patterns were scanned from 3 to 70° in 2θ with a scanning step of 3° min–1 on a Bruker D8 ADVANCE X-ray diffractometer operated at 40 kV and 40 mA with the Cu Kα radiation.
Major elements in silicate minerals were determined by using X-ray fluorescence spectrometry (XRF) method. Before analysis, all the samples were calcined at 900 °C for 90 min to remove organic matter and carbonate. After this, approximately 0.50 g of each treated sample was weighed, mixed with 4.00 g Li2B4O7, and fused into glass discs at 1200 °C. The major elements were measured from the glass discs using a Rigaku ZSX100e X-ray fluorescence spectrometer. The analytical precision of the major element content was better than 1% (relative standard deviation).
Specific surface area measurements of abraded silicates were performed by BET technique with N2 adsorbate gas. Nitrogen adsorption-desorption were measured at 77 K using an ASAP 2020 Surface Area & Pore Size Analyzer (Micromeritics Instrument Corporation). Prior to the measurement, samples were degassed in a vacuum at 200 °C for 12 h.
Calculation of homolysis rate of Si–O bonds in silicate minerals
For the Si–O bond itself, the homolysis rate can be determined by the energy levels of both homolytic (Eho) and heterolytic (Ehe) dissociations. These were previously calculated to be 478 and 250 kJ mol–1 (refs. 80,81), respectively. In terms of the bridge oxygen bond between [SiO4] tetrahedrons in a certain silicate structure, the homolysis rate of the Si–O bonds is affected by the inhomogeneity of their coordination environments. The homolysis rate decreases greatly with an increase in the inhomogeneity of Si–O bonds because a dipole will be formed within the Si–O bond in the inhomogeneous coordination environment82,83, which favors the heterolytic dissociation when the Si–O bond ruptures84. Thus, the Boltzmann factor (wmineral) of Si–O bond dissociation in a certain silicate mineral and temperature can be defined as (Eq. 1):
where \({p}_{{ho}}^{{mineral}}\) and \({p}_{{he}}^{{mineral}}\) denote the homolysis and heterolysis rate of Si–O bonds in a silicate mineral, respectively; \(\triangle E\) denotes the energy change induced by the specific crystal structure; \(n\) is the number of bridge oxygen; k is the Boltzmann constant, and T is the temperature in K. We employ first principles based on Crystal Orbital Hamilton Population (COHP)85 to estimate \(\triangle E\), which is defined as the standard deviation of COHP values of all Si–O bonds in a silicate crystal. The calculations of COHP and Si–O bonds were performed using wave functions generated by density functional theory (DFT) using the Vienna Atomic Simulation Package (VASP)86 along with the projector augmented wave (PAW) method87. We consider a common case for the cleavage of Si–O bonds in nature that happens at room temperature (298.15 K). All the silicates used in this study (Table 1) were modeled in unit cell and calculated using Perdew, Burke, and Ernzerhof (PBE) function88 with an energy cutoff at 400 eV for the plane wave basis. The relative homolysis rates were the normalization of \({p}_{{ho}}^{{mineral}}\) to \({p}_{{ho}}^{{{{{{\rm{quartz}}}}}}}\).
Calculation of ROS production of silicate rocks
The ROS production of a certain silicate rock was calculated by accumulating the ROS production of each mineral according to the weighting factors (Eqs. 2–4).
where P·OH-R and PH2O2-R are the production of ·OH and H2O2 of a certain silicate rock, respectively; P·OH-M and PH2O2-M are the production of ·OH and H2O2 of a certain mineral, respectively; i denotes a certain mineral in the silicate rock; and fCIPW is the percentage of a certain mineral in the silicate rock calculated using the Cross-Iddings-Pirsson-Washington (CIPW) Norm.
Estimation of ROS production in the Himalayan Mountains
The Himalayan Mountains are a modern analog to the oxygen-producing factory in the Mesoarchean (Fig. 3). In mountainous terrains, ROS production is a function of the denudation rate, which can be estimated simply based on the amount of discharged sediment in the drainage basin.
