
Abstract
Search for novel electronically ordered states of matter emerging near quantum phase transitions is an intriguing frontier of condensed matter physics. In ruthenates, the interplay between Coulomb correlations among the 4d electronic states and their spin-orbit interactions, lead to complex forms of electronic phenomena. Here we investigate the double layered Sr3(Ru1−xMnx)2O7 and its doping-induced quantum phase transition from a metal to an antiferromagnetic Mott insulator. Using spectroscopic imaging with the scanning tunneling microscope, we visualize the evolution of the electronic states in real- and momentum-space. We find a partial-gap at the Fermi energy that develops with doping to form a weak Mott insulating state. Near the quantum phase transition, we discover a spatial electronic reorganization into a commensurate checkerboard charge order. These findings bear a resemblance to the universal charge order in the pseudogap phase of cuprates and demonstrate the ubiquity of charge order that emanates from doped Mott insulators.
Similar content being viewed by others

Introduction
The bilayer ruthenate, Sr3Ru2O7, has a complex quasi-two-dimensional electronic structure due to the rotation of the bulk RuO6 octahedra that leads to the reconstruction of the Fermi surface (Fig. 1a). As a result, multiple electronic bands cross the Fermi level, revealed by angle resolved photoemission spectroscopy (ARPES)1 and de Haas–van Alphen (dHvA)2 studies. While naively, the extended 4d Ru-orbitals as compared to 3d orbitals are expected to make it a weakly correlated metal, Sr3Ru2O7 is one of the most strongly renormalized heavy d-electron material systems, demonstrated by the heavy flat electronic bands in previous ARPES1 and scanning tunneling microscopy (STM)3 experiments. Signatures of the inherent electronic correlations are further emphasized by the impact of minute perturbations on its electronic ground state. The most prominent is the magnetic field-tuned quantum critical behavior and emergent electronic nematic order4,5,6 with spin density wave instability7 seen in transport and neutron scattering.

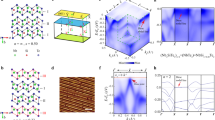
Structure and phase diagram of Sr3(Ru1−xMnx)2O7. a Crystal structure of Sr3(Ru1−xMnx)2O7 and top view of the SrO cleaving plane along with the sublayer RuO6 octahedra. The Mn dopants replace the Ru atoms at the center of the octahedra. The dashed square shows the (1 × 1) tetragonal unit cell, while the larger solid square shows the (√2 × √2)R45° unit cell created by the rotational distortion of the octahedra. b Schematic phase diagram of Sr3(Ru1−xMnx)2O7 showing the doping-temperature (right-axis) transition from a paramagnetic metal to an antiferromagnetic Mott insulator10. The blue circles represent the doping dependence of the spectroscopic local density of states at EF measured at T = 13 K (left axis) normalized to value at 200 meV. The blue square is extracted from ref. 18. c Constant current topographic image of a 5% Mn sample taken at −100 mV and −500 pA showing an atomically ordered surface (Scale bar is 30 Å). The inset shows a magnified image of the same area. d Fourier transform (plotted as the modulus) of the topography in c. Both the Bragg peaks at (±2π/a, 0) and (0, ±2π/a) and satellite reflections at (±π/a, ±π/a) are seen. e Topographic image taken on the x = 1% (1 V, 150 pA), x = 5% (1 V, 150 pA), and x = 10% (1 V, 2500 pA) samples respectively (scale bar is 50 Å). At higher bias voltages, the Mn impurity sites appear as bright spots showing the progression of impurity density from 1 to 10%
Doping acts as another pertinent non-thermal tuning parameter of the electronic states of Sr3Ru2O7. At a few percent of Mn replacing Ru, Sr3(Ru1−xMnx)2O7 undergoes a metal to Mott-insulating quantum phase transition (QPT), accompanied at lower temperatures by an E-type antiferromagnetic (AFM) order with a wave vector QAFM = (π/2a, π/2a) (Fig. 1b)8,9,10 analogous to the AFM structure of FeTe11. Resonant X-ray scattering (REXS) indicates the intensity and correlation length (not exceeding 160 nm) of the AFM order to decrease at lower temperatures (T « TAFM), perhaps providing indications of a competing order10,12. While the origin of the insulating state remains unclear, the Coulomb correlations between the localized 3d Mn orbitals and the relatively extended 4d Ru orbitals may play a crucial role in the metal to insulator transition. X-ray diffraction indicates the RuO6 octahedral-rotation to decrease with doping and be completely suppressed near x = 0.213,14. On the other hand, X-ray absorption experiments15 reveal an unexpected 3+ valence of the Mn suggesting that Mn doping does not introduce holes to the Fermi surface, rather tunes the electronic states likely by structural distortions as well as enhanced Coulomb correlations with increased doping. Such a structural distortion clearly impacts the electronic structure and makes it susceptible towards magnetic instabilities16. Indeed, previous electron microscopy and spectroscopy experiments have shown, in real space, the extreme sensitivity of the Mott-type metal–insulator transition to applied mechanical stress/strain17.
