
Abstract
The species of the genus Nannochloropsis are unique in their maintenance of a nucleus-plastid continuum throughout their cell cycle, non-motility and asexual reproduction. These characteristics should have been endorsed in their gene assemblages (genomes). Here we show that N. oceanica has a genome of 29.3 Mb consisting of 32 pseudochromosomes and containing 7,330 protein-coding genes; and the host nucleus may have been overthrown by an ancient red alga symbiont nucleus during speciation through secondary endosymbiosis. In addition, N. oceanica has lost its flagella and abilities to undergo meiosis and sexual reproduction, and adopted a genome reduction strategy during speciation. We propose that N. oceanica emerged through the active fusion of a host protist and a photosynthesizing ancient red alga and the symbiont nucleus became dominant over the host nucleus while the chloroplast was wrapped by two layers of endoplasmic reticulum. Our findings evidenced an alternative speciation pathway of eukaryotes.
Similar content being viewed by others

Introduction
Species in the genus Nannochloropsis are promising for industrial applications because they accumulate large quantities of polyunsaturated fatty acids1,2. They also show promising characteristics allowing convenient genetic improvement, which include, for example, a monoploid nucleus and asexual reproduction, as revealed by mutation3, homologous recombination4, and preliminary genome sequencing5. They are easy to genetically manipulate. Various species of the genus Nannochloropsis are able to perform homologous recombination4 and genome editing6,7. The species in genus Nannochloropsis are mainly used as feed for fish larvae and rotifers, and as food additives for human nutrition. They have also evolved gradually into the models for both industrial applications and biological research8,9.
Genus Nannochloropsis comprises six known species that inhabit marine, freshwater and brackish environments10,11. Recently, a phylogenetic analysis of the concatenated 18S ribosomal RNA (rDNA) and rbcL for Nannochloropsis taxon revealed a new species, N. australis, and erected a new genus, Microchloropsis, comprising M. salina and M. gaditana, which were modified from N. salina and N. gaditana, respectively12. Whether they belong to Nannochloropsis or Microchloropsis, the species in these taxa are assigned to class Eustigmatophyceae, phylum Ochrophyta, superphylum Heterokonta (or Stramenopiles)10. Heterokonta refers to flagellated cells with two differently shaped flagella. Of the species in Heterokonta, the colored alga-like species in phylum Ochrophyta include diatoms and golden, brown, and yellow-green algae, among others. However, all of the species in the genera Nannochloropsis and Microchloropsis are small, nonmotile, and spherical in shape13; they can be distinguished only by their rbcL and rDNA sequences14,15,16. The algae of genus Nannochloropsis are able to build up high concentrations of diverse pigments, such as astaxanthin, zeaxanthin and canthaxanthin17 and have chlorophyll a but completely lack chlorophylls b and c18.
The genomes of two species in the genus Nannochloropsis, N. gaditana19,20 and N. oceanica21,22, have been sequenced on the Illumina platform. Their genomes vary between 28.5 and 29.0 Mb in size and are characterized by a high density of genes, low intron content, short spacers between genes19 and few repetitive sequences21. In addition, a comparison of the genomes of all six species in the genus Nannochloropsis revealed an extreme case of dose expansion among lipid biosynthesis genes23. These genomes are all sequenced on Illumina platforms, and have not been scaled up to pseudochromosomes with the newly commercialized technologies, including single-molecule real-time (SMRT) sequencing24 and Hi-C technology25,26, among others. Such scenario is detrimental to identify completely the genes each genome.
We noted in particular that the ultrastructure of N. oculata is unusual; a nucleus-plastid continuum exists throughout its cell cycle27, but the basal body and flagellum observable in green algae28 are absent. Considering comprehensively the above findings, we suspected that an invaded red algal nucleus may have overthrown the host protist nucleus during evolution, i.e., a red algal nucleus has occupied the host cytoplasm, and the host nucleus has been expelled. Unfortunately, these unique characteristics have not been deciphered genetically and evolutionarily.
In this study, we sequenced the genome of N. oceanica with a real-time DNA sequencing method and scaled up the primary assembly with the chromatin conformation information revealed by Hi-C data, aiming to look for evidence supporting our assumptions. A genome assembly of 29.3 Mb containing 32 chromosome-scale scaffolds (pseudochromosomes) was obtained. The genome contains 7330 protein-coding genes, and most of them may originate from an ancient red alga symbiont nucleus through secondary endosymbiosis during speciation. We also found that N. oceanica has adopted a genome reduction strategy during speciation; it has lost its flagella and abilities to undergo meiosis and sexual reproduction. We propose that N. oceanica emerged through the active fusion of a host protist and a photosynthesizing ancient red alga and the symbiont nucleus became dominant over the host nucleus, while the chloroplast was wrapped by two layers of endoplasmic reticulum. Our findings evidenced an alternative speciation pathway of eukaryotes.
Results
Genome assembly and characterization
We sequenced the genome of N. oceanica with a third-generation sequencing technique. The quality reads were assembled into contigs that were then grouped, ordered and oriented into pseudochromosomes based on Hi-C revealed chromatin conformational information. In total, 3.31 G raw PacBio reads (4.3 kb in average length, ~113-fold of the genome) were generated on the Sequel System, and 773,137 clean reads were obtained. The PacBio reads were self-corrected and assembled with Canu and polished using PacBio reads and Illumina reads (~105-fold of genome), and then assembled into 129 contigs with a contig N50 of 664.75 kb. The longest contig was 1.54 Mb in length. The assembled genome was 29.3 Mb in total length. The GC content of the genome was 54.01%. To evaluate the integrity of the assembly, the assembly was assessed with 303 Benchmarking Universal Single Copy Orthologs (BUSCO)29,30 genes from eukaryota ortholog database, of which 264 (87.1%) were annotated and 255 (84.2%) were intact. In order to anchor the 129 contigs into pseudochromosomes, the Hi-C library was constructed and ~425.8 million reads were generated on Illumina platform. In total, ~201.7 million valid interaction reads were used to build the interaction matrices and draw the heatmap. Eighty six contigs were ordered as trunks which contained 26,395,858 bp (~93.84%) and the others were reinserted in between the trunks to maximize the amount of linkage. As results, 128 contigs were ordered (28,129,022 bp, ~95.99%) and these contigs have high-confidential orientations within each chromosome-scale group as were calculated with weighted directed acyclic graph (WDGA). With the assistance of Hi-C data, the primary genome assembly was scaled up to 32 pseudochromosomes (pseudochromosomal N50 = 1.14 Mb), including 30 nuclear, one mitochondrial, and one chloroplast pseudochromosomes (Table 1, Supplementary Tables 1–4, Fig. 1a–c). The possible errors revealed by the heatmap showing interactions among the pseudochromosomes (Fig. 1b) may result from the contigs containing high portions of repeats rather than from the order and orientation of the contigs themselves.