The ROS production in the Himalayan Mountains was calculated via multiplying experimental ROS capability of the unit mass of quartz and feldspar by the total weight of the two minerals in the suspended sediments delivered by the Ganges/Brahmaputra River. The ROS-producing capability of quartz and feldspar in this study was estimated at the measured values (Supplementary Table 3). The ROS-producing capability of quartz is: H2O2, 1.62 × 10–7 mol g–1; ·OH, 1.31 × 10–8 mol g–1; O2, 8.79 × 10–9 mol g–1. The ROS-producing capability of feldspar is: H2O2, 5.19 × 10–8 mol g–1; ·OH, 3.18 × 10–8 mol g–1; O2, 5.86 × 10–10 mol g–1. Quartz accounts for about 20% in suspended sediments in the Ganges/Brahmaputra River89. In consideration of the dissolution and transformation of feldspar, the original amount of feldspar was obtained via multiplying the amount of quartz by the mass ratio (4.18) of the two minerals in the upper continental crust. In the upper continental crust, the total content of feldspar (67%) is the sum of orthoclase (20.1%), albite (33.0%), and anorthite (13.9%), while the content of quartz is 16.1% (ref. 90), and thereby the mass ratio is 4.18 (=67% / 16.1%). The estimated ROS production is not relevant to the dissolved O2 in the modern waters, even the radicals generated in the interaction between dissolved O2 and Fe(II).
The average sediment discharge of the Ganges/Brahmaputra River is 1.67 × 1015 g yr–1 (ref. 91), and the ROS production from quartz and feldspar in the Himalayan Mountains was estimated to be 1.79 × 108 mol yr–1 (Supplementary Table 4).
Estimation of ROS production in the emerged land surface in the Archean
At the world-wide scale, the continental erosion can remove the topographic expression produced by tectonic uplift, and the dominant part is played by mechanical erosion92. As Fig. 3 shows, the thick, silica-rich (less-dense) crust formed in the Mesoarchean, and thereby the cratonic crust experienced large positive buoyancy and a greater isostatic uplift relative to the surrounding thin and mafic (more dense) oceanic crust53. As the Ce anomalies in detrital zircons revealed58, the crust became progressively thicker and reached a maximum average thickness of 55 to 65 km at ca. 3.2 Gyr, which implies that a high topography similar to today’s Himalayan Mountains might have been built on the coeval continent. Thus, it is possible to calculate the continental erosion by using the equitation (Eq. 5) developed by Harrison (ref. 92).
where dYD/dt is the mechanical denudation rate (m Myr–1), \(\bar{Y}\) is average elevation, and y is runoff (cm yr–1).
Taking the Singhbhum Craton as an example, the subaerial relief would have varied between 350 and 1100 m (ref. 53), and the average elevation (675 m) is close to that of today’s Asia, suggesting that the rate of continent erosion in the Mesoarchean is also at a relatively high level. If the coeval precipitation is assumed to be similar to that in Asia (41 cm yr–1), the estimated erosion rate is 69.84 m Myr–1. Based on the land exposure at 3.2 Ga (~4%) reported by Flament et al.61, the calculated Mesoarchean continent area is ~2.22 × 1013 m2 and the volume of bedrock removed by mechanical erosion is ~1.55 × 109 m3 yr–1. And if the calculation is based on the rapid continental growth model of Korenaga et al.62 (i.e., the land exposure at 3.2 Ga is ~7%), the calculated Mesoarchean continent area is ~3.88 × 1013 m2 and the volume of bedrock removed by mechanical erosion is ~2.71 × 109 m3 yr–1. Considering that the bulk density of the Mesoarchean continent is 2870 kg m–3 (ref. 53), the calculated mechanical erosion quantity in the Mesoarchean is 4.45 × 109~7.79 × 109 tons per year. Thus, the corresponding ROS production was roughly calculated to be 3.92 × 108 ~6.71 × 108 mol yr–1 on the basis of the ROS-producing capability of felsic minerals (Supplementary Table 5). Further, the change of ROS production in Archean continental weathering was estimated according to the two continental growth models (Supplementary Fig. 2). If the continental surface area at 3.2 Gyr is closed to the present-day level (27.5%) (ref. 63), the estimated ROS production is 2.63 × 109 mol yr–1 by following the procedures motioned above.
Data availability
The authors declare that the main data supporting the findings of this study are available at the following Data Repository link: https://doi.org/10.17632/gw85sx33p7.1. Source data and Supplementary dataset are provided with this paper.
References
Farquhar, J., Bao, H. & Thiemens, M. Atmospheric influence of Earth’s earliest sulfur cycle. Science 289, 756–758 (2000).
Pavlov, A. A. & Kasting, J. F. Mass-independent fractionation of sulfur isotopes in Archean sediments: strong evidence for an anoxic Archean atmosphere. Astrobiology 2, 27–41 (2002).