At the border of AFM and Mott insulating QPT, emergent electronic instabilities are frequently discovered. Using spectroscopic imaging with the STM, we investigate the doping-induced metallic to AFM and Mott-insulating QPT in Sr3(Ru1−xMnx)2O7 to explore whether broken electronic symmetry states emerge near the QPT. We find a partial gap at low Mn concentration that evolves with doping into an inhomogeneous full gap. Near the QPT, we find a spatial reorganization of the electronic states into a commensurate checkerboard charge order. These findings provide further evidence for the ubiquity of these electronically ordered states in doped Mott insulators.
Results
Real-space crystal and electronic structure of Sr3(Ru1−xMnx)2O7
Figure 1c shows a topographic STM image of single crystal Sr3(Ru1−xMnx)2O7 with x = 5% cleaved in situ in our ultrahigh-vacuum STM exposing the SrO surface. The Fourier transform (FT) of the topography reveals atomic Bragg peaks at the corners of the map corresponding to (±2π/a, 0) and (0, ±2π/a) (Fig. 1d). Note that, throughout this paper, we use tetragonal notation with lattice constant a ~ 3.9 Å. Additional satellite reflections at (±π/a, ±π/a) correspond to the doubling of the bulk unit cell owing to the rotational distortion of the RuO6 octahedra (Fig. 1a, d) as previously observed3,14. The Mn dopants can be identified by their bias-dependent signatures in the topographic images (Fig. 1c). They can be best visualized at a sample bias V ~ 1 V as bright spots for different doped samples (Fig. 1e). Counting the number of Mn dopants on the surface reveals a systematic downshift (~2%) with a lower surface doping as compared to the expected bulk (see Supplementary Figures 1–3 and Supplementary Note 1).
The local impact of Mn-dopants on the spatial electronic structure, over a range of doping spanning the QPT, can be visualized through the spectroscopic imaging shown in Fig. 2. At low Mn concentration (x = 1%; much below the QPT) the local electronic density of states (LDOS) reveal a particle-hole asymmetric V-shaped partial-gap of ~± 30 meV near the Fermi energy at T = 13 K (Fig. 2a–c). The density of states is similar to what has been previously observed in the undoped parent compound18. In particular, a small hump at EF previously identified as a van-Hove singularity1,18, is also present in our spectra obtained away from Mn dopants (Fig. 2c and Supplementary Figure 4 and Supplementary Note 2). The effect of the Mn, at this low concentration, shows modifications of the local gap predominantly near EF (see Fig. 2b, c and see Supplementary Figure 4 and Supplementary Note 2). Conductance maps dI/dV (r, V) ≡ g(r, V) locally at locations r and at a constant sample bias V, displayed in Fig. 2b, reveal that, in this dilute limit the Mn-dopants spatially modify the LDOS within a length scale of 20 Å.

