
Abstract
COVID-19 appeared as a highly contagious disease after its outbreak in December 2019 by the virus, named SARS-CoV-2. The threat, which originated in Wuhan, China, swiftly became an international emergency. Among different genomic products, spike protein of virus plays a crucial role in the initiation of the infection by binding to the human lung cells, therefore, SARS-CoV-2’s spike protein is a promising therapeutic target. Using a combination of a structure-based virtual screening and biochemical assay, this study seeks possible therapeutic candidates that specifically target the viral spike protein. A database of ~ 850 naturally derived compounds was screened against SARS-CoV-2 spike protein to find natural inhibitors. Using virtual screening and inhibitory experiments, we identified acetyl 11-keto-boswellic acid (AKBA) as a promising molecule for spike protein, which encouraged us to scan the rest of AKBA derivatives in our in-house database via 2D-similarity searching. Later 19 compounds with > 85% similarity with AKBA were selected and docked with receptor binding domain (RBD) of spike protein. Those hits declared significant interactions at the RBD interface, best possess and excellent drug-likeness and pharmacokinetics properties with high gastrointestinal absorption (GIA) without toxicity and allergenicity. Our in-silico observations were eventually validated by in vitro bioassay, interestingly, 10 compounds (A3, A4, C3, C6A, C6B, C6C, C6E, C6H, C6I, and C6J) displayed significant inhibitory ability with good percent inhibition (range: > 72–90). The compounds C3 (90.00%), C6E (91.00%), C6C (87.20%), and C6D (86.23%) demonstrated excellent anti-SARS CoV-2 spike protein activities. The docking interaction of high percent inhibition of inhibitor compounds C3 and C6E was confirmed by MD Simulation. In the molecular dynamics simulation, we observed the stable dynamics of spike protein inhibitor complexes and the influence of inhibitor binding on the protein’s conformational arrangements. The binding free energy ΔGTOTAL of C3 (−38.0 ± 0.08 kcal/mol) and C6E (−41.98 ± 0.08 kcal/mol) respectively indicate a strong binding affinity to Spike protein active pocket. These findings demonstrate that these molecules particularly inhibit the function of spike protein and, therefore have the potential to be evaluated as drug candidates against SARS-CoV-2.
Similar content being viewed by others


Introduction
A novel coronavirus infection epidemic (COVID-19) was identified in December 2019 in Wuhan city, Hubei province, when the first case manifested with pneumonia-like symptoms in seafood and live animal markets in China 1. The symptoms of this disease in patients included fever, difficulty in breathing, malaise, dry cough, and dyspnea2. Talking, coughing, and sneezing contribute to the spread of COVID-19 because infected people expel infectious virus particles into the air3,4. Betacoronavirus, the genus to which SARS-CoV-2 belongs, is a member of the family Coronaviridae. Also included in the genus Betacoronavirus are other human coronaviruses like SARS-CoV (which caused the SARS outbreak in 2003) and MERS-CoV. (Responsible for the Middle East respiratory syndrome outbreak in 2012)5.
Coronaviruses are non-segmented, encapsulated viruses that have 26–32 kb of single-stranded RNA (ssRNA). The transmembrane spike (S) glycoprotein, projecting from the viral surface, mediates coronavirus entrance into host cells. Spike (S) comprises two functional subunits, S1 and S2, that collaborate to connect to the angiotensin-converting enzyme 2 (ACE2) receptor on the host cell and fuse the viral and cellular membranes (S2 subunit)6. After attaching to ACE2 in the “open” conformation, one or more receptor binding domains (RBDs) of S-protein acquire “up” conformation, thus, the virus particle can fuse with the human host cell membrane by a process known as cleavage into S1 and S2 subunits7. TM protease serine 2 (TMPRSS2), a type 2 TM serine protease localized on the host cell membrane, activates the Spike protein upon binding to the receptor, hence facilitating viral entrance into the cell8. As the virus enters the human lung cell, they release its RNA and use the host machinery to make its copies9. Attachment of the SARS-CoV-2 virus to host cell receptors requires the RBD S1 subunit of spike protein, therefore by specifically inhibiting this subunit, virus's access to the host cell can be restricted, and consequently viral genome's replication within the host cell can be impeded10. SARS-CoV-2 antibodies that have been discovered primarily target the trimeric S glycoproteins, and most of them can recognize epitopes in the RBD that bind the ACE2 receptor11. Additional drugs, that target the Spike protein RBD-ACE2 interface and obstruct this protein–protein interaction, could be COVID-19 treatment options12,13. To combat this highly contagious infectious agent, researchers may search for new compounds that can precisely block the RBD and hACE2 interaction using bioinformatics tools, in silico methods, and inhibition assay14,15. Up to date there is, no specific medications that have been approved to treat the distinct coronavirus infection. However, clinical trials of synthetic drug compounds have been started to evaluate coronavirus treatments16. Mostly synthetics drugs in clinical trials are inhibiting the viral protein 17. Overcoming the issue of SARS-CoV-2 genome change and viral resistance to medicines and vaccines over time remains a challenge18,19,20,21.
