
Abstract
Uterine sarcomas are rare but deadly malignancies without effective treatment. Immunotherapy is a promising new approach to treat these tumors but has shown heterogeneous effects in sarcoma patients. With the goal of identifying key factors for improved patient treatment, we characterized the tumor immune landscape in 58 uterine sarcoma cases with full clinicopathological annotation. Immune cell characterization revealed the overall prevalence of FOXP3+ cells and pro-tumor M2-like macrophages. Hierarchical clustering of patients showed four tumor type-independent immune signatures, where infiltration of FOXP3+ cells and M1-like macrophages associated with favorable prognosis. High CD8+/FOXP3+ ratio in UUS and ESS correlated with poor survival, upregulation of immunosuppressive markers, extracellular matrix (ECM)-related genes and proteins, and YAP activation. This study shows that uterine sarcomas present distinct immune signatures with prognostic value, independent of tumor type, and suggests that targeting the ECM could be beneficial for future treatments.
Similar content being viewed by others

Introduction
Uterine sarcomas are a heterogeneous group of rare neoplasms comprising 3–4% of all uterine malignancies1. Despite their rareness, they are responsible for considerable mortality and morbidity by frequently recurring and metastasizing distantly2. Uterine sarcomas are classified into various histopathological subtypes, and treatment is subtype-specific3. These subtypes include the most frequently diagnosed, uterine leiomyosarcoma (LMS), and rarer subtypes such as low-grade endometrial stromal sarcoma (ESS) and high-grade endometrial stromal sarcoma including YWHAE-FAM22 translocation-bearing tumors (YFAM), adenosarcoma, and undifferentiated uterine sarcoma (UUS)1,2,4. Current treatments include surgery, chemotherapy, radiotherapy, and hormone therapy. However, their poor effectiveness highlights the urgent need for new therapies2,5,6
Immunotherapies are promising treatments that exploit the immune system to eliminate cancer cells. Immune evasion is essential for cancer cells to survive and is achieved by manipulating immune checkpoint pathways, normally used to maintain self-tolerance7. Checkpoint inhibitors have shown promising results in the treatment of various tumor types but the efficiency of these treatments in uterine sarcomas remains low8.
The success of immunotherapies relies on the ability to manipulate the tumor immune microenvironment (TIME) into an anti-tumorigenic state. Checkpoint inhibitor treatments can accomplish this, but their efficacy depends on the expression of targetable immune regulatory proteins (IRPs) and the cellular composition of the TIME. Among these IRPs, the PD-1/PD-L1 pathway, IDO1, and B7-H4 have sparked special interest7,8.
The immune cell repertoire of the TIME is another determining factor for the efficacy of immunotherapies. Tumors rich in CD8+ cytotoxic T lymphocytes (CTLs), frontline defensive cells for targeting and killing tumor cells, often are associated with good prognosis and immunotherapy response9. Moreover, targeting exhausted CTLs can enhance antitumor immunity10. Other promising targetable components of the TIME include regulatory T cells (Treg), CD4+ T cells, and macrophages. The immune inhibitory function of Tregs is generally considered to have pro-tumorigenic effects11,12. Similarly, although macrophages are presented as a phenotypic spectrum, anti-inflammatory or M2-like macrophages are generally associated with poor prognosis while pro-inflammatory or M1-like macrophages are a marker of good prognosis in various cancer types13,14.
In this study, we aimed to characterize the TIME in uterine sarcomas in order to identify immune signatures with therapeutic potential.
Results
Patient cohort
Clinicopathologic characteristics are presented in Supplementary Table 1. A total of 58 malignant tumor cases were included, comprising 13 leiomyosarcomas (LMS), 16 endometrial stromal sarcomas (ESS), 26 undifferentiated uterine sarcomas (UUS), and 3 YWHAE-FAM22 endometrial stromal sarcomas (YFAM). In addition, there were 14 benign leiomyomata controls (LM). All the samples were obtained from primary tumor cores of patients that had not received any prior treatment. The mean follow-up time for LMS was 4.99 years (range 0.55–16.42), for ESS 10.88 years (range 2.93–20.67), for UUS 5.02 years (range 0.09–20.38), and for YFAM 11.16 years (range 2.61–18.79). For LMS 11/13 (84.6%) patients were deceased at last follow-up, for ESS 2/16 (12.5%), for UUS 22/26 (84.6%), and for YFAM 2/3 (66.7%).
Multiplex immunofluorescence reveals T cell and macrophage heterogeneity across sarcoma subtypes
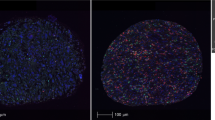
To characterize the TIME in uterine sarcomas, tumors were analyzed for T cell and macrophage infiltrates, and for the expression of immune regulatory molecules. Characterization of T cells included the CTL marker CD8 and the Treg marker FOXP3 (Fig. 1a). Besides cells expressing a single marker, co-expression of CD8+ and FOXP3+ was also detected. Double CD8+FOXP3+ cells have been described as Treg cells with phenotypic resemblance to classical CD4+FOXP3+ Treg cells15,16, however, for the purposes of this paper, they have been analyzed separately. Infiltration of T cells was heterogeneous within each tumor type, with no specific type-dependent differences (Fig. 1b–e). The exception was CD8+FOXP3-cells, which were detected in roughly half of ESS and UUS cases (Fig. 1f, g) but were completely absent in all other cases.