Protein-encoding gene models on each pseudochromosomes of N. oceanica. Mitochondrial and chloroplast pseudochromosomes are presented linearly but they can be circulated as their two ends contain the repeat assemblies of sequences. a The pseudochromosomes with gene models marked with vertical bars. The green bars represent gene models in the sense strand, while the yellow ones in the anti-sense strand of DNA. b The heatmap showing the interaction between 30 nuclear pseudochromosomes. c The Venn diagram of functional genes annotated against NT, NR, BLASTX, and BLASTP databases. d The similarity between nuclear pseudochromosomes. Less homozygosity is found among these pseudochromosomes, which is one of the characteristics of N. oceanica, genome reduction
Gene modeling and homolog searching identified 7330 nonredundant protein-coding genes. The exons of these genes were 483.7 bp in average. The introns of these genes were 370.9 bp in average. The number of exons in each gene was 2.87. Different numbers of non-protein-coding miRNA, tRNA, rRNA, and snRNA genes were also predicted, which in total accounted for 0.0674% of the genome. The repeated sequences occupied ~19.52% of the genome (Table 1; Supplementary Tables 5–8).
The genome assembly of N. oceanica contained 7330 protein-coding genes, which were 2084 bp in average. Unfortunately, a portion of these genes were annotated as putative/hypothetical protein-coding genes. It was interesting to note that 1030 genes (14.05%) contained 2354 transposons, ~2.26 transposons each. This result was different from those in higher plants where retrotransposons are the greatest contributors to the size expansion of the genome21. The transposons in the genome of N. oceanica were fewer in number, indicating reduction of N. oceanica genome (Table 1; Supplementary Table 9; Fig. 1d). The high-quality genome assembly ensured that searching for homologous genes and phylogenetic analyses of both genomes and genes can be carried out appropriately.
Phylogenetic position of N. oceanica within Heterokonta
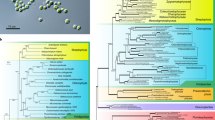
The phylogenetic tree of 18S ribosomal RNA gene (rDNA) sequences representing Rhodophyta, Chromista, Viridiplantae, and Glaucophytes, among others, showed that Nannochloropsis were at the top of the current Chromista (Fig. 2c; Supplementary Fig. 1), which were similar to some peridinin-containing dinoflagellates evolved through tertiary endosymbiosis. The nuclei involved in diverse endosymbioses belong to different protists. The positions of Nannochloropsis on the phylogenetic tree of 18S rDNA were as expected and demonstrated that the nuclei of Nannochloropsis are host ones. It was expected that the vast majority of Nannochloropsis protein-coding genes could be placed at the position of rDNA. However, we found that the whole-genome-based microevolution of Heterokonta showed a parallel relationship between Nannochloropsis and the microalgae derived through the second endosymbiosis (Fig. 2a), indicating that the vast majority of Nannochloropsis protein-coding genes are phylogenetically different from rDNA. This discrepancy implied that Nannochloropsis nuclei have different origins from those of other Chromista.

Phylogeny of Nannochloropsis and their relatives in different taxa deduced from nuclear genome (a), chloroplast genome (b), 18S ribosomal RNA gene (18S rDNA, c) and ribulose bisphosphate carboxylase large chain gene (rbcL) (d). a The phylogenetic tree of species inferred from the OrthoFinder using the genome protein sequences each species from the whole genome. b The phylogenetic tree of species inferred from the OrthoFinder using the chloroplast genome sequences each species. c The phylogenetic trees for 18S rDNA. The tree shows the consensus tree topology inferred by Bayesian analysis using alignments of 18S genes from NCBI. The scale bar indicates the nucleotide substitutions per site. This consensus topology derived from 512 trees, lnL = 22033.73. d The phylogenetic trees for rbcL protein. The tree shows the consensus tree topologies inferred by Bayesian analysis using alignments of rbcL proteins from NCBI. Scale bars represent 0.1 amino acid substitutions per site. In total, 439 aligned amino acid sites were analyzed. This consensus topology derived from 726 trees, α = 0.47 (0.41 < α < 0.56), pI = 0.0019 (0.0000007 < pI < 0.0059) and lnL = 10364.8
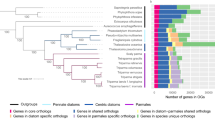
We calculated the percentages of homologous genes of two diatoms, one golden microalga and two Nannochloropsis in E. huxleyi, G. theta (nucleus) and two single-cell red algae (Fig. 3a). We found that the percentages of genes homologous to those of red algae are similar among Nannochloropsis and other Chromista, while the percentages of genes homologous to those of Haptophytes and Cryptophytes are obviously different (Fig. 3a). The percentages of Nannochloropsis genes with homologs in Haptophytes and Cryptophytes are obviously lower than those of diatoms and golden microalgae, indicating that a portion of Nannochloropsis genes have been replaced by those from the symbiont nucleus. When we calculated the microevolutionary tree, OrthoFinder simultaneously generated a list showing the homologs among species used to calculate the tree. We selected three species, N. oceanica, A. anophagefferens, and P. tricornutum, as data points; their genome sequences are currently available, and their positions on the 18S rDNA phylogenetic tree are appropriate for determining the sources of genes. We counted the homologs among N. oceanica, A. anophagefferens, and P. tricornutum. Of 7330 protein-coding genes of N. oceanica, 2867 genes were unique, and 4463 genes were homologous to A. anophagefferens (656) or P. tricornutum (1329) only or to both of them (2478) (Fig. 3b). Judging from the number of homologous genes, the nucleus of N. oceanica is more similar to that of P. tricornutum and, thus, more distant from that of A. anophagefferens (1329 vs. 656). Except for the microevolutionary tree, the OrthoFinder calculated also the phylogenetic trees for the homologs using BLAST, MAFFT, and FastTree as the tools and green algae and Glaucophytes as outgroup. A gene of N. oceanica is from the host nucleus if it is phylogenetically near its homolog from A. anophagefferens (Pattern I; Fig. 3c), while a gene of N. oceanica is from the symbiont nucleus if it is phylogenetically far from its homolog in A. anophagefferens (Patterns II and III, Fig. 3c). In order to clarify the evolutionary relationships of 2478 N. oceanica proteins with homologs in both A. anophagefferens and P. tricornutum, we aligned the amino acid sequences of these proteins one by one by setting that a cluster must contain only two sequences, and found that 645 (26%) belonged to Pattern I, 635 (26%) to Pattern II, and 722 (29%) to Pattern III. The remaining 476 cannot be categorized into these patterns; they are encoded by multiple copy genes (Fig. 3c). The genes in pattern I were phylogenetically near A. anophagefferens, indicating their host nuclear origin, while those in patterns II and III were phylogenetically far from A. anophagefferens, indicating their symbiont nuclear origin. Judging again from the number of genes homologous to those of both A. anophagefferens and P. tricornutum, Nannochloropsis nuclei are different from those of A. anophagefferens (1357 vs. 645). Therefore, we believe that the Nannochloropsis nuclei derive mainly from symbiont nuclei.

Homologous protein-coding genes of N. oceanica found among a few representative microalgal species. a The percentages of homologous genes of P. tricornutum, T. pseudonana, A. anophagefferens, N. oceanica, and N. gaditana found in the nuclear genomes of E. huxleyi, G. theta, C. merolae, and G. sulphuraria. b The homologous protein-coding genes of N. oceanica found in A. anophagefferens but not in P. tricornutum, in P. tricornutum but not in A. anophagefferens and in both of them. c The number and percentage of the homologous genes of N. oceanica found in both A. anophagefferens and P. tricornutum (2478 in total) can be further partitioned into three patterns (I through III) and multi-copy genes. The pattern I contains the genes phylogenetically near to those of A. anophagefferens, while patterns II and III contain the genes phylogenetically far from those of A. anophagefferens
The protoplasts of Chromista are believed to have a single ancient origin31,32. However, we found that the phylogenetic position of class Eustigmatophyceae is unusual. As indicated by the phylogenetic tree of the rbcL, families Eustigmatophyceae and Dictyochophyceae clustered separately from single-cell red algae and Chromista (Fig. 2d; Supplementary Fig. 2). The whole-genome-based microevolutionary analysis showed that Nannochloropsis formed a clade outside the single-cell red algae (Fig. 2b). These findings demonstrated that the symbiont carries an ancient red algal chloroplast. Accordingly, we believe that the symbiont is an ancient red alga that is phylogenetically distant from the current single-cell microalgae.