To get beyond this obstacle small, naturally occurring substances with inhibitory properties could prevent the SARS CoV-2 spike protein from interacting with the hACE2 receptor22,23. When compared to synthesized medications, naturally occurring or derived substances typically have less side effects and are more effective in blocking enzymes and proteins24. Natural substances have been refined by the elements to engage specifically with biological targets while having a low immunological response and good absorption25. So, this study aims to discover novel potential inhibitors against SARS CoV-2 spike protein from naturally derived compound (AKBA) (Fig. 1) derivatives by applying 2D similarities searching via computational tools and testing by inhibition assay, which is a crucial step of drug discovery. The AKBA is a naturally derived compound from Boswellia serrata and have uses against antiviral and chronic inflammatory diseases of the lung 26,27. The inhibition activity of AKBA is reported for many diseases (Brain, Lungs, and Viral infection)28,29,30,31. So based on potent inhibition activity of AKBA in this study we aim to discover potential inhibitors from its derivatives against spike protein receptor binding domain.

Some already reported compounds for anti-viral activities.
Material and methods
Structure retrieval of SARS CoV-2 spike protein
The reported crystal structure of spike protein receptor binding domain (RBD) bound with human ACE2 receptor (6M0J, resolution = 2.45 Å) was obtained from Protein Data Bank (RCSB) (https://www.rcsb.org/)32. Molecular Operating Environment (MOE) version 2022.02 built loop modeler algorithm with Amber14: EHT (Amber ff14SB combined with the EHT) forcefield was applied to model the missing residues in the 3D structure of RBD33. With the help of MOE’s Loop builder feature, we added the missing loops to the spike protein chain, and important terminals like start and end (C-N) were labeled34. The MOE’s Quick Prep module modified the protein file by adding missing parameters (hydrogen bonds, angle parameters, partial charges, and Vander Wal forces) to the receptor binding domain35 using Amber: EHT14 forcefield36.
Screening of inhibitors candidates by docking
For searching inhibitors from natural sources, an in-house collection of ~ 850 compounds was docked at the receptor binding domain using MOE Dock tool37. Preceding docking, partial charges and hydrogen were added in each compound in the database via MMFF94x forcefield in MOE, and their structures were minimized with RMS gradient of 0.5 kcal/mol/Å38. The docking site for compounds was selected from the protein–protein interface where the human ACE2 receptor binds with the Spike protein RBD domain. We applied the MOE's Triangle Matcher approach39 and the London dG Scoring system40 with optimal 100 postures, and the GBVI-WSA dg scoring technique was used to determine the final 30 postures for each chemical. Afterward, the docked library was ranked based on the top docking score (i.e., highly negative) and binding position. The binding interactions of compounds at RBD were quantitively calculated by Protein–Ligand Interaction Fingerprints (PLIF) of MOE41. The schematic workflow of this study is presented in Fig. 2.

Schematic workflow of design study.