Wille, M. et al. Evidence for a gradual rise of oxygen between 2.6 and 2.5 Ga from Mo isotopes and Re-PGE signatures in shales. Geochim. Cosmochim. Acta 71, 2417–2435 (2007).
Anbar, A. D. et al. A whiff of oxygen before the great oxidation event? Science 317, 1903–1906 (2007).
Kendall, B. et al. Pervasive oxygenation along late Archaean ocean margins. Nat. Geosci. 3, 647–652 (2010).
Stüeken, E. E., Catling, D. C. & Buick, R. Contributions to late Archaean sulphur cycling by life on land. Nat. Geosci. 5, 722–725 (2012).
Planavsky, N. J. et al. Evidence for oxygenic photosynthesis half a billion years before the great oxidation event. Nat. Geosci. 7, 283–286 (2014).
Satkoski, A. M., Beukes, N. J., Li, W., Beard, B. L. & Johnson, C. M. A redox-stratified ocean 3.2 billion years ago. Earth Planet. Sci. Lett. 430, 43–53 (2015).
Frei, R. et al. Oxidative elemental cycling under the low O2 Eoarchean atmosphere. Sci. Rep. 6, 1–9 (2016).
Wang, X. et al. A Mesoarchean shift in uranium isotope systematics. Geochim. Cosmochim. Acta 238, 438–452 (2018).
Ostrander, C. M. et al. Fully oxygenated water columns over continental shelves before the great oxidation event. Nat. Geosci. 12, 186–191 (2019).
Nabhan, S., Marin-Carbonne, J., Mason, P. R. D. & Heubeck, C. In situ S-isotope compositions of sulfate and sulfide from the 3.2 Ga Moodies Group, South Africa: a record of oxidative sulfur cycling. Geobiology 18, 426–444 (2020).
Brüske, A. et al. The onset of oxidative weathering traced by uranium isotopes. Precambrian Res. 338, 105583 (2020).
Ono, S. Early evolution of atmospheric oxygen from multiple-sulfur and carbon isotope records of the 2.9 Ga Mozaan Group of the Pongola Supergroup, Southern Africa. South Afr. J. Geol. 109, 97–108 (2006).
Slotznick, S. P. et al. Reexamination of 2.5-Ga “whiff” of oxygen interval points to anoxic ocean before GOE. Sci. Adv. 8, eabj7190 (2022).
Lalonde, S. V. & Konhauser, K. O. Benthic perspective on Earth’s oldest evidence for oxygenic photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 112, 995–1000 (2015).
Sumner, D. Y., Hawes, I., Mackey, T. J., Jungblut, A. D. & Doran, P. T. Antarctic microbial mats: a modern analog for Archean lacustrine oxygen oases. Geology 43, 887–890 (2015).
Kasting, J. F., Liu, S. C. & Donahue, T. M. Oxygen levels in the prebiological atmosphere. J. Geophys. Res. Oceans 84, 3097–3107 (1979).
Haqq-Misra, J., Kasting, J. F. & Lee, S. Availability of O2 and H2O2 on pre-photosynthetic Earth. Astrobiology 11, 293–302 (2011).
Crowe, S. A. et al. Atmospheric oxygenation three billion years ago. Nature 501, 535–538 (2013).
Kim, J. D., Yee, N., Nanda, V. & Falkowski, P. G. Anoxic photochemical oxidation of siderite generates molecular hydrogen and iron oxides. Proc. Natl. Acad. Sci. U.S.A. 110, 10073–10077 (2013).
Liu, W. et al. Anoxic photogeochemical oxidation of manganese carbonate yields manganese oxide. Proc. Natl. Acad. Sci. U.S.A. 117, 22698–22704 (2020).
Hao, J. et al. Anoxic photochemical weathering of pyrite on Archean continents. Sci. Adv. 8, eabn2226 (2022).
He, H. et al. An abiotic source of Archean hydrogen peroxide and oxygen that pre-dates oxygenic photosynthesis. Nat. Commun. 12, 6611 (2021).
Fubini, B., Giamello, E., Volante, M. & Bolis, V. Chemical functionalities at the silica surface determining its reactivity when inhaled. Formation and reactivity of surface radicals. Toxicol. Ind. Health 6, 571–598 (1990).
Fukuchi, T. A mechanism of the formation of E′ and peroxy centers in natural deformed quartz. Appl. Radiat. Isot. 47, 1509–1521 (1996).
Balk, M. et al. Oxidation of water to hydrogen peroxide at the rock–water interface due to stress-activated electric currents in rocks. Earth Planet. Sci. Lett. 283, 87–92 (2009).