Simple protein import mechanism into the plastid
We have extensively searched the genome of N. oceanica obtained in this study and those of N. gaditana, P. tricornutum, C. merolae, and G. sulphuraria, which were published previously, for the protein components of translocons (or complexes) mediating protein import across the four membranes covering their plastids using the identified proteins33,34 as the queries. We found that the major components of SELMA complexes were absent in the two species of Nannochloropsis, and a few residual proteins had no predictable bipartite topogenic signal except sDer1-1 and sHsp70 of N. oceanica (Table 2). We also found that TOC75, the main component of the import complex across the outer membrane of plastids, was absent in N. oceanica, while its alternative, Omp85, existed (Table 3). In addition, the main component of the import complex across the inner membrane of plastids, Tic110, was found to exist in N. oceanica but not in N. gaditana, which may be due to the incompleteness of its genome sequence. All these findings indicated that N. oceanica and N. gaditana have either evolved a different protein import system from that of P. tricornutum or do not need these complexes at all.
Nonmotility of N. oceanica
The flagellum is an axial 9 × 2+2 structural assembly that includes substructures such as dynein arms, radial spokes, and a central pair complex. Jang et al.35 described the existence of flagellum-associated genes in motile (mainly dinoflagellates), motile small (cryptophytes, euglenophytes, stramenopiles, and haptophytes), and nonmotile (stramenopiles, haptophytes, diatoms, and green algae) protists and found a nearly two-fold differential between motile and nonmotile protists (104 vs. 55). We found that the total numbers of flagellum-associated genes in N. oceanica and N. gaditana were 29 and 17, respectively (Table 4 and Supplementary Data 1), which explained to some extent the two most salient characteristics of Nannochloropsis species, their small size and nonmotility. The loss of flagellum-associated genes also reflected the reduction of Nannochloropsis genomes. From being motile to being small and motile to being completely nonmotile, the number of movement-associating genes is reduced exponentially. Nannochloropsis have lost their ability to move because their number of movement-associating genes is approximately one-fourth that of motile protists and approximately one-half that of small and nonmotile protists.
Loss of sexual reproduction
From mating types in yeast to different sexes in plants and animals and from a single gene to sex chromosome systems36,37,38,39 sex determination mechanisms have been intensively investigated. The sexual reproduction and characteristics of organisms can be extremely complex; however, mating, cell fusion, and meiosis are among the basic processes that are absolutely involved in sexual reproduction. We have tried to identify the mating and cell fusion-associating genes found in yeast40 and diatoms (phylogenetically closer to Nannochloropsis)41. Unfortunately, we found that this gene set is extremely diverse; thus, it is difficult to uncover the homologs of yeast and diatom cell mating and fusion genes among other organisms, including N. oceanica and N. gaditana. We turned to the second basic process involved in sex determination, the meiosis, focusing on the meiosis-specific genes developed as a sex detection toolkit42,43.
During evolution, the function of a gene may be replaced by other genes, and a gene may evolve a new function. We assumed that a meiosis-specific gene was truly absent and not replaced by an unidentified gene if it existed in a relatively wide taxonomic range of species. Of the nine meiosis-specific genes documented early42,43, Rec8 was not identified in the two species of genus Nannochloropsis. A gene homologous to Rec8 is found in their genomes; however, it is phylogenetically similar to Rad21, a mitosis-associating but not specific gene. The remaining eight meiosis-specific genes were identified with BLASTp or tBLASTn and by phylogenetic analysis in a yeast, two green algae, a brown alga and a haptophyte among the currently available genome sequences (Table 5; Supplementary Data 1). Although they do not completely encompass all species reproducing sexually, these species represent a relatively wide taxonomic range of species. Eight meiosis-specific genes are conserved among these species, indicating that they cannot be replaced and are not substitutable in meiosis. We failed to identify three meiosis-specific genes (Hop1, Dmc1, and Mnd1) in the genome of N. oceanica and two (Hop1 and Hop2) in the genome of N. gaditana (Table 5). These findings (the absence of meiosis-specific and mating-associating genes) indicated that species in the genus Nannochloropsis have lost their ability to reproduce sexually.
Discussion
Heterokonta (or Stramenopiles) refers to the type of motile life cycle stage at which the flagellated cells possess two differentially shaped flagella. The typical classes in this infrakingdom10 include the colored Ochrophyta (alga-like) and colorless Pseudofungi and Bigyra phyla. The classes in the alga-like Ochrophyta include Dictyochophyceae (axodines), Bacillariophyceae (diatoms), Chrysophyceae (golden algae), Eustigmatophyceae, Phaeophyceae (brown algae), and Xanthophyceae (yellow-green algae), among others, while those in Pseudofungi include Oomycetes (water molds), among others44. However, the two most prominent characteristics, motile and flagellated cells, have never been observed among species of genus Nannochloropsis, class Eustigmatophyceae. This discrepancy between classification and observation prompted us to rethink the phylogenetic position of genus Nannochloropsis within phylum Ochrophyta.
We found that the phylogenetic positions of Nannochloropsis indicated by their 18S rDNA were different from those indicated by their whole genomes. The former implied an identical origin way to modern Chromista, while the latter implied an unusual way different from that of modern Chromista. If the nuclei of modern Chromista belong to diverse host protists, those of Nannochloropsis should belong to a protist different from all of them. We found that Nannochloropsis contained a similar number of red algal genes with diatoms and golden microalgae but obviously less genes of Haptophytes and Cryptophytes than diatoms and golden microalgae, indicating that a portion of Nannochloropsis genes should have been replaced by those from the symbiont nucleus. The positions of N. oceanica, A. anophagefferens, and P. tricornutum on phylogenetic trees are appropriate for determining the sources of genes; a gene of N. oceanica is from the host nucleus if it is phylogenetically near its homolog from A. anophagefferens, while a gene of N. oceanica is from the symbiont nucleus if it is phylogenetically far from its homolog in A. anophagefferens. We found that the nucleus of N. oceanica is more distant from that of A. anophagefferens. N. oceanica has more genes from the symbiont, thus its nucleus derived mainly from symbiont nucleus during speciation. As shown on rbcL phylogenetic and the whole-genome-based microevolutionary trees, the symbiont carries an ancient red algal chloroplast. Therefore, we believe that the symbiont is an ancient red alga that is phylogenetically distant from the current single-cell microalgae.
The widely accepted understanding is that a protist engulfed and enslaved a red alga and discarded its nucleus but returned its chloroplast during secondary endosymbiosis, yielding cryptophytes, haptophytes, and heterokonts. During the speciation of Nannochloropsis, it seems that the host nucleus contributed only a small portion of the genes, including the rDNA, while the symbiont nucleus contributed a large portion of the genes. In other words, the symbiont nucleus has overthrown the host nucleus. We propose that Nannochloropsis evolutionarily emerged through the active fusion of a host protist and a symbiont photosynthesizing protist, forming a triad of two nuclei and a plastid; the two nuclei were covered by independent nuclear membranes and shared endoplasmic reticulum, while the plastid was covered by two layers of endoplasmic reticulum outside the two membranes derived from the primary endosymbiosis; and the two nuclei interacted with each other, the symbiont nucleus finally won the competition, and the host nucleus was overthrown during the speciation of Nannochloropsis (Fig. 4). The cell fusion mode of speciation requires no evolution of new structure and function, which should have helped Nannochloropsis to thrive during speciation.