2D-similarity searching
Comparison of fingerprints is the most common approach to discover the structural similarities between compounds. Because of good interactions and top docking score, 3-acetyl 11-keto-β-boswellic acid (AKBA) was taken to search its structurally similar compounds using 2D similarity searching approach. To calculate the 2D-similarity searches for AKBA, the “Tanimoto coefficient” 42 and MACCS (Molecular Access System) structural keys (166 bits of MOE)43 was used with > 85% cutoff. First, we calculate the MACCS fingerprint for all compounds in the in-house database and search the fingerprints for similar compounds, then those with > 85% similarity with AKBA were selected.
Estimation of drug-like characteristics and pharmacokinetics of compounds
The drug-like behavior of compounds, as well as their physiochemical characteristics and pharmacokinetics, were calculated by a server SwissADME44 to study the compounds’ modes of absorption, toxic profiles, metabolic pathways, and distribution45. By using ProTox-II server46, various toxicity parameters of compounds, such as rat oral median lethal dose (LD50), hepatotoxicity, carcinogenicity, mutagenicity, immunotoxicity, and cytotoxicity were predicted. The canonical SMILES (Simplified Molecular Input Line Entry System) of each compound was supplied to the servers to compute the relevant properties. The allergenicity of selected compounds was predicted using the server CHAIPred47 because some of the small inhibitors after binding to a specific protein can elicit immune response48.
Spike protein inhibition bioassay
The selected natural compounds obtained through virtual screening and by 2D similarity searches were tested by inhibitory assay of the spike protein. By using the Spike protein kit assay (BPS Bioscience/Tebu-bio, Offenbach, Germany), the inhibitory efficacy of drugs was measured following the described procedure49. A colorimetric assay was used to evaluate the strength of the interaction between RBD and ACE250. The assay was started by adding 50 µL of ACE2/well into a 96-well plate followed by an hour of incubation at room temperature (25 °C) with gentle shaking. After one hour, the 96-well plate was washed 3 times with 1× immune-buffer 1, 100 µL of buffer/well, followed by adding 100 µL blocking buffer-2/well and incubated at room temperature (25 °C) with shaking for 10 min, and later washed with the same washing buffer 1. All the compounds were dissolved in 10% DMSO and tested at a concentration of 0.5 mM. Additionally, as a positive control, 30 µL of blocking buffer 2 was utilized in each well. Moreover, a 96-well plate with 50 µL of blocking buffer 2 in each well was used as a blank and incubated for an hour at ambient temperature. The test (compounds) wells and the positive control wells received 20 µL of spike S1 (0.0625 n/µL) each well after the third time incubation. The 96-well plate was incubated once more for an hour at 25 °C while being shaken, after which 100 µL of antibody 1 that had been HRP-labeled (dissolved in blocking buffer 2) was added to each well and incubated for an hour at room temperature (25 °C), after which the plate was washed with washing buffer 1. Ultimately, 100 µL of colorimetric HRP substrate per well were employed and incubated at room temperature until the positive control turned blue51. The reading was taken at 450 nm after the blue color developed and 100 µL of 1N HCl had been added to each well. The following calculation was used to calculate the percent inhibition using the SoftMax Pro and Excel software programs.