Nilges, M. J., Pan, Y. & Mashkovtsev, R. I. Radiation-induced defects in quartz. III. Single-crystal EPR, ENDOR and ESEEM study of a peroxy radical. Phys. Chem. Miner. 36, 61–73 (2009).
Heubeck, C. An early ecosystem of Archean tidal microbial mats (Moodies Group, South Africa, ca. 3.2 Ga). Geology 37, 931–934 (2009).
Schoonen, M. A. A. et al. Mineral-induced formation of reactive oxygen species. Rev. Mineral Geochem. 64, 179–221 (2006).
Hurowitz, J. A., Tosca, N. J., McLennan, S. M. & Schoonen, M. A. A. Production of hydrogen peroxide in Martian and lunar soils. Earth Planet. Sci. Lett. 255, 41–52 (2007).
Hendrix, D. A., Port, S. T., Hurowitz, J. A. & Schoonen, M. A. Measurement of OH* generation by pulverized minerals using electron spin resonance spectroscopy and implications for the reactivity of planetary regolith. GeoHealth 3, 28–42 (2018).
Edgar, J. O., Gilmour, K., White, M. L., Abbott, G. D. & Telling, J. Aeolian driven oxidant and hydrogen generation in Martian regolith: The role of mineralogy and abrasion temperature. Earth Planet. Sci. Lett. 579, 117361 (2022).
Kita, I., Matsuo, S. & Wakita, H. H2 generation by reaction between H2O and crushed rock: An experimental study on H2 degassing from the active fault zone. J. Geophys. Res. Solid Earth 87, 10789–10795 (1982).
Stone, J., Edgar, J. O., Gould, J. A. & Telling, J. Tectonically-driven oxidant production in the hot biosphere. Nat. Commun. 13, 4529 (2022).
Catling, D. C. & Zahnle, K. J. The Archean atmosphere. Sci. Adv. 6, eaax1420 (2020).
Edgar, J. O., Gould, J. A., Badreshany, K. & Telling, J. Mechanochemical generation of perchlorate. Icarus 387, 115202 (2022).
Horwell, C. J., Fenoglio, I. & Fubini, B. Iron-induced hydroxyl radical generation from basaltic volcanic ash. Earth Planet. Sci. Lett. 261, 662–669 (2007).
Lv, Y. et al. Photoreductive dissolution of iron (hydr)oxides and its geochemical significance. ACS Earth Space Chem. 6, 811–829 (2022).
Kaupp, G. Mechanochemistry: the varied applications of mechanical bond-breaking. Cryst. Eng. Comm. 11, 388–403 (2009).
Tossell, J. A. A comparison of silicon-oxygen bonding in quartz and magnesian olivine from X-ray spectra and molecular orbital calculations. Am. Mineral. 62, 136–141 (1977).
Mysen, B. O., Virgo, D. & Seifert, F. A. The structure of silicate melts: implications for chemical and physical properties of natural magma. Rev. Geophys. 20, 353–383 (1982).
Catling, D. C., Zahnle, K. J. & McKay, C. P. Biogenic methane, hydrogen escape, and the irreversible oxidation of early earth. Science 293, 839–843 (2001).
Dhuime, B., Wuestefeld, A. & Hawkesworth, C. J. Emergence of modern continental crust about 3 billion years ago. Nat. Geosci. 8, 552–555 (2015).
Kump, L. R. & Barley, M. E. Increased subaerial volcanism and the rise of atmospheric oxygen 2.5 billion years ago. Nature 448, 1033–1036 (2007).
Tang, M., Chen, K. & Rudnick, R. L. Archean upper crust transition from mafic to felsic marks the onset of plate tectonics. Science 351, 372–375 (2016).
Sizova, E., Gerya, T., Stüwe, K. & Brown, M. Generation of felsic crust in the Archean: a geodynamic modeling perspective. Precambrian Res. 271, 198–224 (2015).
Guo, M. & Korenaga, J. Argon constraints on the early growth of felsic continental crust. Sci. Adv. 6, eaaz6234 (2020).
Roerdink, D. L., Ronen, Y., Strauss, H. & Mason, P. R. D. Emergence of felsic crust and subaerial weathering recorded in Palaeoarchaean barite. Nat. Geosci. 15, 227–232 (2022).
Hickman, A. H. & Van Kranendonk, M. J. Early earth evolution: evidence from the 3.5–1.8 Ga geological history of the Pilbara region of Western Australia. Episodes 35, 283 (2012).