Real space structure of electronic states in Sr3(Ru1−xMnx)2O7. Topographic images, conductance maps at selected energies taken on the same areas as their corresponding topographies (with the same bias voltage and setpoint current), and spectra taken on and off of Mn impurity sites for a–c x = 1% (150 mV, 500 pA) (scale bar is 15 Å), d–f x = 5% (200 mV, 500 pA) (scale bar is 20 Å), and g–i x = 10% (300 mV, 2500 pA) (scale bar is 25 Å). Mn impurity sites are marked with a blue circle. The blue arrows emphasize the widening of the gap around Fermi energy (EF) with increased doping. All measurements were taken at T = 13 K
Increasing the Mn-doping towards the QPT reveals a striking spatial reorganization of the local electronic states. Figure 2d–f displays a topographic image and the LDOS, showing the evolution of the partial-gap in the x = 5% sample. Conductance maps g(r, V) reveal a bi-directional long-range checkerboard charge modulations (Fig. 2e); particularly V = −40 meV and V = −100 meV maps embedded within the long-wavelength inhomogeneity. The absence of this charge ordering in the real space images of the x = 1% sample (Fig. 2b), as well as in earlier STM experiments on the undoped3,18 and Mn-doped compound at 100 K14, indicate its origin not to be related to any surface reconstruction, rather to an electronic instability induced by Mn-doping at low temperatures. Further indication of the electronic nature of the ordering phenomenon can be concluded by the absence of any structural peaks, corresponding to the observed modulations, in neutron scattering8, in recent low energy electron diffraction (LEED)14, and in non-resonant hard X-ray scattering12. The observed charge modulations, along the Ru–O bond direction with a periodicity of 2a (corresponding to a wave vector q = (±π/a, 0) and (0, ±π/a) ≡ Q*) therefore corroborate an emergent electronic charge ordering near the metal-to-Mott-insulator phase transition.
At even higher doping (x = 10%) (Fig. 2g–i), the STM spectra reveals strong inhomogeneity with areas of V-shaped partial gaps mixed with a complete, yet inhomogeneous, gapping of the Fermi surface into a U-shaped insulating gap of ~100 meV (Fig. 2i and Supplementary Figure 5). This is indicative of a close proximity to the metal-to-Mott insulator transition19. The evolution of the gap with doping resembles the opposite trend of what is seen in the hole-doped, spin–orbit-driven, Mott insulator Sr3(Ir1−xRux)2O719. The conductance maps g(r, V) (Fig. 2h) reveal inhomogeneous short-range modulations, equally populated along the two atomic directions with their periodicity locked to 2a. The glassy nature of the observed charge order could be a consequence of the larger electronic inhomogeneity introduced by the Mn-dopants and the transition into the Mott insulating state.
It is important to emphasize that the doping-induced metal to insulator transition evaluated from the surface LDOS at T = 13 K (blue points in Fig. 1b) closely follows that extracted from bulk resistivity measurements (gray area in Fig. 1b)10. The slight offset may be a result of the lower doping on the surface as compared to the bulk. This may also explain the slight enhancement of the octahedral distortion on the surface of Sr3(Ru1−xMnx)2O7 as compared to the bulk20.
Momentum-space electronic structure of Sr3(Ru1−xMnx)2O7
To visualize the momentum-space structure of these modulations and their doping dependence, we display in Fig. 3a–f (raw data in Supplementary Figure 6) the real-space conductance maps and selected FTs at several energies taken on a larger surface area for optimal momentum-space resolution. For x = 1%, in addition to the Bragg peaks at (±2π/a, 0) and (0, ±2π/a) and the octahedral rotation satellite peaks at (±π/a, ±π/a), the FTs show dispersive features (q2, q7, and q8) along the high symmetry directions, corresponding to quasiparticle interference (Fig. 3a, b) (here we follow the same notations as reference3), similar to those previously seen in the STM studies of 1% Ti-doped Sr3Ru2O73. The extracted dispersions along (±π/a, ±π/a) are shown in Fig. 3g (more detail in Supplementary Figure 7 and Supplementary Note 3). While no clear peak is observed at the Q* location in this 1% sample, there is a faint and broad intensity in the FT around this wave vector (q1 in Fig. 3b, whose dispersion is difficult to extract due to its proximity to q7 and their weak intensity), which locally originates only from areas with Mn dopants (more detail in Supplementary Note 2).