a IF staining of a representative example of a tumor with infiltrated T cells (left), and its corresponding H&E staining (right), and representative images of single and double staining for CD8 and FOXP3 (bottom). Upper scale bars indicate 100 μm and lower panel scale bar indicates 20 μm. b–f Quantification of T cell densities shows heterogeneity in overall CD8+ (b), overall FOXP3+ (c), CD8+FOXP3+ (d), and CD8-FOXP3+ (e) in leiomyoma (LM), leiomyosarcoma (LMS), endometrial stromal sarcoma (ESS), undifferentiated uterine sarcoma (UUS), and YFAM translocation-bearing ESS. f CD8+FOXP3- cells are found in ESS and UUS but not in LM or LMS tumors. g Proportion of T cell types in individual patients. h Representative IF staining of macrophages (left), their corresponding H&E staining (right), and representative examples of single and double CD68- and CD163-expressing cells (bottom). Upper scale bars indicate 100 μm and lower panel scale bar indicates 20 μm. i, j Quantification of macrophage densities shows heterogeneity in overall CD68+ CD163− (i) and CD68+ CD163+ (j) infiltration. k Percentage of macrophages with M2-like phenotype. Plots indicate average ± s.d.
Macrophages were characterized using the CD68 and CD163 markers, allowing quantification of M1-like (CD68+ CD163−) and M2-like (CD68+CD163+) populations (Fig. 1h). Macrophage infiltration was heterogeneous within each tumor type, with no specific type-dependent differences (Fig. 1i, j). Yet, anti-inflammatory M2-like macrophages were more prevalent than M1-like macrophages in all tumor types, with most tumors presenting over 60% M2-like macrophages (Fig. 1k).
The expression of potentially targetable IRPs, including PD-1, PD-L1, B7-H4, and IDO1, was also characterized by immunofluorescence (Fig. 2a). Expression of PD-1, B7-H4, and IDO1 was heterogeneous in the different tumor types (Fig. 2b–d) with LMS tumors presenting a higher B7-H4 expression than ESS and UUS. Expression of PD-L1 was not detected in any group (Fig. 2e, simultaneously stained control tissue showed expected positive staining, Supplementary Fig. 1a). Further classification of cells co-expressing PD-1 with CD8 and FOXP3, indicative of T cell exhaustion or activation, revealed that PD-1 is expressed in a subset of FOXP3+ but not in CD8+ cells (Fig. 2f, simultaneously stained control tissue showed CD8+ PD-1+ expressing cells, see Supplementary Fig. 1b). Moreover, the proportion of PD-1-expressing FOXP3+ cells was generally low (Fig. 2g; 7.0% in CD8+FOXP8+ and 3.9% in CD8-FOXP3+). Interestingly, a large proportion of PD-1+ cells did not express either CD8 or FOXP3. As CD8-FOXP3+ and CD8-FOXP3-PD-1+ cells are likely to be CD4+ Treg and exhausted or activated CD4+ T cells, respectively, we assessed the CD4+ cell infiltration in these tumors by immunohistochemistry (Fig. 2h). Infiltration of CD4+ cells was heterogeneous in all tumor types and positively correlated with CD8-FOXP3+ and CD8-FOXP3-PD-1+ cell infiltration (Fig. 2i, j), suggesting that a considerable proportion of these cells are CD4+.

a Representative IF staining of an IRPs-expressing tumor (left), its corresponding H&E staining (right), and representative images of cells expressing PD-1, B7-H4, and IDO1 (bottom). Upper scale bars indicate 100 μm and lower panel scale bar indicates 20 μm. b–e Quantification of IRP-expressing cells shows a heterogeneous expression of PD-1+ (b), IDO1+ (c), B7-H4+ (d), whereas PD-L1 is not detected in any tumor group (e). f Percentage types of PD-1 expressing cells. g PD-1 expression in FOXP3+ cells. h Representative examples of IHC staining for CD4+ T cells representing high and low infiltration tumors. Scale bar indicates 100 μm. i Quantification of CD4+ T cell density shows heterogeneity in CD4+ expression in all tumor types. j Scatter plot shows the significant correlation between IHC stained CD4+ and IF stained CD8-FOXP3+ and CD8-FOXP3-PD-1+ T cells (r indicates Pearson correlation coefficient). Plots indicate average ± s.d. Significance is indicated as **(P < 0.01).
Uterine mesenchymal tumors can be divided into four intrinsic immune cell subtypes
To better understand the nature of the immune cell microenvironment, we performed unsupervised clustering of individual patients based on immune cell infiltration and expression of IRPs. This analysis resulted in clustering of patients based on the tumor immune cell infiltrate rather than the specific tumor type (Fig. 3a). Three distinct immune cell groups were identified. These immune groups were defined by macrophages, FOXP3+ IRP negative, and IRP together with CD8+ cells (Fig. 3b). Patient clustering revealed two major clusters. One cluster included tumors with high immune cell infiltration (hot tumors; four LMS, three ESS, six UUS, and three LM). A second major cluster generally presented low immune cell infiltrates (cold tumors; nine LMS, 13 ESS, 20 UUS, and three YFAM). Clustering of cold tumors further revealed three specific subclusters. These included a FOXP3-high (immune excluded; one LMS, four ESS, one UUS, one YFAM, and two LM), a group of tumors expressing IRPs (immune ignored; four LMS, four ESS, 11 UUS, and eight LM), and a third subgroup expressing either FOXP3+PD-1+ or some IRP/CD8+ markers (nonspecific; four LMS, five ESS, eight UUS, two YFAM, and one LM). These results again indicate that immune cell infiltrates are largely independent of the current tumor subtype classification.