Molecular dynamic simulations
The compounds which show high inhibition rate were further subjected to MD simulation to confirm their binding stability with protein. For this MD simulation, we chose a reference-inhibited system spike protein receptor binding domain bound with hACE2 receptor PDB ID (6MOJ). The AMBER22 software package was used to run molecular dynamics simulations52. Topology and coordinate files for each system were generated in AMBER22’s LEap module using the ff19SB residue-specific force field53. To simplify the simulation of protein–ligand interactions, the ff19SB force field parameterized the residues of the protein. Monovalent OPC ions (Na+ and Cl−) were added to each system and the NI++ elements were treated with the divalent OPC ion library to maintain electrostatic neutrality in the systems54. To get reliable simulation results, it is necessary to neutralize the system’s charge by adding counterions. When it comes to creating an optimal environment for protein–ligand interactions, water molecules play a vital role. Because of their importance to the simulations' hydration and stability, an octahedral OPC-water model was used to solvate the systems. A buffer area comprised of water molecules extending out to 12 Å from the protein surface was created. Prior to the MD production run, the minimum energy state was achieved by a two-step energy reduction method. By reorganizing the atoms in the system, we can reduce the amount of stress and steric conflicts that exist36. The steepest descent algorithm, a gradient-based optimization method, was initially applied for 50,000 steps55. It iteratively adjusts the positions of the atoms in the system by following the negative gradient of the potential energy until a local minimum is reached. After the initial steepest descent minimization, another 25,000 iterations with the conjugate gradient approach were performed52. Another optimization method that quickly reaches a local minimum by merging data from earlier iterations is the conjugate gradient method. During the process of minimizing the energy, the water molecules were free to move while the protein residues were held in place. This calms the protein–ligand complex by allowing water molecules to equalize around the protein. Before the dynamic production runs, the systems must be gradually equilibrated at the desired temperature and pressure. Using a Langevin thermostat, the systems were gradually heated from a lower temperature to the target simulation temperature (300 K) at 400 ps52. The Langevin thermostat maintains the system heated by subjecting them to random forces that are supposed to represent their interaction with a heat bath. During heating, the kinetic energy was adjusted for dynamic propagation using the Langevin thermostat with a collision frequency value of 2.0 ps for the harmonic oscillators. This makes for an easily achieved target temperature in the system. Any potential steric conflicts were weeded out by the SHAKE algorithm before the dynamic production run55. SHAKE is a constraint algorithm that keeps hydrogen-atom covalent bonds stable, letting the simulation run for a longer period. After heating, density adjustment was performed in a 400 ps run with the same protocol. This permits water molecules to settle around the protein–ligand complex and completes the equilibration of the system at the desired temperature. Subsequently, each system was equilibrated for 5000 ps at 300 K under the NPT ensemble, which means the number of particles (N), Pressure (P), and Temperature (T) were kept constant. During this equilibration, no restrictions were placed on the arrangement of the atoms, so the system was free to investigate all possible phases. The production MD run, the meat of the simulations in which data for analysis and insights into protein–ligand interactions are obtained, could begin once the systems had equilibrated. Each system underwent a production run of 200 ns56. This duration of simulation is optimized for capturing the protein–ligand complex’s dynamics and producing statistically significant results57. An 8 Å cutoff distance was utilized in order to compute non-bonded interactions, such as van der Waals and electrostatic interactions, efficiently. Beyond this point, interactions are ignored, which allows the simulation to run faster without compromising accuracy. At 10 ps, all system’s trajectory was observed throughout the production phase. These trajectories capture the atomic positions and velocities of the protein and inhibitors ligand pair, which can be examined to determine the changes in the system.
MD production evaluation
CPPTRAJ, a tool in AMBER 22, was used to examine the simulated trajectories at 1 ps intervals58. Root Mean Square Fluctuation (RMSF) and Root Mean Square Deviation (RMSD) were computed for all systems containing Cα atoms using Eqs. (2) and (3).
In RMSD calculation, N = the number of atoms, mi = mass of atoms, Xi = the target atom i vector coordinate, Yi = the reference atom i vector coordinate, and M = the total mass.
By averaging the positions of the atoms in the input frames denoted by x, we were able to get the RMSF of the atom i of interest. Using the (Rg) Radius of gyration, we were able to calculate the locations and velocities of the atoms at each time step59.
Principal component analysis
Using AMBER 22’s CPPTRAJ module, principal component analysis (PCA) was used to determine the protein’s slow and efficient motions. First, Eq. (4) gives us the predicted covariance matrix C, and its component Cij.
C atoms’ ith and jth cartesian coordinates are designated by xi and xj, respectively; the average ith and jth atom’s coordinates during the duration of the ensemble are written as xi and xj, respectively.
The location-based covariance matrix of each system was computed using the 3-D spatial parameters of each system’s production trajectory on a time scale with ten motion patterns. Principal Components were obtained by firstly computing the covariance matrix C. The eigenvalue and eigenvector of the matrix were then calculated after the matrix was diagonalized. The resultant principal components (PCs) characterize the motion descriptively, whereas the eigenvalues quantify that description The proportional impact of each eigenvector was included. To examine their changes, the first two PCs were displayed on a graph.