Momentum space structure of electronic states in Sr3(Ru1−xMnx)2O7. Real space conductance maps and corresponding Fourier transforms (FT) (plotted as the modulus) at selected energies measured on a, b x = 1% (150 mV, 500 pA), c, d x = 5% (−100 mV, −500 pA), and e, f x = 10% (100 mV, 2500 pA) Mn doped samples (scale bar is 50 Å). The Q* = (±π/a, 0) and (0, ±π/a) peak, QAFM = (π/a, π/a), and quasiparticle interference peaks q1, q2, q7, and q8 are all circled in the FTs. For the 10% sample, the Bragg peak intensities are too weak at these energies (inside the gap) to be seen but their location is marked by a white cross. All FTs are four-fold symmetrized but all the structures are visible in the unsymmetrized data (see Supplementary Figure 6). All measurements were taken at T = 13 K. g, h Energy dependency of the q value of the peaks and features for the 1, 5, and 10% samples. For the 5% Mn sample, Q* was extracted from two maps with a set-point bias of −100 meV (solid red diamonds) and 200 meV (open red diamonds). The q values and error bars are extracted from the fits (see Supplementary Figures 7–10 and Supplementary Notes 3 and 4)
In contrast, for x = 5%, the FTs indicate distinct sharp and non-dispersive peaks at Q* = (±π/a, 0) and (0, ±π/a) that correspond to a real-space modulation with a periodicity of 2a (Fig. 3d, h). In addition to the Q* modulations, we also observe strong non-dispersive modulations coinciding with the AFM ordering wavevector, QAFM = (±π/2a, ±π/2a) (Fig. 3d, g and Supplementary Figures 8–10 and Supplementary Note 4). In principle, without a spin-polarized tip, STM is not sensitive to a magnetic contrast. Our experiments indicate that the AFM order also has a charge component, which was suggested by previous REXS experiments10. Moving into the Mott state (x = 10%) reveals the Q* modulations to broaden, yet dominate and remain locked to the same commensurate momentum, independent of doping (Fig. 3f, h and Supplementary Figure 8 and Supplementary Note 4).
These experiments, unsurprisingly, indicate strong interplay between spin and charge instabilities. Close to the QPT (x ~ 5%) the system is susceptible to long-range charge ordering at Q* and QAFM (Fig. 3) as well as a competing short-range AFM ordering at QAFM (as revealed by previous REXS10,12). Signs of such a competition can also be seen in the suppressed AFM correlation length with decreasing temperature in REXS10,12. This presents an exotic electronic phase in which charge and spin orders simultaneously coincide at the same wavevector. With further doping, however, the charge ordering dominates at Q*, whereas a long-range E-type AFM order (with negligible charge contribution, as seen in our STM data (Fig. 3f)) dominates at QAFM8,12. It is important to note that although STM is primarily sensitive to charge order, some forms of orbital orders can also be detected through the STM tip sensitivity to different orbital wave functions21,22 and therefore orbital contribution to QAFM cannot be omitted as a possible scenario.