a Unsupervised hierarchical clustering of mesenchymal uterine tumors based on the expression of immune markers. b IF images of tumors representing the four immune groups represented in the unsupervised hierarchical clustering. Scale bar indicates 100 μm. c Kaplan–Meier curves of patients with hot and cold tumors defined in (a). d Kaplan–Meier curves of patients grouped based on the four distinct immune groups defined in (a).
Finally, we assessed the prognostic value of these patient groups, excluding the benign cases. Comparing patient overall survival between hot and cold tumors did not show any major differences in overall patient survival (Fig. 3c). Similarly, overall survival (OS) did not show statistical differences when cold tumors were further divided into the three subgroups (Fig. 3d).
Correlation with clinicopathologic data shows that FOXP3+ T cell and M1-like macrophage infiltration are associated with improved overall survival
We next evaluated the relationship between OS and individual immune cell infiltrates. Considering all cases, improved OS was significantly associated with the presence of FOXP3+ cells (Fig. 4a–c) but not with CD8+ or CD4+ cells (Fig. 4d, e and Supplementary Fig. 2a). Moreover, an abundance of M1-like macrophages was associated with better OS, while M2-like macrophages or the M1/M2 ratio showed no association (Fig. 4f and Supplementary Fig. 2b, c). Dividing the cases by tumor subtype, the FOXP3+ survival improvement was observed in UUS, and as a nonsignificant trend in ESS (p = 0.0269 and p = 0.0954, respectively), but not in LMS (Supplementary Fig. 3). The M1-like macrophage prevalence was associated with survival improvement, specifically in UUS patients (p = 0.0296, Supplementary Table 2). Neither PD-1, IDO1, nor B7-H4 expression showed general or tumor type-specific prognostic value (Supplementary Fig. 4a–c and Supplementary Table 3). However, absence of PD-1-expression in FOXP3+ cells was a marker of good prognosis (Fig. 4g–j). Contrarily, the infiltration of PD-1+ cells that do not express either CD8 or FOXP3 was associated with improved OS (Fig. 4k).

a–k Kaplan–Meier curves indicating patient overall survival. Patients were grouped based on median density of specific cell subtypes regardless of diagnosis, excluding benign leiomyomas. Median survival is indicated for each patient group. a–e Kaplan–Meier curves of patients grouped based on distinct T cell markers indicate FOXP3 as a marker of good prognosis. f Kaplan–Meier curves based on M1-like type macrophage density show CD68+ CD163- cells to be a marker of improved survival. g–j Kaplan–Meier curves based on distinct FOXP3+ cell populations classified according to their PD-1 expression, show that only PD-1-negative FOXP3+ cell infiltration is associated with good prognosis. k Kaplan–Meier curves indicating overall survival based on density of CD8-FOXP3-PD-1+ cells. l Kaplan–Meier curves showing overall survival based on CD8+FOXP3-/FOXP3+ ratio (CFR) including ESS and UUS cases (Low TILs group corresponds to CD8+FOXP3- and FOXP3+ density below the 40th percentile).
In other tumor types, the CD8+/FOXP3+ ratio (CFR) has been shown to have a stronger association with patient survival than individual cell types17,18,19. We investigated the impact of this ratio on survival in ESS and UUS as these contained both CD8+ and FOXP3+ cells. Cases with few tumor-infiltrating lymphocytes (Low TILs) were considered as a third group. Low CFR (<1) (i.e., tumors with a larger proportion of FOXP3+ than CD8+ cells) was associated with significantly improved survival, compared to both CFRHigh and low TILs (Fig. 4l). This association was stronger in ESS than in UUS (Supplementary Fig. 5a, b). Together, these data show that infiltration of FOXP3+ cells (either as an absolute density or CFR), M1-like macrophages, and CD8-FOXP3-PD-1+ cells (i.e., presumed CD4+PD-1+ cells), are associated with improved overall survival, suggesting that these cells are involved in modulating tumor aggressiveness.
High CFR associates with ECM signaling genes
To further understand the biological underpinnings of the observed survival differences, we first compared the expression of immune marker and cytokine genes between CFRHigh and CFRLow or Low TILs. CFRHigh tumors presented upregulation of markers associated with immunosuppression such as the T cell exhaustion markers HAVCR2 (TIM3) and ENTDP1 (CD39), and immune evasion markers including the inhibitor of macrophage phagocytosis function CD47 and CD274 (PD-L1) (Supplementary Fig. 6a). However, no significant differences were observed in the expression of cytotoxicity markers, while the T cell activation marker CD69 was upregulated in CFRHigh tumors. Cytokine gene expression showed the anti-inflammatory HGF as the most highly upregulated gene, as well as TGFB1 and FGF7, upregulated in CFRHigh. Several pro-inflammatory cytokine genes such as IL2 and TNFSF13B (BAFF) were instead downregulated in these tumors (Supplementary Fig. 6b). These results suggest that CFRHigh tumors present generally more anti-inflammatory and immune-escaping characteristics compared to CFRLow tumors.