The hypothetical route of N. oceanica speciation through cellular fusion and nuclear haploidization. The symbiont nucleus may have overthrown the host nucleus during speciation of Nannochloropsis. The cell fusion mode of speciation needs no evolution of new structure and function, which should aid to fast speciation of Nannochloropsis. Instead of free floating, the plastid homing in the host cytoplasm may be enveloped by either one or two layers of cytoplasmic reticulum, forming a continuum with nucleus and dividing simultaneously with nucleus
We can find more clues among previous studies. For example, the nucleus and plastid of N. oculata accompany each other throughout the whole cell cycle by forming a nucleus-plastid continuum covered by a membrane with ribosomes on the outer surface27. This observation supports our hypothesis. The existence of barrel-shaped nuclear pole bodies27 in N. oculata evidence the ancient nature of the nuclei of Nannochloropsis, which may explain the unusual position of Nannochloropsis on the rbcL phylogenetic tree. In addition, it has been hypothesized previously that the two outermost membranes of complex plastids may both originate from the endoplasmic reticulum45.
Cell fusion and nuclear competition may avoid the formation of complex plastids and the requirement of new protein targeting systems. Complex plastids are believed to be veiled by four membranes originating during secondary endosymbiosis45,46. Early studies have demonstrated that translocons (or complexes) help to import proteins synthesized on the cytoplasmic ribosomes into the periplastid compartment and the stroma of the chloroplast47,48,49. From the outermost (possibly derived from the host endoplasmic reticulum) to the second outermost (periplastid membrane) and the inner plastid membranes, Sec61, SELMA, and TIC complexes have been suggested to import preproteins across these membranes; however, the TOC complex in the outer plastid membrane has not yet been found among complex plastids50. Instead of the TOC complex, a homolog of TOC75 (Omp85) has been demonstrated to mediate the translocation of proteins across the outer membranes of complex plastids51, and a Der complex has been proposed to replace TOC52. In addition, the genes encoding SELMA complex components are believed to be recycled from the red alga symbiont, and their corresponding proteins are synthesized in the cytoplasm and imported either into the periplastid compartment or the plastid stroma. These proteins are called preproteins and are marked by a bipartite topogenic signal at their N-terminals. A bipartite topogenic signal consists of a signal peptide for crossing Sec61, a transient peptide for crossing the periplastid membrane and a special amino acid residue between them for targeting the plastid stroma50. If the two outermost membranes of chloroplasts are derived from the endoplasmic reticulum, the evolution of complex preprotein transportation systems can be avoided. As was strongly expected, the plastids of N. oceanica and N. gaditana had not evolved a protein import system similar to that of P. tricornutum.
If N. oceanica and N. gaditana do not need these complexes, they must have adopted a membrane system different from that of P. tricornutum during evolution. Secondary endosymbiosis is always believed to be a passive process in which the host protist engulfs a photosynthesizing alga and enslaves its plastid but discards its mitochondria. This widely accepted mechanism hypothesized that: the newly appearing chloroplast must be covered by four membranes. The outermost membrane possibly originated from the endoplasmic reticulum, while the periplastid membrane possibly originated from the host cellular membrane, and the two interior membranes were inherited from the first endosymbiosis; a new system must import proteins synthesized in the cytoplasm into the periplastid compartment and the stroma of plastids; and the genes of the enslaved nucleus must undergo horizontal transfer to the host nucleus on a large scale. It seems that this mechanism has received strong support from the studies carried out in some Chromista47,48,49. However, this mechanism also has exceptions, including, for example, the absence of the components of the protein import system in many other species, including N. oceanica and N. gaditana as analyzed in this study. In our assumption of N. oceanica speciation, the chloroplast of N. oceanica was enveloped outside by two layers of endoplasmic reticulum, which is homogeneous to that found in the cytoplasm. Sec61 and ERAD should be involved in transportation into and out of the endoplasmic reticulum45. Although some modifications may exist, the protein transportation system of Nannochloropsis was inherited from the symbiont, e.g., an ancestral single-cell red alga. This evolutionary path would readily explain the speciation of Nannochloropsis.
The two nuclei fused to make a hybrid nucleus, which then reduced its genetic information by haploidization. During this kind of evolution, diverse structures and functions may have been lost through either selection or genetic drift or both, including motility and sexual reproduction. The flagellum plays important roles in competition for nutrients by enabling rapid movement, and thus, rapid finding, capture, and ingestion of prey. It also helps autotrophic and mixotrophic species sense and receive solar light. It is known that all species in the genus Nannochloropsis are extremely small, structurally simple and nonmotile13. The advantages and/or disadvantages of these characteristics in evolution remain unclear. However, these characteristics may imply the loss of structures, thus marking the reduction of the Nannochloropsis genome. In this study, we found that most flagellum-associating genes were lost, providing at least one example of such change during the speciation of N. oceanica.
The genome reduction of N. oceanica has also caused the loss of its other functions, including sexual reproduction. Neither ploidy variation nor morphological mating and cellular fusion has been documented among the species in the genus Nannochloropsis to date. As was expected, the function of sexual reproduction has been lost during the speciation of N. oceanica; an incomplete set of meiosis-specific genes were identified in the genome of N. oceanica. We once tried to determine the existence of sexual reproduction among microalgae with their genome sequences available by setting the number of meiosis-specific genes ≥ 653. This early set threshold is roughly in accordance with what we found in this study. With the increase in available genome sequences, we believe that molecular characterization of the reproductive strategies of many organisms will become feasible and convenient.
Methods
Microalgal cultivation and DNA isolation
The marine microalga N. oceanica was provided by Key Laboratory of Mariculture of the Chinese Ministry of Education, Ocean University of China. The algae were cultivated in f/2 medium (pH 7.8, salinity 30)54 prepared with natural seawater and autoclaved at 121 °C for 30 min. DNA was isolated with the cetyltrimethylammonium bromide (CTAB) method55.
Genome sequencing
The whole genome of N. oceanica was sequenced on the PacBio Sequel System (https://www.pacb.com/products-and-services/pacbio-systems/sequel/) based on the single-molecule real-time (SMRT) sequencing technology. The template library was constructed using SMRTbell Template Prep Kit 1.0 (product code 100-259-100) and SMRTbell Damage Repair Kit (product code 100-465-900). Following the procedure described in the PacBio brochure to prepare > 20 kb template using BluePippin™ size-selection system (15–20 kb cutoff) for Sequel™ Systems, the quality DNA was fragmented with g-TUBE (covaries, 520079) and concentrated with AMPure® PB magnetic beads with the fragments eluted with the Pacific Biosciences® Elution Buffer. The fragments were damage-repaired with ExoVII, end-repaired with End Repair Mix and ligated with the blunt adapter. After removing away the failed ligation products with ExoIII and ExoVII, the ligation products were purified twice with AMPure® PB Beads, and subjected to size selection using BluePippin™ Size-Selection System. The yielded fragments were bead-purified, damage-repaired, and used as the ~20 kb SMRTbell templates. The templates were annealed with primers and bound to DNA polymerase using the PacBio DNA/Polymerase Kit and magnetic beads, and loaded onto PacBio Sequel™ Systems to read the sequence of templates.