Origin and structure of the observed charge order
To provide insights into the origins of the emergent broken symmetry states at Q* and QAFM, we compute the bare susceptibility χ(q, ω) using a 12 orbital tight-binding model23, fit to match the Fermi surface measured by ARPES on Sr3Ru2O7 (see Methods and Supplementary Figure 11). The calculations of the bare susceptibility, χ(q, 0), whose peaks correspond to the wavevectors with strong Fermi surface nesting, reveal broad features around Q* = (π/a, 0) and QAFM = (π/2a, π/2a), which are enhanced with increased Mn-doping (see Supplementary Figure 11). As previously shown, the effect of Mn-dopants on the Sr3Ru2O7 band structure can be partly accounted for by the suppression of the RuO6 octahedral rotation in the tight-binding band structure24. As doping increases, the suppressed rotational-distortion (modeled as suppression in the hopping parameter24) reduces the coupling between the dxz and dyz orbitals making the α2 band more square-like. This itself enhances the nesting at Q* and QAFM, leading to an increase in χ(q, 0) (see Supplementary Figure 11). Our theoretical analysis therefore indicates that a broad Fermi surface nesting does exist near Q* and QAFM near x ~ 5%. However, we would like to emphasize that nesting alone cannot explain the emergent charge order at Q* and it will require enhanced anisotropic electron–phonon (e–p) coupling and/or strong Coulomb correlations to induce a commensurate charge ordering instability. Recently, strong e–p coupling have been observed at the charge ordering wavevectors in YBa2Cu3O6.625, Bi2Sr2CaCu2O8+x26, and NbSe227. In addition, a significant softening in the temperature dependence of the B1g phonon mode has also been observed to coincide with the metal to Mott insulator transition in Ca3Ru2O7 pointing towards the possibility of similar physics in Mn-doped Sr3Ru2O7. Inelastic resonant X-ray or neutron scattering28,29 can probe the momentum dependence of the e–p coupling and such future experiments may offer a more complete picture of the origin of the observed instabilities in Sr3(Ru1−xMnx)2O7.
Finally, we examine the structure and symmetry of the charge ordering at Q*. Figure 4a shows a zoom-in of the conductance map for the x = 10% sample (white box in Fig. 3e). While the charge order can be clearly observed, the long-wavelength inhomogeneity in the conductance map obscures the detail of the ordering patterns. To visualize the real space character of these patterns, we Gaussian-filter the Q* peaks in the raw unsymmetrized FTs (see Supplementary Figure 12) and inverse Fourier transform back into real space. Figure 4b shows the charge order map corresponding to Fig. 4a. The figure displays a mixture of spatially inhomogeneous checkerboard and stripe patterns. To quantitatively distinguish between the two glassy orders, we compute the amplitudes of the two orthogonal components of the charge order (Fig. 4c, d) and calculate their cross-correlation (Fig. 4e) (see Supplementary Figure 13 and Supplementary Note 5). In a checkerboard order, the two orthogonal components are locally correlated (positive cross-correlation). A stripe order leads to a local anti-correlation between the two orthogonal charge ordering amplitudes. The positive cross-correlation observed as a function of bias (Fig. 4e) indicates globally a four-fold symmetric bi-directional charge order.

Real-space structure of the emergent charge order. a Real space conductance map for x = 10% sample, corresponding to the area in the white box of Fig. 3e. b Inverse Fourier transform (FT) of the FT (preserving the phase) for x = 10% sample after isolating the Q* peaks using a Gaussian profile filter (see Supplementary Note 5). The process enhances the glassy checkerboard charge order seen in a by suppressing the long wavelength inhomogeneity. c, d Spatial amplitude of the charge order along the two orthogonal directions (Y and X), respectively. Higher intensity corresponds to higher strength of the charge order along the specific (Y or X) direction (scale bar is 20 Å). e Cross-correlation of the two orthogonal charge order amplitude maps (see Supplementary Figure 13) as a function of bias. The positive correlation is an indication of a glassy checkerboard order
Discussion
In conclusion, our experiments reveal non-dispersive commensurate charge order instability to emerge near the metal to AFM Mott insulator transition in Sr3Ru2O7. These findings draw similarities to the ubiquitous charge order seen in STM and X-ray experiments on cuprates30,31,32,33,34,35,36,37. While the metal to Mott insulator transition in Sr3Ru2O7, which is likely related to structural distortion and enhanced Coulomb correlations by 3d Mn replacing 4d Ru, is quite different than the charge doping-induced Mott insulator to metal transition in cuprates, it is quite intriguing that a charge-ordered state with similar real-space patterns exists near the Mott transition of both systems. Finally, similar charge modulation patterns on strong spin–orbit coupled Sr2IrO4 have recently been found near its doping-induced Mott insulator to metal transition with a similar periodicity to our Q*38. Overall, all these experiments, independent of their detailed intrinsic electronic structure, point to ubiquitous tendencies towards some form of charge order emerging at the border of Mott insulators.