Van Kranendonk, M. J. et al. Making it thick: a volcanic plateau origin of Palaeoarchean continental lithosphere of the Pilbara and Kaapvaal cratons. Geol. Soc. Lond. Spec. Publ. 389, 83–111 (2015).
Johnson, T. E. et al. Secular change in TTG compositions: implications for the evolution of Archaean geodynamics. Earth Planet. Sci. Lett. 505, 65–75 (2019).
Chowdhury, P. et al. Magmatic thickening of crust in non–plate tectonic settings initiated the subaerial rise of Earth’s first continents 3.3 to 3.2 billion years ago. Proc. Natl. Acad. Sci. U.S.A. 118, e2105746118 (2021).
Lee, C.-T. A. et al. Regulating continent growth and composition by chemical weathering. Proc. Natl. Acad. Sci. U.S.A. 105, 4981–4986 (2008).
Hawkesworth, C. J., Cawood, P. A., Dhuime, B. & Kemp, T. I. S. Earth’s continental lithosphere through time. Annu. Rev. Earth Planet. Sci. 45, 169–198 (2017).
Wiemer, D., Schrank, C. E., Murphy, D. T., Wenham, L. & Allen, C. M. Earth’s oldest stable crust in the Pilbara Craton formed by cyclic gravitational overturns. Nature Geosci. 11, 357–361 (2018).
Campbell, I. H. & Davies, D. R. Raising the continental crust. Earth Planet. Sci. Lett. 460, 112–122 (2017).
Tang, M., Chu, X., Hao, J. & Shen, B. Orogenic quiescence in Earth’s middle age. Science 371, 728–731 (2021).
Zegers, T. E., de Wit, M. J., Dann, J. & White, S. H. Vaalbara, Earth’s oldest assembled continent? A combined structural, geochronological, and palaeomagnetic test. Terra Nova 10, 250–259 (1998).
Anderson, S. P. Breaking it down: Mechanical processes in the weathering engine. Elements 15, 247–252 (2019).
Flament, N., Coltice, N. & Rey, P. F. The evolution of the 87Sr/86Sr of marine carbonates does not constrain continental growth. Precambrian Res. 229, 177–188 (2013).
Korenaga, J., Planavsky, N. J. & Evans, D. A. D. Global water cycle and the coevolution of the Earth’s interior and surface environment. Philos. Trans. A Math. Phys. Eng. Sci. 375, 20150393 (2017).
Cawood, P. A. & Hawkesworth, C. J. Continental crustal volume, thickness and area, and their geodynamic implications. Gondwana Res. 66, 116–125 (2019).
Armstrong, R. L. & Harmon, R. S. Radiogenic isotopes: the case for crustal recycling on a near-steady-state no-continental-growth Earth. Philos. Trans. R. Soc. Lond. A Math. Phys. Sci. 301, 443–472 (1981).
Hao, J., Sverjensky, D. A. & Hazen, R. M. Mobility of nutrients and trace metals during weathering in the late Archean. Earth Planet. Sci. Lett. 471, 148–159 (2017).
Molnar, P., Anderson, R. S. & Anderson, S. P. Tectonics, fracturing of rock, and erosion. J. Geophys. Res. Earth Surf. 112, 1–12 (2007).
Donaldson, J. A. & de Kemp, E. A. Archaean quartz arenites in the Canadian shield: examples from the Superior and Churchill Provinces. Sediment. Geol. 120, 153–176 (1998).
Long, D. G. F. Archean fluvial deposits: a review. Earth Sci. Rev. 188, 148–175 (2019).
Singh, P. & Mukherjee, S. Chemical signature detection of groundwater and geothermal waters for evidence of crustal deformation along fault zones. J. Hydrol. 582, 124459 (2020).
Cinti, D., Pizzino, L., Voltattorni, N., Quattrocchi, F. & Walia, V. Geochemistry of thermal waters along fault segments in the Beas and Parvati valleys (north-west Himalaya, Himachal Pradesh) and in the Sohna town (Haryana), India. Geochem. J. 43, 65–76 (2009).
Aeschbacher, M., Graf, C., Schwarzenbach, R. P. & Sander, M. Antioxidant properties of humic substances. Environ. Sci. Technol. 46, 4916–4925 (2012).
Farquhar, J., Zerkle, A. L. & Bekker, A. Geological constraints on the origin of oxygenic photosynthesis. Photosyn. Res. 107, 11–36 (2011).