Generation of short reads for genome correction
In order to collect the Illumina paired-end reads, the possible degradation and contamination of genomic DNA were observed in 1% agarose gel, the purity of the genomic DNA was determined on NanoPhotometer® (IMPLEN, CA, USA) and the concentration was measured with Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). The genomic DNA survived quality control was used to construct the short fragment library following the TruSeq DNA Sample Preparation Guide (Illumina, 15026486 Rev. C). The procedure includes mainly the steps of DNA fragmentation, end-repairing, base A tailing, adaptors ligation, recovering of DNA with desirable sizes from gel, and PCR amplification of the recovered DNA. The amplification products are used as the libraries for sequencing once they survived the quality checking. In brief, the amplification products were quantitated on Qubit2.0, and the size range of the amplification products was determined with Agilent 2100. If the fragment size ranged as the expected, the library was accurately quantified with Bio-RAD CFX 96 real-time quantitative PCR thermocycler and Bio-RAD KIT iQ SYBR GRN Q-PCR thermocycler. The quality library was sequenced on HiSeq X Ten Platform set at PE150 program with paired-end reads obtained.
Genome assembling
The reads exported by Sequel™ Systems were quality evaluated with the in-built High Quality Region Finder (HQRF) which identifies the longest high-quality regain each read generated by a singly-loaded DNA polymerase according to the ratio of signal to noise. Upon being generated by the system, all bases were marked with exclamation point in order to make the format perfect. The quality reads (or regions) were marked with 0.8, while the low quality ones were marked with zero.
The quality reads obtained were assembled into contigs using Canu (v1.5 https://github.com/marbl/canu)56 by setting the parameters as the following: canu -pacbio-raw nanoc.subreads.bam.fasta -p nanoc -d nanoc -canu genomeSize=40m gridEngineMemoryOption=“-l vf=MEMORY”. Canu uses the all-versus-all overlap information to correct individual reads. It selects these overlaps in a two-step manor, global and local filtrations. The global filtration identifies the targets where a read may provide correction support, and the local filtration allows a read to accept or reject the correction evidences provided by other reads. At the trimming stage, Canu identifies the region of reads without correction supports, trimming or splicing the reads into their longest regions with correction supports. These regions were subjected to a final identification of sequencing errors, and used to construct the best overlap graph as the output contigs and make a summary statistics.
The errors in the primary assembly were identified and corrected with blasr (v5.1, https://github.com/PacificBiosciences/blasr)57 and Arrow (v2.2.1), a tool built in Smrt Link (https://downloads.pacbcloud.com/public/software/installers/smrtlink_5.0.1.9585.zip). First, the PacBio reads were mapped to the raw contig using blasr with the parameter: --bam --bestn 5 --minMatch 18 --nproc 4 --minSubreadLength 1000 --minAlnLength 500 --minPctSimila rity 70 --minPctAccuracy70 --hitPolicy randombest --randomSeed 1, and then the consensus sequences were obtained and variants were called via Arrow with the default parameters.
The consensus genome was subjected to a final round of base-error correction (polish) by referring to the Ilumina reads with BWA (v0.7.9a) and Pilon (v1.22, https://github.com/broadinstitute/pilon)58. The Illumina paired-end reads were mapped to the contigs by BWA (parameter, -k 30), and Pilon 1.22 (default parameters) uses this alignment to correct the assembly. The quality of the genome sequence obtained was further evaluated with BUSCO (v2, http://busco.ezlab.org/)59 (default parameters) based on a benchmark of 303 conserved eukaryota ortholog database.
Construction of Hi-C library
Algal cells (0.2 g, fresh weight) were harvested through centrifugation and fixed with 2% formaldehyde to crosslink the DNA and proteins at a final concentration of 1% (v/v) and at room temperature for 15 min. The crosslinking was quenched with glycine at a final concentration of 0.2 M through incubation at room temperature for 5 min. The algal cells were collected and resuspended in cold 1× PBS. The cells were rinsed again and the cell pellet was flash-frozen in liquid nitrogen and stored at −80 °C.
The crosslinked algal cells were lysed in Hi-C lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% Igepal CA630) supplemented with the protease inhibitors (Sigma, P8340) on ice for > 15 min. The debris (mainly nuclei) was collected through centrifugation (2500 × g, 5 min), washed with ice-cold Hi-C lysis buffer, gently resuspended in 0.5% sodium dodecyl sulfate (SDS) and incubate at 62 °C for 5–10 min, and then quenched with 10% Triton X-100 (Sigma, 93443) at 37 °C for 15 min. The chromatin was digested with Mbo I (New England Biolabs, R0147) overnight at 37 °C. The chromatin digestion was ceased by incubating at 62 °C for 20 min.
The cohesive ends of DNA were filled in and biotin labeled with DNA polymerase I Large (Klenow) Fragment (NEB, M0210) and biotin-14-dATP (Life Technologies, 19524-016) at 37 °C for 45 min. The DNA fragments were ligated proximately with T4 DNA ligase (NEB, B0202) at room temperature for 4 h. Crosslink was reversed by degrading the protein with proteinase K (NEB, P8102) when SDS was present at 55 °C for 30 min.
In order to extract HiC information through high-throughput sequencing using Illumina sequencers, the biotinylated DNA was sheared to sizes varying between 300 and 500 bp in length with Covaris LE220 (Covaris, Woburn, MA) (volume 130 μL in a Covaris microTUBE; fill level 10; duty cycle 15; PIP 500; cycles/burst 200; time 58 s). The sheared DNA was eluted and verified to be successfully sheared through agarose gel electrophoresis. The sheared DNA was purified with AMPure XP beads (Beckman Coulter, A63881). The DNA fragments in the range of 300–500 bp retained on the beads were eluted, quantified by Qubit dsDNA High Sensitivity Assay (Life Technologies, Q32854), and run on a 2% agarose gel to verify successful size selection.
The sheared DNA fragments were end-repaired with NEB T4 PNK (NEB, M0201), NEB T4 DNA polymerase I (NEB, M0203), and NEB DNA polymerase I Large (Klenow) Fragment (NEB, M0210) at room temperature for 30 min, and NEB Klenow exo minus (NEB, M0212) at 37 °C for 30 min. The DNA fragments containing biotin labels were pulled down and the biotin from unligated ends were removed away by adding to the reaction the pretreated Dynabeads MyOne Streptavidin T1 beads (Life Technologies, 65602). The Illumina indexed adapter was added to the DNA fragments attained on the beads, which were further polished with NEB Klenow exo minus (NEB, M0212) and ligated with an Illumina indexed adapter with NEB DNA Quick ligase (NEB, M2200) at room temperature for 15 min.
The HiC library was constructed by amplifying the innate one directly off of the T1 beads through PCR using Illumina primers and protocol, which was separated from the beads, purified with AMpure XP beads and eluted from the beads into 1× Tris buffer, yielding an in situ HiC library. The library was quantified and sequenced on an Illumina HiSeq 4000 Platform (San Diego, CA, USA) which reads 150 bp paired ends of DNA fragments.