Subsequently, to capture other differences between these tumors, we compared the general gene expression of CFRHigh and all other tumors, which revealed 267 differentially expressed genes (Supplementary Data 1). Specifically, comparing CFRHigh and CFRLow tumors resulted in 524 differentially expressed genes, indicating that these tumors differ considerably at the gene expression level. Contrarily, transcriptomic analysis of tumors expressing high and low M1-like macrophages showed 18 differentially expressed genes, indicating that these tumor groups are transcriptionally similar (Supplementary Data 2). Then, we conducted a pathway analysis to uncover major pathway alterations between CFRHigh and other tumors. Major alterations were found in pathways that included extracellular matrix organization, proteoglycans in cancer, regulation of cell adhesion, and integrin-mediated signaling (Fig. 5a). Similarly, independently comparing tumors with high vs low CD8+ cells or tumors with high vs low FOXP3+ cells, showed ECM-related pathways as the most significantly altered ones (Supplementary Fig. 7). MCODE analysis for protein–protein interactions showed two densely connected neighborhoods (Fig. 5b). Gene ontology enrichment analysis of these two MCODE neighborhoods linked MCODE1 with non-integrin membrane–ECM interactions, integrin pathway, and laminin interactions, and MCODE2 with smooth muscle contraction, muscle contraction, and actin cytoskeleton organization. These results indicate that differences in tumor CFR are related to ECM-cell adhesion and signaling at the transcriptional level.

a Metascape pathway analysis reveals the most significantly altered pathways in the CFRHigh group (CD8+FOXP3−/FOXP3+ >1) and their interactions. b MCODE-identified neighborhoods of densely connected proteins based on upregulated genes in the high CFR group showing their corresponding enriched Gene Ontologies. c Heat map of gene expression of significantly altered genes in the CFR-defined groups with patient clustering indicating their corresponding ECM signature as defined by Binzer-Panchal et al. d Ingenuity pathway analysis (IPA) indicates the potential upstream regulators of the differential gene expression between CFR high and low tumors. e Connectivity Map analysis shows potential gene expression-altering drugs for the upregulated genes in the CFRHigh compared to CFRLow groups. f Expression of 22 YAP-TEAD target genes in CFR groups and Low TILs. Significance is indicated as *(P < 0.05), **(P < 0.01), ***(P < 0.001).
Previously, we identified an ECM gene signature in a subgroup of UUS tumors characterized by a trend towards an increased incidence of lymphovascular invasion and very poor patient outcome20. Comparing tumor CFR and ECM signature revealed that five out of eight CFRHigh tumors presented the ECM gene signature (Fig. 5c). Moreover, 75% (six out of eight) of CFRHigh cases presented lymphovascular invasion compared to 62% (8 out of 13) of the other cases. Finally, to further uncover the biological characteristics of CFRHigh tumors, we identified likely upstream regulators and drugs targeting the genes upregulated in these tumors by Ingenuity Pathway Analysis and Connectivity Map, respectively. These analyses revealed TNF, TGFB1/2, PDGF-BB, and TEAD as upstream regulators (Fig. 5d), all of which are involved in tissue fibrosis21,22,23. Furthermore, verteporfin and chlorphenesin were uncovered as potential gene perturbing drugs (Fig. 5e). Interestingly, verteporfin is a suppressor of the YAP-TEAD complex downstream of ECM-integrin signaling, and chlorphenesin is a muscle relaxant24,25. Thus, to investigate whether the YAP-TEAD complex was differentially activated in the different CFR groups, we compared the expression of known YAP-TEAD target genes26. Out of 22 target genes, eight were significantly upregulated in CFRHigh compared to CFRLow tumors, indicating higher YAP-TEAD activity (Fig. 5f). Altogether, these results suggest that there is a link between ECM signaling and infiltration of distinct T cell populations in uterine sarcomas.
CFRLow tumors show reduced ECM protein expression and YAP nuclear localization
To validate the ECM gene expression differences, we analyzed the protein expression of collagen I, VI, fibronectin, and MMP14, as these proteins are significantly enhanced in tumors with the ECM gene signature. In UUS tumors, the expression of collagen I, collagen VI, and fibronectin was lower in CFRLow than in the CFRHigh group, although fibronectin was not significant (p = 0.0699) (Fig. 6a). Similarly, the expression of these proteins was lower in CFRLow ESS tumors compared to CFRHigh, although MMP14 expression was similar (Fig. 6b). To validate the possible implication of YAP signaling in defining the tumor CFR, we compared the YAP nuclear to the cytoplasmic ratio in the different CFR-based groups. In conjunction with ECM protein expression and YAP-TEAD target gene expression, the proportion of nuclear YAP was higher in CFRHigh compared to CFRLow and Low TILs, significantly in UUS tumors (Fig. 6c, d). Together, these data support the correlation between ECM expression, YAP activation, and high CD8+/low FOXP3+ infiltration in ESS and UUS, suggesting a mutual regulation of these components in the tumor microenvironment.