Characterization of hydrogen bonds
Here, we used the hbond function in CPPTRAJ to look at the interactions between the targeted Spike protein and the molecules of concern. By examining all the system’s 200 ns excursion, the typical length, their distance, and angle of the hydrogen bonds formed with the object being targeted and ligand were identified. The bond angle between the protein–ligand’s electron acceptor and donor atoms was fixed at 120 degrees and the cutoff distance was 3.5 Å60.
Binding free energy calculations
The Generalized Born accessibility algorithms (MMGB-SA) implemented in AMBER22 were used to calculate the binding free energy of the selected inhibitors concerning the Spike protein RBD61. Equation (5) was used to get the free energy of the Spike protein complex and its free APO form.
The combined energy of the protein and the inhibitor, denoted by “GR + L,” and the free energy of binding, designated by “Gbind,” is represented by Eq. (6).
Equation (4) shows the components of the two systems’ total energy “ΔG”. The “Ebond” term represents the energy associated with the bonds and dihedral angles, while “Evdw” represents van der Waals energy. “Eelec” signifies the electrostatic energy, and “GGB” and “GSA” represent the contributions of the polar and non-polar solvents (usually water). Finally, “T” represents the absolute temperature of both systems, and SS is used to calculate the protein's entropy.
Data analysis
MOE 2022.02 and Blender were used to create graphs and visuals. Origin-Pro was used to extract the ensemble of lowest energy structures for each system, and those structures were then utilized to generate all graphs.
Results
Structural details of receptor binding domain of spike protein
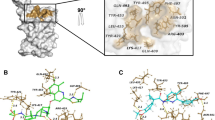
The receptor binding domain (RBD) of the SARS CoV-2 spike protein (PDB ID: 6M0J) was taken from the protein databank RCSB, and the in-house compound’s database was docked at the protein–protein interface where RBD interacts with the hACE2 receptor. The residues of the hACE2 receptor (GLN24, ASP30, ASP38, TYR41, TYR41, GLN42, GLN42, TYR83, LYS353, LYS353, and ASP30) interact with RBD of spike protein (residues: ASN487, LYS417, TYR449, THR500, THR500, GLY446, TYR449, ASN487, GLY502, GLY496 and LYS417) to stabilize protein–protein interaction through hydrogen bonds and salt bridges. The detailed interaction of spike protein and hACE2 receptor is given in (Fig. 3).

The 2D interactions diagram shows the crystal structure of the SARS-CoV-2 spike protein attached to human ACE2 receptor (PDB ID:6M0J).
Selection of inhibitors by virtual screening and 2D-similarity searching
The virtual screening of ~ 850 compounds was performed at the active pocket of SARS CoV-2 spike RBD. To select the natural products, the docked library was ordered by its docking score, and acetyl 11-keto-β-boswellic acid (AKBA) was identified among the top listed compounds with the highest docking score and good docked position. Therefore, AKBA was tested against spike protein in the in-vitro inhibition assay, where AKBA demonstrated significant percentage inhibition against RBD (80.56%). This in-vitro result compelled us to search other AKBA-like compounds in the in-house database, therefore 2D-similarity searching was conducted by MOE using the MACCS structural keys and a Tanimoto coefficient 51. There are 166 different structural substructures or patterns in MACCS made up of atoms other than hydrogen. The default value of 85% overlap was chosen for this search. The inhibitor AKBA with good inhibition against spike protein was selected as a query. By 2D-similarity search, we obtained 19 derivatives of acetyl 11-keto-β-boswellic acid (AKBA) (Table 1), their binding at RBD was examined, and their physicochemical properties and drug-likeness was estimated.