Methods
Sample preparation and characterization
The single crystals of Sr3(Ru1−xMnx)2O7 used in our experiments were grown at RIKEN laboratory using the floating zone technique. These single crystals were obtained under 1 MPa atmosphere of argon gas containing 10% oxygen. Small flat samples were cut down to ~(2 × 2 × 0.5 mm3) and attached to aluminum plates using H20E conducting epoxy. Aluminum cleaving posts were then attached to samples perpendicular to the a–b cleaving plane using H74F non-conducting epoxy. Samples were cleaved at room temperature in ultrahigh vacuum by knocking the post off, and then immediately transferred in situ to the custom RHK Pan STM head, which had been cooled down to T ~ 13 K. STM topographies were taken in constant current mode and dI/dV measurements were performed using a standard lock-in technique with a reference frequency of 0.921 kHz. PtIr tips were used in all experiments. Tips were prepared prior to each experiment on a Cu(111) surface that had been treated with several rounds of sputtering and annealing, and then placed into the microscope head to cool. The sample and Cu were placed next to each other inside the microscope head so as to reduce exposure and preserve tip structure when moving from one to the other. The data presented in this paper were collected from approximately 3 successfully cleaved samples for each doping percentage (1, 5, 7.5, and 10%). There were negligible differences measured between different cleaves and between different areas on a sample.
Crystal structure
All representations of the crystal structure seen here or in the accompanying supplementary material were produced using VESTA 339.
Tight binding calculations
To calculate the bare susceptibility, we start from a 12 orbital tight-binding model that contains t2g orbitals, the bilayer structure, and the spin–orbit coupling. The hopping parameters are determined by fitting the Fermi surfaces observed in ARPES. Details can be found in ref. 23. The resulting Hamiltonian in the momentum space can be expressed as
where \({\mathrm{\Phi }}_k = \left( {d_{k,s,k_z}^{yz},d_{k,s,k_z}^{xz},d_{k, - s,k_z}^{xy},d_{k + Q,s,k_z}^{yz},d_{k + Q,s,k_z}^{xz},d_{k + Q, - s,k_z}^{xy}} \right)\) are the electron annihilation operators, k is the in-plane momentum, kz is the out-of-plane momentum associated with the bilayer structure, Q = (π, π) is the wavevector associated with the octahedral rotation, and s is the spin index. The matrix element hk(kz) can be found in ref. 23. The retarded bare susceptibility in a multiorbital system can in general be written as a tensor of
where \(W_{l_1,l_2,l_3,l_4}^{\alpha ,\beta } = \psi _{l_1s}^{\alpha \ast }(k)\psi _{l_2s\prime }^\beta (k + q)\psi _{l_3s\prime }^{\beta \ast }(k + q)\psi _{l_4s}^\alpha (k)\), and \(\psi _{ls}^\alpha (k)\) is the component of the eigenvector of the eigenstate α projected on the state with orbital l and spin s. This bare susceptibility expressed in the form of Lindhard function with extra matrix elements takes into account the scatterings between different orbitals in a 12 orbital model. To perform the momentum sum over the Lindhard function, we have used a k-mesh of (Nx × Ny = 250 × 250 points) in all the calculations. In the normal state, the system still has the spin rotational symmetry in the presence of the spin–orbit coupling. As a result, \(\chi _{l_1,l_2,l_3,l_4}^{s,s\prime }\left( {q,\omega } \right)\) is the same for any combination of (s, s′), and consequently we can omit (s, s′). Finally, we compute
which exhibits peaks at wavevectors corresponding to the Fermi surface nesting.