Johnson, A. C. et al. Experimental determination of pyrite and molybdenite oxidation kinetics at nanomolar oxygen concentrations. Geochim. Cosmochim. Acta 249, 160–172 (2019).
Lasaga, A. C. Chemical kinetics of water-rock interactions. J. Geophys. Res. 89, 4009–4025 (1984).
Robbins, L. J. et al. Trace elements at the intersection of marine biological and geochemical evolution. Earth Sci. Rev. 163, 323–348 (2016).
Anbar, A. D. & Knoll, A. H. Proterozoic ocean chemistry and evolution: a bioinorganic bridge? Science 297, 1137–1142 (2002).
Zahnle, K., Claire, M. & Catling, D. The loss of mass-independent fractionation in sulfur due to a Palaeoproterozoic collapse of atmospheric methane. Geobiology 4, 271–283 (2006).
Joo, S. H., Feitz, A. J., Sedlak, D. L. & Waite, T. D. Quantification of the oxidizing capacity of nanoparticulate zero-valent iron. Environ. Sci. Technol. 39, 1263–1268 (2005).
Cohn, C. A., Pak, A., Strongin, D. & Schoonen, M. A. Quantifying hydrogen peroxide in iron-containing solutions using leuco crystal violet. Geochem. Trans. 6, 47 (2005).
Luo, Y.-R. Comprehensive handbook of chemical bond energies. (CRC Press, 2007).
Murashov, V. V. & Demchuk, E. Surface sites and unrelaxed surface energies of tetrahedral silica polymorphs and silicate. Surf. Sci. 595, 6–19 (2005).
Raymond, M. Electric-field-gradient calculations in the aluminum silicates (Al2SiO5). Phys. Rev. B 3, 3692–3702 (1971).
Tossell, J. A. & Cohen, R. E. Calculation of the electric field gradients at ‘tricluster’-like O atoms in the polymorphs of Al2SiO5 and in aluminosilicate molecules: models for tricluster O atoms in glasses. J. Non-Cryst. Solids 286, 187–199 (2001).
Rincón, L., Mora, J. R., Torres, F. J. & Almeida, R. On the activation of σ-bonds by electric fields: a Valence Bond perspective. Chem. Phys. 477, 1–7 (2016).
Dronskowski, R. & Bloechl, P. E. Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617–8624 (1993).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mate. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Goldberg, E. D. & Griffin, J. J. The sediments of the northern Indian Ocean. Deep Sea Res. Oceanogr. Abstr. 17, 513–537 (1970).
Taylor, S. R. & McLennan, S. M. The geochemical evolution of the continental crust. Rev. Geophys. 33, 241–265 (1995).
Milliman, J. D. & Meade, R. H. World-wide delivery of river sediment to the oceans. J. Geol. 91, 1–21 (1983).
Harrison, C. G. A. Rates of continental erosion and mountain building. Geol. Rundsch. 83, 431–447 (1994).
GEOROC Compilation: Rock Types, https://doi.org/10.25625/2JETOA.
Acknowledgements
We thank Huifang Xu, H. Henry Teng, Peter A. Cawood for helpful discussions and suggestions. H. He and J. Zhu were supported by National Science Fund for Distinguished Young Scholars (Grant No. 41825003), National Natural Science Foundation of China (Grant No. 41921003), CAS Key Research Program of Frontier Sciences (Grant No. QYZDJ-SSW-DQC023). X. Wu was supported by National Natural Science Foundation of China (Grant No. 42202037), Director’s Fund of Guangzhou Institute of Geochemistry, Chinese Academy of Science (2022SZJJT-05) and Project funded by China Postdoctoral Science Foundation (Grant No. 2022M713164). Y. Li was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB41000000). This is contribution No. IS-3344 from GIGCAS.
Author information
Authors and Affiliations
Contributions
H.H and J.Z, led the projects and designed the experiments; X.W performed the experiments with assistance from Y.Y, L.M, X.L and S.L; H.H, J.Z, X.W, H.X and X.L analyzed the data; X.W, J.Z, H.H, Y.L and K.K wrote the manuscript with input from H.X.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Jihua Hao and Jon Telling for their contribution to the peer review of this work. Primary Handling Editors: Joe Aslin and Clare Davis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, X., Zhu, J., He, H. et al. Geodynamic oxidation of Archean terrestrial surfaces. Commun Earth Environ 4, 132 (2023). https://doi.org/10.1038/s43247-023-00789-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-023-00789-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