Analysis of binding interactions at RBD of spike protein
The binding modes of 19 AKBA derivatives were analyzed which showed that these molecules exhibit good scores and mediate good docking interaction. The AKBA docking score at spike protein receptor binding domain is − 5.8732 kcal/mol and mediates three hydrogen bonds with TYR505, TYR473 and GLN409 of RBD at 2.06 Å, 1.14 Å and 3.09 Å, respectively. Among those selected compounds, C-6C exhibited the highest docking score of − 5.73 kcal/mol, followed by C-6F, C6F, C3, A6, C6E, C6D, C6B, A5, A1, A4, C6A, A7, A3, and A2 which have docking score in range of − 5.55 to − 4.73 kcal/mol. The compound A1 interacts with residues of spike protein TYR453 and ARG403 through hydrogen bond, while A2 mediates H-bonds with SER494 and TYR453 to be stabilized at RBD interface. The compounds A3 and A4 make two hydrogen bonds with GLY496 and ASN501, and GLY496 and THR500, respectively. While A5 forms multiple H-bonds and hydrophobic (π-H) interactions with ASN501, GLY496, and TYR505. Similarly, A6 mediates numerous hydrogen bonds with GLY496 (2X) and TYR449 to stabilize its binding. The compounds A7 and C3 bind with GLY504 and VAL503, and GLY496 and TYR505, respectively by two hydrogens that firms the ligand in the active pocket. Similarly, C6A is attached at the RBD by mediating two H-bonds with GLY496 and TYR505. Moreover, C6B also forms multiple H-bonds with TYR453, TYR505 (2X), and TYR449, and C-6C forms two H-bonds with TYR449 and TYR453 which holds the ligand firmly. While C6D makes two H-bonds with ARG403 and GLY496 and a π-H bond with TYR505, and C6E forms two H-bonds with TYR505 and TYR453. Likewise, C6F makes a π-H bond with TYR495 and two H-bonds with TYR449 and GLY496 indicating ligand’s firmness in the protein complex. Whereas C6G makes an H-bond and a π-H bond with TYR 449 and TYR495, respectively, and C6H makes two H-bonds with GLY502 and THR500. The compound C6I interacts with spike protein through two π-H bonds with TYR449 and TYR495, and a H-bond with TYR505, while C6J interacts with THR500 and TYR449 through H-bonds. Lastly, the compound C6K also makes two π-H bonds with TYR495, TYR505 and a H-bond with GLY496. The docking predicted interaction of these compounds at molecular level, reflecting firm binding of compounds at RBD interface (Table S1 and Fig. 4).

3D molecular docking interaction of selected compounds against spike protein receptor binding domain PDB ID: 6MOJ.
Pharmacokinetics and drug-likeness of selected compounds
For the drug’s dose, availability at the predicted region of action, and bonding to the target of therapeutic concern to possess pharmacokinetic and drug-like qualities44. Therefore, the pharmacokinetic and drug-like characteristics were estimated for the selected inhibitors ligand against spike protein (RBD). The selected compounds have molecular weights (MW) ranging from 556.82 to 748.79 g/mol. All these inhibitor compounds possess the number of hydrogen bond acceptors (NHBAs) < 10, and the number of hydrogen bond donors (NHBDs) < 5. Also, the number of rotatable bonds (NRB) is less than 10 for the chosen compounds (Tables 2 and S2). The predicted polarity and topological surface area (TPSA) are in an acceptable range from 46.53 to 100.38 Å2 (a molecule with a TPSA value less than 140 Å2 has a high probability of good absorption or oral bioavailability). The LogP o/w (octanol–water partition coefficient) value is up to 5, and the obtained LogP score of compounds indicated good absorbency or lipophilicity (LogS) score ranging from − 7.75 to − 9.96 showed the solubility of molecules. All the compounds obey Lipinski's rule of five. It is pertinent to mention that a compound violating any Lipinski rule results in only molecular weight (MW) > 500 Da. Although Lipinski’s Rule of Five is a helpful guideline, it should be noted that it is not infallible and that many well-marketed medications do not meet all its requirements62,63. Indeed, molecules with molecular weights above 500 Da may still be deemed drug-like provided they possess additional desirable characteristics, making the molecular weight criterion a flexible one64,65. Natural compounds such as those derived