HiC information aided scaling-up of the genome assembly
A read was considered to be clean if it contained < 5 bases from adaptor; > 50% of bases with phred quality value of < 19; and < 5% of unknown base (N). The clean reads were first aligned on the genome assembly using the bowtie 2 (v2.2.3; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)60. The parameters used for bowtie were as the following: --very-sensitive -L 20 --score-min L,−0.6,−0.2 --end-to-end --reorder --rg-id BMG --phred33-quals -p 5. The unmapped reads were mainly composed of the chimeric regions spanning across the ligation junction. Following the philosophy of the Hi-C, the ligation site of an unmapped read was detected with HiC-Pro (v2.7.8; https://github.com/nservant/HiC-Pro)61 using an exact matching procedure and aligned its 5′ fractions back onto the genome. The results of both mapping steps were merged into a single alignment file, while the low mapping quality and multiple hitting reads and singletons were discarded.
The genome of N. oceanica was sequenced with the third-generation sequencing method. The contigs obtained cannot be scaled up to scaffolds; only one library was constructed. In this case, contig and scaffold are identical in meaning. The valid interaction pairs were used to build the interaction matrices and scale up the primary genome assembly in contigs to chromosome-scale scaffolds (hereafter pseudochromosomes) with LACHESIS26 (https://github.com/shendurelab/LACHESIS). The tool first clustered the contigs into chromosomal groups with the agglomerative hierarchical clustering algorithm and then ordered and oriented the contigs of each chromosomal group into pseudochromosomes. LACHESIS ordered the contigs by building a graph and extracting the longest path as the trunk (the highest-confidence order of the contigs in a chromosomal group). Those contigs excluded by the trunk were reinserted according to the linkage information between adjacent contigs. The ordered contigs were oriented by building a weighted, directed and acyclic graph (WDAG)26. The weight was calculated as the log-likelihood of Hi-C links between adjacent contigs25, and the orientation each contig each chromosomal group was predicted according to the maximum likelihood path through WDAG.
N. gaditana may contain 30 chromosomes, one chloroplast, and one mitochondrial chromosomes62. Accordingly, we set CLUSTER_N = 30 for LACHESIS. The tool then full-range scanned the five key parameters, including CLUSTER_MIN_RE_SITES [15,2000], CLUSTER_MAX_LINK_DENSITY [1,10], CLUSTER_NONINFORMATIVE_RATIO [1,v 10], ORDER_MIN_N_RES_IN_TRUNK [15,2000], and ORDER_MIN_N_RES_IN_SHREDS [15,2000]. All other parameters were set as the default. After about 2000 trials, a pseudochromosome candidate was picked up if it contained > 95% of the contig (in total length) and its corresponding trunk contained > 90% of the contigs (in total length). A single pseudochromosome was considered as the best if it contained 100% of the contig (in total length) and its corresponding trunk contained > 93% of the contigs (in total length). Finally, the tool yielded 29 nucleic pseudochromosomes, one chloroplast, and one mitochondrial pseudochromosomes.
To determine the accuracy of the scaling-up results, we cut the pseudochromosomes predicted by Lachesis into bins with 100 kb equal length and constructed the heatmap based on the interaction signals that were revealed by valid mapped read pairs between bins. The matrix was produced by Hic-Pro and then visualized as the heatmap to show the diagonal patches of strong linkage.
The plastid and mitochondrial genomes were clustered as two contig singletons, and both had long overlapping regions at both ends. This helped us to circularize their genomes. In addition, gene modeling and annotation characterized sets of genes shared by the chloroplasts and mitochondria of other species. To simplify chromosomal drawing, the chloroplast and mitochondrial genomes were linearized (Fig. 1a).
Identification of repeat sequences
The tandem and interspersed repeats were recognized and annotated with RepeatProteinMask and RepeatMasker (v1.323, http://www.repeatmasker.org/) according to their sequence similarity with the deposited ones in Repbase63 (http://www.girinst.org/repbase/) and predicted as models with RepeatModeler (open-1.0.8, http://www.repeatmasker.org/RepeatModeler/) and de novo documented and annotated with RepeatMasker (v1.323, http://www.repeatmasker.org/). All the parameters were default. The tandem repeats were also predicted and annotated directly with TRF software64 built in RepeatMasker.
Repeats that were predicted and annotated in different ways were integrated and documented as nonredundant repeats. The tRNAs were predicted using tRNAscan-SE65 (http://lowelab.ucsc.edu/tRNAscan-SE/). Other noncoding RNAs, for example, rRNA, snRNA and miRNA, were identified by homologous searching for sequences deposited in the Rfam database (http://rfam.xfam.org/). All the parameters were default.
Protein-encoding gene modeling and annotation
The genes in the genome assembly were modeled (predicted) following the philosophy described previously66. Such gene modeling procedure integrated the evidences from ab initio gene predictors based on Hidden Markov Models, spliced transcripts from RNA-seq data available currently and orthologous proteins from closely related species. Trinity (v2.4.0, default parameters)67 predicted and joined the potential transcripts. PASA (v2.1, https://github.com/PASApipeline/PASApipeline/wiki)68 extracted the open reading frames (ORFs) from these potential transcripts. These ORFs were directly used to optimize (train) the parameters of Augustus (v3.3, http://bioinf.uni-greifswald.de/augustus/), SNAP 2006-07-28 (http://archive.broadinstitute.org/mpg/snap/) and GlimmerHMM (v4.33, http://ccb.jhu.edu/software/glimmerhmm/) but not GeneMark-ES (v4.33, http://opal.biology.gatech.edu/GeneMark/) which used the genome assembly with the repetitive sequences marked to obtain an initial set of gene models69,70. Augustus was optimized for the parameters by eightfold cross-validation. SNAP and GlimmerHMM was optimized for the parameters by one round of training with the training set. GeneMark-ES did not require an explicit training data set. Orthologous protein sequences from Nannochloropsis gaditana20, Phaeodactylum tricornutum71, Chlamydomonas reinhardtii72, and Chlorella variabili73 from DOE-JGI (https://jgi.doe.gov) were mapped against the masked genome using tblastn 2.2.28 (https://blast.ncbi.nlm.nih.gov/Blast.cgi)74 (e-value = 1e−5), and then Genewise (v2.2.0, https://www.ebi.ac.uk/~birney/wise2/) was used to obtain the evidence of spliced protein-encoding gene. All the results were integrated into a single high-confidence gene model set with EVidenceModeler (v1.1.1, http://evidencemodeler.github.io/)75 using a weight set (PROTEIN GeneWise 5 ABINITIO_PREDICTION SNAP 1 ABINITIO_PREDICTION Augustus 5 ABINITIO_PREDICTION GlimmerHMM 1 ABINITIO_PREDICTION GeneMark.hmm 1 TRANSCRIPT assembler 10).
These genes were functionally annotated by homologous searching against Swiss-Prot (https://web.expasy.org/docs/swiss-prot_guideline.html), NCBI nt (https://www.ncbi.nlm.nih.gov/nucleotide/), NCBI nr (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz), Pfam (http://xfam.org/), eggnog (http://eggnogdb.embl.de/), GO (http://geneontology.org/page/go-database), and KEGG (http://www.genome.jp/kegg/).