a, b Representative IHC images and quantification of indicated protein expressions show higher expression in the CFRHigh group in UUS (a) and in ESS patients (b). Scale bars indicate 100 μm. c, d Representative IHC images of YAP expression and quantification of its nuclear/cytoplasmic ratio shows a higher ratio in the CFRHigh patient group in UUS (c) and in ESS patients (d). Upper panels scale bars indicate 100 μm and lower panels scale bars indicate 20 μm. Plots indicate average ± s.d. Significance test is indicated *(P < 0.05), **(P < 0.01).
Discussion
Uterine sarcomas are aggressive and rare mesenchymal tumors that currently lack effective treatment. The biology of sarcomas and their interaction with their microenvironment is complex and influences the potential utility of immunotherapy. There is a critical need to understand the sarcoma immune microenvironment and the molecular mechanisms behind it, in order to identify new therapies.
In the work reported here, we have used multiplex immunofluorescence to identify TIL and macrophage populations across a variety of uterine sarcomas and to investigate the expression of potentially targetable IRPs. Further, we have used transcriptomic and protein level data to identify potential pathways involved in the observed TIME differences. These experiments have led to several surprising findings.
First, the immune microenvironment is largely independent of tumor type. Previous studies have shown that immune cell accumulation is associated with diverse sarcoma subtypes, where dominant infiltration of TAMs was observed in UUS and undifferentiated pleomorphic sarcomas27,28. In line with these reports, our results indicate that the immune microenvironment depends on factors other than the tumor type. This is seen when considering the infiltration of individual immune cell types and seen again in our identification of intrinsic immune cell subgroups. This finding carries an important translational relevance; namely, that immunotherapy trials should be performed with a well-planned translational component, so that observed responses can be related to the TIME in those tumors. These hypothetical predictive biomarkers may then be “agnostic” to the sarcoma subtype and should be evaluated as such. This could greatly assist with the clinical management of sarcomas, where a large number of histopathologic subtypes can make traditional “subtype-specific” trials difficult. Instead, trials should focus on the varieties of TIME and intrinsic immune cell subgroup present.
Second, we have demonstrated that the nature of the immune cell infiltrate has an influence on overall survival. The presence of FOXP3+ cells and M1-like macrophages was associated with a better prognosis. This association was observed in both tumors enriched with CD8− and CD8+FOXP3+ cells. Classifying tumors according to their CD8 to FOXP3 ratio (CFR) indicated that low CFR is associated with improved survival. This result is counterintuitive, given that in most tumors it is the presence of CD8+ cells that is associated with a better prognosis. Our results indicate that CD8+ infiltration plays a much less important role in uterine sarcomas. The current knowledge of FOXP3+ cell function in uterine sarcomas is limited. However, our data indicate that these cells play a central role in uterine sarcoma progression and are associated with better prognosis. Contrarily, a previous study identified FOXP3+ Treg cells to associate with high tumor grade and poor survival in a cohort including various soft tissue sarcoma types29, suggesting that our findings are limited to uterine sarcomas or that the detected FOXP3+ cells are not functionally Tregs. The good prognosis associated with FOXP3+ cells opposes the general tumor-promoting role of these cells, but similar observations have been made in colorectal cancer, head and neck carcinoma, and Hodgkin lymphoma, among others30. One possible explanation for these effects is that a significant proportion of the FOXP3+ cells detected are not suppressive Treg cells, rather non-suppressive cells with low expression of FOXP3, which secrete pro-inflammatory cytokines, and are associated with better prognosis in colorectal cancer31. This hypothesis is supported by the expression of pro-inflammatory factors observed in high FOXP3-expressing CFRLow uterine sarcoma tumors. As these non-Treg FOXP3+ cells lack the expression of CD45RA, including this marker for further characterization of this cell population may shed light on the function of FOXP3+ cells in uterine sarcomas and other tumors.
Third, the results from our transcriptomic analysis indicate that pathways such as extracellular matrix organization, proteoglycans in cancer, regulation of cell adhesion, and integrin-mediated signaling are associated with the observed differences in the TIME. Predicted drug regulators of these altered genes highlighted verteporfin and chlorphenesin. Verteporfin is an inhibitor of the YAP-TEAD transcription factor complex32. There is currently a clinical trial planned for the use of liposomal verteporfin in the treatment of recurrent glioblastoma multiforme, and previous reports indicate that verteporfin can modulate the TIME through PD-L1 inhibition33. Although our findings show that uterine sarcomas present low PD-L1 expression, independent responses of the YAP-TEAD inhibition may lead to the regulation of the TIME in these tumors.
Finally, analysis of protein expression confirms the role of the ECM regulatory proteins in the observed differences in immune cell infiltration. The aggressive nature of CFRHigh tumors may emanate from the enhanced ECM expression and YAP activation observed through their immune regulatory and invasion-promoting functions. Accumulation of CD8+ T cells occurs in collagen-rich areas34. This indicates that, although the density of CD8+ cells may be enhanced in collagen-rich tumors, their motility and function may be impaired. Moreover, the phenotype-modulating function of the ECM has been shown to promote M2 polarization in macrophages and TGFβ-dependent Treg differentiation35,36. Thus, the lower expression of ECM and TGFB1 in high FOXP3+ CFRLow tumors further supports the notion that these cells may not have a Treg function.