As the doping is introduced, the octahedral rotation is reduced. The leading effect of this octahedral rotation is to enable hopping between different orbitals on nearest neighbor sites. The details of this model are discussed in ref. 23. The reduced octahedral rotation results in less hybridization between the dxz and dyz orbitals. Note that the current model cannot capture the insulating behavior in the paramagnetic state.
Data availability
The authors declare that the main data supporting the findings of this study are available within the article and its Supplementary Information files. Extra data are available from the corresponding author upon request.
References
Tamai, A. et al. Fermi surface and van Hove singularities in the itinerant metamagnet Sr3Ru2O7. Phys. Rev. Lett. 101, 026407 (2008).
Borzi, R. A. et al. de Haas–van Alphen effect across the metamagnetic transition in Sr3Ru2O7. Phys. Rev. Lett. 92, 216403 (2004).
Lee, J. et al. Heavy d-electron quasiparticle interference and real-space electronic structure of Sr3Ru2O7. Nat. Phys. 5, 800 (2009).
Rost, A. W. et al. Thermodynamics of phase formation in the quantum critical metal Sr3Ru2O7. Proc. Natl Acad. Sci. USA 108, 16549 (2011).
Grigera, S. A. et al. Magnetic field-tuned quantum criticality in the metallic ruthenate Sr3Ru2O7. Science 294, 329 (2001).
Rost, A. W., Perry, R. S., Mercure, J. F., Mackenzie, A. P. & Grigera, S. A. Entropy landscape of phase formation associated with quantum criticality in Sr3Ru2O7. Science 325, 1360 (2009).
Lester, C. et al. Field-tunable spin–density–wave phases in Sr3Ru2O7. Nat. Mater. 14, 373 (2015).
Mesa, D. et al. Single-bilayer E-type antiferromagnetism in Mn-substituted Sr3Ru2O7: neutron scattering study. Phys. Rev. B 85, 180410 (2012).
Mathieu, R. et al. Impurity-induced transition to a Mott insulator in Sr3Ru2O7. Phys. Rev. B 72, 92404 (2005).
Hossain, M. A. et al. Mott versus Slater-type metal–insulator transition in Mn-substituted Sr3Ru2O7. Phys. Rev. B 86, 41102 (2012).
Bao, W. et al. Tunable (δπ, δπ)-type antiferromagnetic order in α-Fe(Te,Se) superconductors. Phys. Rev. Lett. 102, 247001 (2009).
Hossain, M. A. et al. Electronic superlattice revealed by resonant scattering from random impurities in Sr3Ru2O7. Sci. Rep. 3, 2299 (2013).
Hu, B. et al. Structure–property coupling in Sr3(Ru1−xMnx)2O7. Phys. Rev. B 84, 174411 (2011).
Li, G. R. et al. Atomic-scale fingerprint of Mn dopant at the surface of Sr3(Ru1−xMnx)2O7. Sci. Rep. 3, 2882 (2013).
Hossain, M. A. et al. Crystal-field level inversion in lightly Mn-doped Sr3Ru2O7. Phys. Rev. Lett. 101, 016404 (2008).
Fang, Z. & Terakura, K. Magnetic phase diagram of Ca2−xSrxRuO4 governed by structural distortions. Phys. Rev. B 64, 20509 (2001).
Kim, T.-H. et al. Imaging and manipulation of the competing electronic phases near the Mott metal–insulator transition. Proc. Natl Acad. Sci. USA 107, 5272 (2010).
Iwaya, K. et al. Local tunneling spectroscopy across a metamagnetic critical point in the bilayer ruthenate Sr3Ru2O7. Phys. Rev. Lett. 99, 057208 (2007).
Dhital, C. et al. Carrier localization and electronic phase separation in a doped spin–orbit-driven Mott phase in Sr3(Ir1−xRux)2O7. Nat. Commun. 5, 3377 (2014).
Hu, B. et al. Surface and bulk structural properties of single-crystalline Sr3Ru2O7. Phys. Rev. B 81, 184104 (2010).