Phylogenetic analysis
The 18S ribosomal RNA (18S rDNA), ribulose bisphosphate carboxylase large chain (rbcL), meiosis-associated and flagellum component genes and genome sequences used in this study were retrieved from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). A set of selected proteins or gene sequences was aligned with ClustalX (v2.0, http://www.clustal.org)76, and the phylogenetic tree of such set of sequences was constructed with MrBayes (v3.2.6, http://nbisweden.github.io/MrBayes/)77. The nuclear and chloroplast genomes selected for microevolution analysis included those of C. merolae78, G. sulphuraia 79, C. reinhardtii72, C. variabilis 73, C. paradoxa80, E. huxleyi81, G. theta82, P. tricornutum71, A. anophagefferen83, and N. gaditana20 from either NCBI (https://www.ncbi.nlm.nih.gov/) or DOE-JGI (https://jgi.doe.gov) and those of N. oceanica were obtained in this study. The OrthoFinder (v2.2.6, https://github.com/davidemms/OrthoFinder/releases)84 generated an orthogroup set showing the protein homologs among the species selected. The gene trees and species tree were inferred using the OrthoFinder workflow, including BLAST, MAFFT, and FastTree with green algae and Glaucophytes as outgroup. From the orthogroup set of all species, the orthogroups of N. oceanica, P. tricornutum, and A. anophagefferens were selected to clarify the similarities of protein homologs among these three species one by one. The protein homologs in orthogroups each were clustered using ClustalO (v1.2.3, http://www.clustal.org)85 by limiting the maximum number of sequences in sub-clusters to two. In this way, the most similar protein homologs between N. oceanica and diatom, between N. oceanica and golden alga and between diatom and golden alga can be identified and counted.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The N. oceanica genome assembly data are available through the NCBI under project number PRJNA503776. This genome assembly has been deposited at DDBJ/ENA/GenBank under accession number GenBank CP038106-CP03813.
References
Sukenik, A., Carmeli, Y. & Berner, T. Regulation of fatty acid composition by irradiance level in the Eustigmatophyte Nannochloropsis sp. J. Phycol. 25, 686–692 (1989).
Boussiba, S. et al. Lipid and biomass production by the halotolerant microalga Nannochloropsis salina. Biomass 12, 37–47 (1987).
Galloway, R. E. Selective condition and isolation of mutants in salt-tolerant, lipid-producing microalgae. J. Phycol. 26, 752–260 (1990).
Kilian, O., Benemann, C. S. E., Niyogi, K. K. & Vick, B. High-efficiency homologous recombination in the oil-producing alga Nannochloropsis sp. Proc. Natl Acad. Sci. USA 108, 21265–21269 (2011).
Pan, K. H. et al. Nuclear monoploidy and asexual propagation of Nannochloropsis oceanica (Eustigmatophyceae) as revealed by its genome sequence. J. Phycol. 47, 1425–1432 (2011).
Verruto, J. et al. Unrestrained markerless trait stacking in Nannochloropsis gaditana through combined genome editing and marker recycling technologies. Proc. Natl Acad. Sci. USA 115, E7015–E7022 (2018).
Wang, Q. et al. Genome editing of model oleaginous microalgae Nannochloropsis sp. by CRISPR/Cas9. Plant J. 88, 1071–1081 (2016).
Weeks, D. P. Homologous recombination in Nannochloropsis: a powerful tool in an industrially relevant alga. Proc. Natl Acad. Sci. USA 108, 20859–20860 (2011).
Gee, C. W. & Niyogi, K. K. The carbonic anhydrase CAH1 is an essential component of the carbon-concentrating mechanism in Nannochloropsis oceanica. Proc. Natl Acad. Sci. USA 114, 4537–4542 (2017).
Hibberd, D. J. Notes on the taxonomy and nomenclature of the algal classes Eustigmatophyceae and Tribophyceae (synonym Xanthophyceae). Bot. J. Linn. Soc. 82, 93–119 (1981).
Fawley, K. P. & Fawley, M. W. Observations on the diversity and ecology of freshwater Nannochloropsis (Eustigmatophyceae), with descriptions of new taxa. Protist 158, 325–336 (2007).
Fawley, M. W., Jameson, I. & Fawley, K. P. The phylogeny of the genus Nannochloropsis (Monodopsidaceae, Eustigmatophyceae), with descriptions of N. australis sp. nov. and Microchloropsis gen. nov. Phycologia 54, 545–552 (2015).
Kandilian, R., Lee, E. & Pilon, L. Radiation and optical properties of Nannochloropsis oculata grown under different irradiances and spectra. Bioresour. Technol. 137, 63–73 (2013).
Andersen, R. A., Brett, R. W., Potter, D. & Sexton, J. P. Phylogeny of the Eustigmatophyceae based upon 18s rDNA, with emphasis on Nannochloropsis. Protist 149, 61–74 (1998).
Bailey, J. C. & Freshwater, D. W. Molecular systematics of the Gelidiales: Inferences from separate and combined analyses of plastid rbcL and nuclear SSU gene sequences. J. Eur. J. Phycol. 32, 343–352 (1997).
Daugbjerg, N. & Andersen, R. A. A molecular phylogeny of the heterokont algae based on analyses of chloroplast-encoded rbcl sequence data. J. Phycol. 33, 1031–1041 (1997).
Lubian, L. M. et al. Nannochloropsis (Eustigmatophyceae) as source of commercially valuable pigments. J. Appl. Phycol. 12, 249–255 (2000).
Manning, W. M. & Starain, H. H. Chlorophyll d, a green pigment of red algae. J. Biol. Chem. 151, 1–19 (1943).
Carpinelli, E. C. et al. Chromosome scale genome assembly and transcriptome profiling of Nannochloropsis gaditana in nitrogen depletion. Mol. Plant 7, 323–335 (2014).
Radakovits, R. et al. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropis gaditana. Nat. Commun. 3, 686 (2012).
Vieler, A. et al. Genome, functional gene annotation, and nuclear transformation of the heterokont oleaginous alga Nannochloropsis oceanica CCMP1779. PLoS Genet. 8, e1003064 (2012).
Liang, C. et al. De novo sequencing and global transcriptome analysis of Nannochloropsis sp. (Eustigmatophyceae) following nitrogen starvation. Bioenerg. Res. 6, 494–505 (2012).
Wang, D. et al. Nannochloropsis genomes reveal evolution of microalgal oleaginous traits. PLoS Genet. 10, e1004094 (2014).
Eid, J. et al. Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 (2009).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Murakami, R. & Hashimoto, H. Unusual nuclear division in Nannochloropsis oculata (Eustigmatophyceae, Heterokonta) which may ensure faithful transmission of secondary plastids. Protist 160, 41–49 (2009).
Nishikawa, K. & Tominaga, N. Isolation, growth, ultrastructure, and metal tolerance of the green alga, Chlamydomonas acidophila (Chlorophyta). Biosci. Biotechnol. Biochem. 65, 2650–2656 (2001).
Waterhouse, R. M. et al. BUSCO Applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2018).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3012 (2015).
Bhattacharya, D., Yoon, H. S. & Hackett, J. D. Photosynthetic eukaryotes unite: Endosymbiosis connects the dots. BioEssays 26, 50–60 (2004).
Yoon, H. S., Hackett, J. D., Pinto, G. & Bhattacharya, D. The single, ancient origin of chromist plastids. Proc. Natl Acad. Sci. USA 99, 15507–15512 (2002).
Stork, S. et al. Distribution of the SELMA translocon in secondary plastids of red algal origin and predicted uncoupling of ubiquitin-dependent translocation from degradation. Eukaryot. Cell 11, 1472–1481 (2012).