The differential YAP activity between tumor groups indicates that YAP signaling may play a role in the distinct behavior of uterine sarcomas. YAP is clearly a central regulator of ECM signaling and is involved in sarcomagenesis37,38. Thus, our data suggests that YAP is involved in uterine sarcoma progression and may have immune regulatory functions. Indeed, tumoral YAP expression prevents the activation of CTLs by inhibiting PRF1 and GZMB expression in pancreatic cancer39.
Altogether, these results show that, independently of tumor type, tumors with an ECM signature present a similar immune microenvironment. Thus, characterizing tumors for specific TME signatures including immune cells and ECM might be a better approach to precision therapy than solely using traditional histopathologic subtypes.
These results have numerous translational implications. First, they indicate that clinical trials evaluating immune therapies will need to include a strong translational component to identify what immune cells underlie any potential observed response. Furthermore, any immune cell signature that can be related to a response should then be evaluated in a tumor agnostic manner, given the heterogeneity observed. The central role of YAP and ECM pathways in these differences indicates that therapies focusing on modulation of the ECM should be considered.
Methods
Patient cohort and central review
The retrospective cohort used in this study contains material from two collaborating centers, the Karolinska University Hospital and Skånes University Hospital. Ethical approval was obtained from the relevant authorities. All cases were reviewed centrally, and the cases of endometrial stromal sarcoma and UUS were analyzed with RT-PCR for detection of the YWHAE-FAM22 translocation. Details of the histopathological review have been described previously at Binzer-Panchal et al.20. Patients gave their written informed consent. This study was approved by Stockholm’s regional ethical review board (Dnr2010/1916-31/1).
Tissue microarray preparation
Formalin-fixed paraffin-embedded (FFPE) primary tumors were punctured at 1 mm diameter in two different sites of the tumor core. Therefore, using two distinct tumor core sites per patient minimized the effect of the inherent heterogeneity of the tumor immune microenvironment. Consecutive cuts of these TMAs were used to conduct different stainings.
Multiplex immunofluorescence
Tissue microarray (TMA) slides were air-dried at room temperature, deparaffinized, and then rehydrated in phosphate-buffered saline (PBS) (P4417 Sigma-Aldrich, St. Louis, MO, USA) for 15 min. Primary antibodies (Supplementary Table 4) were diluted in 0.3% Triton X-100 (X100, Sigma-Aldrich, St. Louis, MO, USA) containing 0.1% NaN3 in PBS pH7.4 and applied to the slides for 16 h (4 °C). Afterward, sections were washed three times in Tris Buffered Saline solution, containing Tween® 20 (0.05%) (TBS-Tween 20, T9039 Sigma-Aldrich, St. Louis, MO, USA) for 15 min each. The sections were then incubated in a Tris–NaCl-blocking buffer (TNB-buffer, FP1020, PerkinElmer, Waltham, MA, USA) for 30 min at room temperature. This was followed by the addition of the secondary fluorescent-labeled antibody mix (Supplementary Table 4) diluted in TNB-buffer and sections were incubated for 90 min at room temperature. Subsequently, the sections were washed three times in 0.05% TBS-Tween 20 for 15 min, in the dark. The sections were then immersed in 70% EtOH for 5 min before being transferred to Sudan Black (1% solution, 70%EtOH) for 10 min and then rinsed in 70% EtOH for about a minute before mounting, using PVA/DABCO medium (ProLong® Gold anti-fade with DAPI, P36931, Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA).
Fluorescent images were obtained using a “VSlide” slide scanning microscope (MetaSystems, Alltlussheim, Germany). The system has a CoolCube 2 camera (12-bit grayscale), a 10x objective, and filter sets for 4′,6-diamidino-2-phenylindole (DAPI) (EX350/50–EM470/40), Fluorescein isothiocyanate (FITC) (EX493/16–EM527/30), Cyanine (Cy) 3 (EX546/10–EM580/30), Cy3.5 (EX581/10–EM617/40), and Cy5 (EX630/20–647/long pass). First, the whole TMA was initially pre-scanned at 2.5x to generate the TMA scanning area. Tissue and focus depth were detected based on the DAPI signal. All tissue-covered areas were scanned using a 10x objective. Finally, the individual images were stitched together (VSlide) to generate a large image of the entire section. After scanning, the images (vsi-files) were extracted to high-quality Iconforge Create Executable Library Data (ims)-files for further analysis using the software Qupath (v.0.2.0-m2). In order to facilitate the analysis, the images were not downsampled to avoid losing any valuable biological information.
Immunofluorescence quantification
Qupath (v.0.2.0-m2) (https://github.com/petebankhead/qupath) open-source software40, was used for single-cell detection on 3 TMAs with IF staining for CD8, FOXP3, PDCD1, CD68, CD163, and DAPI. TMA slides that hold 80 of the cases were dearrayed, and then detected.
Unsupervised patient clustering
The average cell densities of two cores from each patient was used to create unsupervised patient clustering. The analysis was conducted in R v.4.0.3 (R Core Team, 2020) with a heatmap.2 package.