Aynajian, P. et al. Visualizing heavy Fermion formation and their unconventional superconductivity in f-electron materials. J. Phys. Soc. Jpn. 83, 61008 (2014).
Aynajian, P. et al. Visualizing heavy fermions emerging in a quantum critical Kondo lattice. Nature 486, 201 (2012).
Lee, W. C., Arovas, D. P. & Wu, C. Quasiparticle interference in the unconventional metamagnetic compound Sr3Ru2O7. Phys. Rev. B 81, 184403 (2010).
Mukherjee, S. & Lee, W. C. Structural and magnetic field effects on spin fluctuations in Sr3Ru2O7. Phys. Rev. B 94, 64407 (2016).
Le Tacon, M. et al. Inelastic X-ray scattering in YBa2Cu3O6.6 reveals giant phonon anomalies and elastic central peak due to charge–density–wave formation. Nat. Phys. 10, 52 (2013).
Chaix, L. et al. Dispersive charge density wave excitations in Bi2Sr2CaCu2O8+δ. Nat. Phys. 13, 952 (2017).
Weber, F. et al. Extended phonon collapse and the origin of the charge–density wave in 2H-NbSe2. Phys. Rev. Lett. 107, 107403 (2011).
Aynajian, P. et al. Energy gaps and Kohn anomalies in elemental superconductors. Science 319, 1509 (2008).
Keller, T. et al. Momentum-resolved electron–phonon interaction in lead determined by neutron resonance spin–echo spectroscopy. Phys. Rev. Lett. 96, 225501 (2006).
Hoffman, J. E. et al. A four unit cell periodic pattern of quasi-particle states surrounding vortex cores in Bi2Sr2CaCu2O8+δ. Science 295, 466 (2002).
Parker, C. V. et al. Fluctuating stripes at the onset of the pseudogap in the high-T c superconductor Bi2Sr2CaCu2O8+x. Nature 468, 677 (2010).
Ghiringhelli, G. et al. Long-range incommensurate charge fluctuations in (Y,Nd)Ba2Cu3O6+x. Science 337, 821 (2012).
Chang, J. et al. Direct observation of competition between superconductivity and charge density wave order in YBa2Cu3O6.67. Nat. Phys. 8, 871 (2012).
Tabis, W. et al. Charge order and its connection with Fermi-liquid charge transport in a pristine high T c cuprate. Nat. Commun. 5, 5875 (2014).
da Silva Neto, E. H. et al. Ubiquitous interplay between charge ordering and high-temperature superconductivity in cuprates. Science 343, 393 (2014).
Comin, R. & Damascelli, A. Resonant X-ray scattering studies of charge order in cuprates. Annu. Rev. Condens. Matter Phys. 7, 369 (2016).
da Silva Neto, E. H. et al. Charge ordering in the electron-doped superconductor Nd2−xCexCuO4. Science 347, 283 (2015).
Battisti, I. et al. Universality of pseudogap and emergent order in lightly doped Mott insulators. Nat. Phys. 13, 21 (2016).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272 (2011).
Acknowledgements
The authors thank Michael Lawler for helpful discussions. P.A. thanks Abhay Pasupathy for helpful technical discussions. P.A. acknowledges support from the U.S. National Science Foundation (NSF) CAREER under Award No. DMR-1654482. S.M. acknowledges support from New Faculty Initiative Grant from IIT Madras under Project No. PHY/16-17/865/NFIG/SHAA.
Author information
Authors and Affiliations
Contributions
J.L., M.K. and I.G. performed the STM measurements and data analysis. Y.K. and Y.T. synthesized and characterized the materials. S.M. and W.-C.L. carried out the theoretical modeling and analysis. P.A. wrote the manuscript. All authors commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leshen, J., Kavai, M., Giannakis, I. et al. Emergent charge order near the doping-induced Mott-insulating quantum phase transition in Sr3Ru2O7. Commun Phys 2, 36 (2019). https://doi.org/10.1038/s42005-019-0138-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-019-0138-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