Hirakawa, Y., Burki, F. & Keeling, P. J. Genome-based reconstruction of the protein import machinery in the secondary plastid of a Chlorarachniophyte alga. Eukaryot. Cell 11, 324–333 (2012).
Jang, S. H., Jeong, H. J., Chon, J. K. & Lee, S. Y. De novo assembly and characterization of the transcriptome of the newly described dinoflagellate Ansanella granifera: Spotlight on flagellum-associated genes. Mar. Genom. 33, 47–55 (2017).
Charlesworth, D. Plant sex chromosome evolution. J. Exp. Bot. 64, 405–420 (2013).
Charlesworth, D. Plant sex chromosomes. Annu. Rev. Plant Biol. 67, 397–420 (2016).
Loidl, J. Conservation and variability of meiosis across the eukaryotes. Annu. Rev. Genet. 50, 293–316 (2016).
Bachtrog, D. et al. Are all sex chromosomes created equal? Trends Genet. 27, 350–357 (2011).
Merlini, L., Dudin, O. & Martin, S. G. Mate and fuse: how yeast cells do it. Open Biol. 3, 130008 (2013).
Russo, M. T. et al. MRP3 is a sex determining gene in the diatom Pseudo-nitzschia multistriata. Nat. Commun. 9, 5050 (2018).
Malik, S. B. et al. An expanded inventory of conserved meiotic genes provides evidence for sex in Trichomonas vaginalis. PLoS ONE 3, e2879 (2008).
Schurko, A. M. & Logsdon, J. M. Jr. Using a meiosis detection toolkit to investigate ancient asexual ‘scandals’ and the evolution of sex. BioEssays 30, 579–589 (2008).
Cavalier-Smith, T. The kingdom Chromista, origin and systematics. In Progress in Phycological Research Vol. 4 (eds Round, F. E. & Chapman, D. J.) 309–347 (Elsevier, Amsterdam, Netherlands, 1986).
Gould, S. B., Maier, U.-G. & Martin, W. F. Protein import and the origin of red complex plastids. Curr. Biol. 25, R515–R521 (2015).
Agrawal, S. & Striepen, B. More membranes, more proteins: complex protein import mechanisms into secondary plastids. Protist 161, 672–687 (2010).
Gentil, J. et al. Origin of complex algae by secondary endosymbiosis: a journey through time. Protoplasma 254, 1835–1843 (2017).
Gross, J. & Bhattacharya, D. Revaluating the evolution of the Toc and Tic protein translocons. Trends Plant Sci. 14, 13–20 (2008).
Maier, U. G., Zauner, S. & Hempel, F. Protein import into complex plastids: cellular organization of higher complexity. Eur. J. Cell Biol. 94, 340–348 (2015).
Sommer, M. S. et al. Der1-mediated preprotein import into the periplastid compartment of Chromalveolates? Mol. Biol. Evol. 24, 918–928 (2007).
Bullmann, L. et al. Filling the gap, evolutionarily conserved Omp85 in plastids of Chromalveolates. J. Biol. Chem. 285, 6848–6856 (2010).
Tonkin, C. J., Kalanon, M. & McFadden, G. I. Protein targeting to the malaria parasite plastid. Traffic 9, 166–175 (2008).
Guo, L. & Yang, G. Predicting the reproduction strategies of several microalgae through their genome sequences. J. Ocean Univ. China 13, 1–12 (2014).
Guillard, R. R. L. & Ryther, J. H. Studies of marine planktonic diatoms. I. Cyclotella nana Hustedt and Detonula confervacea Cleve. Can. J. Microbiol. 8, 229–239 (1962).
Couch, J. A. & Fritz, P. J. Isolation of DNA from plants high in polyphenolics. Plant Mol. Biol. Rep. 8, 8–12 (1990).
Koren, S. et al. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238 (2012).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
Simão, F. A. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Corteggiani Carpinelli, E. et al. Chromosome scale genome assembly and transcriptome profiling of Nannochloropsis gaditana in nitrogen depletion. Mol. Plant 7, 323–335 (2014).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 25, 4.10.1–4.10.14 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: search and contextual analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57 (2016).
Salojärvi, J. et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 49, 904–912 (2017).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011). (7).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Majoros, W. H. et al. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Stanke, M. et al. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Bowler, C. et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456, 239–244 (2008).
Merchant, S. S. et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318, 245–250 (2007).
Blanc, G. et al. The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell 22, 2943–2955 (2010).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25 (2004).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Ronquist, F. et al. MRBAYES 3.2: efficient Bayesian phylogenetic inference and model selection across a large model space. Syst. Biol. 61, 539–542 (2012).
Nozaki, H. et al. A 100%-complete sequence reveals unusually simple genomic features in the hot-spring red alga Cyanidioschyzon merolae. BMC Biol. 5, 28 (2007).
Schonknecht, G. et al. Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science 339, 1207–1210 (2013).
Price, D. C. et al. Cyanophora paradoxa genome elucidates origin of photosynthesis in algae and plants. Science 335, 843–847 (2012).
Read, B. A. et al. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 499, 209–213 (2013).
Curtis, B. A. et al. Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature 492, 59–65 (2012).
Gobler, C. J. et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc. Natl Acad. Sci. USA 108, 4352–4357 (2011).
Emms, D. M. & Kelly, S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Acknowledgements
This study was financially supported by National Key R&D Program of China (2018YFD0900305 and 2018YFD0901506), The Marine S & T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (Qingdao) (2018 SDKJ0406-3), and The Fundamental Research Funds for The Central Universities (201762017). The authors sincerely thank Weifei Yang, Yezhen Chen, Guori Gao, Sanyang Liu, Tao Yan, Ning Wang, Yanfang Gu, Yunlong Zhang, Ying Xin, Yuanyuan Li, and Jiao Zhang of ANOROAD GENOME (Beijing, China) for their assistances in sequencing the genome and suggestions and assistances in data processing.
Author information
Authors and Affiliations
Contributions
L.G. carried out culture and collected the algae, processed the genome data, and drafted the paper. As the corresponding author, G.Y. contributed to designing and coordinating of the project, and writing the paper; J.X. and K.P., two professors who work on N. oceanica, discussed the topics described in this paper and provided their assistance in paper writing and data processing. X.R. and S.P. contributed to the SMRT sequencing and data processing. Other authors including S.L., Z.Z., H.L., and S.W. contributed to the microalgal culture, data processing among others.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, L., Liang, S., Zhang, Z. et al. Genome assembly of Nannochloropsis oceanica provides evidence of host nucleus overthrow by the symbiont nucleus during speciation. Commun Biol 2, 249 (2019). https://doi.org/10.1038/s42003-019-0500-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-019-0500-9
This article is cited by
-
An efficient method for constructing a random insertional mutant library for forward genetics in Nannochloropsis oceanica
Journal of Oceanology and Limnology (2024)
-
The genomes of Vischeria oleaginous microalgae shed light on the molecular basis of hyper-accumulation of lipids
BMC Biology (2023)
-
Nannochloropsis artificial chromosomes (NannoACs) loom on the horizon
Journal of Oceanology and Limnology (2023)
-
Genomic adaptation of the picoeukaryote Pelagomonas calceolata to iron-poor oceans revealed by a chromosome-scale genome sequence
Communications Biology (2022)
-
Zeocin treatment significantly elevated transformation efficiency of Nannochloropsis oceanica
Journal of Applied Phycology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