Gene expression analysis
The RNA expression arrays method has been previously described20. Briefly, RNA from each sample was used to generate amplified and biotinylated sense-strand cDNA from the entire expressed genome according to the Sensation Plus FFPE Amplification and WT Labeling Kit (P/N 703089, Rev.4 Thermo Fisher Scientific Inc., Life Technologies). GeneChip ST Arrays (GeneChip Human Gene 2.1 ST Array Plate) were hybridized, washed, stained, and finally scanned with the GeneTitan Multichannel (MC) Instrument, according to the GeneTitan Instrument User Guide for Expression Array Plates (PN 702933, Thermo Fisher, Scientific Inc., Life Technologies).
The RNA raw data were normalized and compared using the free Affymetrix Expression Console Software provided by Thermo Fisher. Pathway analysis of differentially expressed genes was conducted by Metascape41.
Immunohistochemistry
The method for immunohistochemistry (IHC) staining of MMP14, collagen I, collagen IV, and fibronectin was previously described20. Briefly, TMA sections were deparaffinized and rehydrated. Antigen retrieval was performed using 10 mmol/L sodium citrate pH 6. Endogenous peroxidase was quenched with 0.6% H2O2 for 10 min, 2 × 5 min PBS (for ImmPRESS kit), or with 0.03% H2O2 for 10 min, 1 min H2O, and 10 min PBS [for Tyramide Signal Amplification (TSA) kit].
The ImmPRESS method was used for MMP14 staining with anti-MT1-MMP (LEM) antibody (Supplementary Table 4). The TSA method was used for collagen I, collagen VI, and fibronectin stainings antibody information in (Supplementary Table 4).
Finally, CD4 and YAP stainings (antibody information in Supplementary Table 4) were performed on tissue sections retrieved at the accredited clinical laboratory of the Department of Pathology, Karolinska University Hospital, Sweden. Staining was performed in the routine pathology laboratory by using an automated Ventana Benchmark Ultra system (Ventana Medical Systems, Tucson, AZ, USA).
Statistical analysis
Descriptive statistics were calculated and presented in tables. For the comparison of the density within groups and intragroup, non-parametric ANOVA (Kruskal–Wallis test) and one-way ANOVA testing were used, respectively. The overall survival (OS) probabilities were estimated and presented by Kaplan–Meier survival curves. The correlation between densities was obtained using a two-tailed Pearson correlation coefficient. All statistical tests were two-sided. P values less than 0.05 were considered statistically significant. Prism v8.0 software (GraphPad) and R programming were used for statistical analysis.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Affymetrix expression array and clinical metadata are available via the Gene Expression Omnibus database repository accession number GSE119043.
References
Abeler, V. M. et al. Uterine sarcomas in Norway. A histopathological and prognostic survey of a total population from 1970 to 2000 including 419 patients. Histopathology 54, 355–364 (2009).
Amant, F., Coosemans, A., Debiec-Rychter, M., Timmerman, D. & Vergote, I. Clinical management of uterine sarcomas. Lancet Oncol. 10, 1188–1198 (2009).
Benson, C. & Miah, A. B. Uterine sarcoma - Current perspectives. Int. J. Women’s Health 9, 597–606 (2017).
D’Angelo, E. & Prat, J. Uterine sarcomas: a review. Gynecologic Oncol. 116, 131–139 (2010).
Novetsky, A. P. & Powell, M. A. Management of sarcomas of the uterus. Curr. Opin. Oncol. 25, 546–552 (2013).
Cuppens, T. et al. Potential targets’ analysis reveals dual PI3K/mTOR pathway inhibition as a promising therapeutic strategy for uterine leiomyosarcomas - An ENITEC Group Initiative. Clin. Cancer Res. 23, 1274–1285 (2017).
Vanderstraeten, A., Luyten, C., Verbist, G., Tuyaerts, S. & Amant, F. Mapping the immunosuppressive environment in uterine tumors: Implications for immunotherapy. Cancer Immunol. Immunother. 63, 545–557 (2014).
Siozopoulou, V. et al. Immune checkpoint inhibitory therapy in sarcomas: Is there light at the end of the tunnel? Cancers 13, 1–20 (2021).
Farhood, B., Najafi, M. & Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J. Cell. Physiol. 234, 8509–8521 (2019).
Sade-feldman, M. et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013 (2018).
Devaud, C., Darcy, P. K. & Kershaw, M. H. Foxp3 expression in T regulatory cells and other cell lineages. Cancer Immunol. Immunother. 63, 869–876 (2014).
Saresella, M. et al. CD4+CD25+FoxP3+PD1− regulatory T cells in acute and stable relapsing-remitting multiple sclerosis and their modulation by therapy. FASEB J. 22, 3500–3508 (2008).
Ma, C. et al. CD163-positive cancer cells are potentially associated with high malignant potential in clear cell renal cell carcinoma. Med. Mol. Morphol. 51, 13–20 (2018).
Barros, M. H. M., Hauck, F., Dreyer, J. H., Kempkes, B. & Niedobitek, G. Macrophage polarisation: an immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 8, 1–11 (2013).
Churlaud, G. et al. Human and mouse CD8+CD25+FOXP3+ regulatory T cells at steady state and during interleukin-2 therapy. Front. Immunol. 6, 2–11 (2015).
Mayer, C. T. et al. CD8+Foxp3+ T cells share developmental and phenotypic features with classical CD4+Foxp3+ regulatory T cells but lack potent suppressive activity. Eur. J. Immunol. 41, 716–725 (2011).
Baras, A. S. et al. The ratio of CD8 to Treg tumor-infiltrating lymphocytes is associated with response to cisplatin-based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. Oncoimmunology 5, 1–7 (2016).
Suzuki, H. et al. Intratumoral CD8+ T/FOXP3+ cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol. Immunother. 59, 653–661 (2010).
Sideras, K. et al. Prognostic value of intra-tumoral CD8+/FoxP3+ lymphocyte ratio in patients with resected colorectal cancer liver metastasis. J. Surgical Oncol. 118, 68–76 (2018).
Binzer-Panchal, A. et al. Integrated molecular analysis of undifferentiated uterine sarcomas reveals clinically relevant molecular subtypes. Clin. Cancer Res. 25, 2155–2165 (2019).
Distler, J. H. W., Schett, G., Gay, S. & Distler, O. The controversial role of tumor necrosis factor α in fibrotic diseases. Arthritis Rheumatism 58, 2228–2235 (2008).
Trojanowska, M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology 47, 4–6 (2009).
Noguchi, S., Saito, A. & Nagase, T. YAP/TAZ signaling as a molecular link between fibrosis and cancer. Int. J. Mol. Sci. 19, 3674 (2018).
Wang, C. et al. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 6, 27–37 (2016).
Kurachi, M. & Aihara, H. Effect of a muscle relaxant, chlorphenesin carbamate, on the spinal neurons of rats. Jpn. J. Pharmacol. 36, 7–13 (1984).
Wang, Y. et al. Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep. 25, 1304–1317.e5 (2018).
Przybyl, J. et al. Macrophage infiltration and genetic landscape of undifferentiated uterine sarcomas. JCI Insight 2, 1–17 (2017).
Chen, L. et al. The immunosuppressive niche of soft-tissue sarcomas is sustained by tumor-associated macrophages and characterized by intratumoral tertiary lymphoid structures. Clin. Cancer Res. 26, 4018–4030 (2020).
Que, Y. et al. PD-L1 expression is associated with FOXP3+ regulatory T-cell infiltration of soft tissue sarcoma and poor patient prognosis. J. Cancer 8, 2018–2025 (2017).
Martin, F., Ladoire, S., Mignot, G., Apetoh, L. & Ghiringhelli, F. Human FOXP3 and cancer. Oncogene 29, 4121–4129 (2010).
Saito, T. et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 22, 679–684 (2016).
Wei, C. & Li, X. The role of photoactivated and non-photoactivated verteporfin on tumor. Front. Pharmacol. 11, 1–15 (2020).
Liang, J. et al. Verteporfin inhibits PD-L1 through autophagy and the STAT1-IRF1-TRIM28 signaling axis, exerting antitumor efficacy. Cancer Immunol. Res. 8, 952–965 (2020).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
Atcha, H. et al. Mechanically activated ion channel Piezo1 modulates macrophage polarization and stiffness sensing. Nat. Commun. 12, 1–14 (2021).
Li, M. O. & Flavell, R. A. TGF-β: a master of all T cell trades. Cell 134, 392–404 (2008).
Fullenkamp, C. A. et al. TAZ and YAP are frequently activated oncoproteins in sarcomas. Oncotarget 7, 30094–30108 (2016).
Ye, S. et al. YAP1-mediated suppression of USP31 enhances NFkB activity to promote sarcomagenesis. Cancer Res. 78, 2705–2720 (2018).
Murakami, S. et al. Yes-Associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene 36, 1232–1244 (2017).
Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 1–7 (2017).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523–1532 (2019).
Acknowledgements
This study was supported by The Swedish Cancer Society (200169 F, 201128Pj, 2018/858, and 2017/473), the Clas Groschinskys foundation (M21139), The Swedish Research Council (2019-01541), and The Radiumhemmets Forskningsfonder (141082). O.G. is supported by a Karolinska Institutet doctoral grant (2–5586/2017), J.G.-M. is supported by Barncancerfonden (TJ2019-0100), and D.S. is supported by a Jonas Söderquist’s scholarship (2432019).
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
Conception and design and development of methodology: J.W.C. Acquisition of data: O.G., J.G.-M., E.H., G.K., L.M.-G., N.M., J.M., A.I., K.L. and J.W.C. Analysis and interpretation of data: O.G., J.G.-M., E.H., D.S., K.L. and J.W.C. Writing, review, and/or revision of the manuscript: O.G., J.G.-M., E.H., D.S., K.L. and J.W.C. Data organization and database construction: O.G., J.G.-M., E.H., J.M., A.I. and J.W.C. Study supervision: K.L. and J.W.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gultekin, O., Gonzalez-Molina, J., Hardell, E. et al. FOXP3+ T cells in uterine sarcomas are associated with favorable prognosis, low extracellular matrix expression and reduced YAP activation. npj Precis. Onc. 5, 97 (2021). https://doi.org/10.1038/s41698-021-00236-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-021-00236-6
This article is cited by
-
Matrix stiffness affects tumor-associated macrophage functional polarization and its potential in tumor therapy
Journal of Translational Medicine (2024)
-
Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment
Nature Communications (2022)