from plants, marine life, and microorganisms can have greater molecular weights than synthetic ones and can be employed as drugs or therapeutic agents. Antibiotics, anti-cancer drugs, and immunosuppressants drugs are a few examples66,67,68. Another crucial factor in determining oral bioavailability is molar refractivity (MR), defined by the Ghose rule 69, and its value, as per the Lipinski rule62, should range from 40 to 130 for fulfilling the drug-likeness criteria by all compounds except A2, A6, and C-6H. Besides, Ghose, Veber70, Egan71, and Muegge72 rules were used to examine the drug-likeness of compounds. The compounds fulfill the rule of Veber and Muegge while the violation is shown by some compounds in the Egan rule (WLOGP > 500) by A2, A5, C6F, and C6G. The pharmacokinetic evaluation showed that these compounds have good absorption in the gastrointestinal tract (GIA) except A4, A5, A6, C6A, C6F, and C6G. None of the compounds can cross the blood–brain barrier (BBBP). Except for A3, and A7 the other compounds do not display features typical of P-glycoprotein substrates. None of the compounds showed inhibitory potential for cytochrome p450 enzymes, CYP1A2 CYP2C9, and CYP2C19, except A4, A6, A7, C6G, and C-6D which show inhibition for CYP2D6. Besides these, all the compounds are non-allergic as predicted by CHAIPred and cannot elicit any kinds of immune response. The toxicity analysis as predicted by server ProTox-II for the selected compounds indicates that these compounds are non-toxic except the A4, A5, A6, C6F, and C6G which show immunotoxicity and only A5 shows cytotoxicity. The computed LD50 values range from 500 to 3300 mg/Kg and most of the ligands fall in predicted toxicity class IV, while compounds A6 and C3 fall in predicted toxicity class V. The pharmacokinetic features of these candidates are encouraging, and they may prove useful as inhibitors in future clinical studies specially A3, A4, C3, C6A, C6B, C6C, C6E, C6H, C6I and C6J which can not violate any rules of Lipinski’s, pharmacokinetics and drugs-like (Tables S3 and S4).
In-vitro validation of our computational results
Furthermore, the inhibitory mechanism of the selected AKBA derivatives was evaluated activity against SARS-CoV-2 spike protein. Among the tested hits, eleven compounds displayed > 50% inhibition of RBD (in the range of 73.00–91.00% inhibition) while rest eight compounds exhibited < 50% inhibition and were declared inactive. The basic skeleton of these compounds is similar while the R group is varied. For instance, the invitro activity demonstrates that compound A1 with methanol substitution is inactive, in contrast, A2 and A3 with methyl 4-methyl benzenesulfonate and, azido methane substitutions, respectively exhibited good inhibitory capability with 73.20% and 73.00% inhibition respectively. While inhibitory potency of A7 was somehow declined (63.00% inhibition) with the substitution of methyl 1-methyl-1H-1,2,3-triazole-4-carboxylate as compared to A2 and A3. Similarly, compound C-3 with prop-1-yne substituent resulted in high inhibitory potential with 90.00% inhibition, however, C6A exhibited < 50% inhibition and was found inactive. On the other hand, C6B with the substitution of 4-methyl-1-(o-tolyl)-1H-1,2,3-triazole exhibited good inhibitory capability against SARS CoV-2 spike protein with 73.60% inhibition. The inhibitory potency of C6C and C6D was enhanced with the substitution of 1-(2-methoxyphenyl)-4-methyl-1H-1,2,3-triazole and 4-methyl-1-(2-(trifluoromethyl)phenyl)-1H-1,2,3-triazole, resulted in 86.20% inhibition of both compounds as compared to C6B. Interestingly, the substitution of 1-(4-methoxyphenyl)-4-methyl-1H-1,2,3-triazole in C3 and C6E further increased its potency against RBD and was found to be the most active molecule with 91.00% inhibition. We observe that compounds C6F, C6H and C6I has < 50% inhibition, while C6G with 1-(3-bromophenyl)-4-methyl-1H-1,2,3-triazole substitution significantly inhibited the protein with 78.00% inhibition. However, the inhibitory effect of C6J declined with the substitution of 1-(4-fluorophenyl)-4-methyl-1H-1,2,3-triazole (68.20% inhibition). While C6k with 4-methyl-1-(4-(trifluoromethyl) phenyl)-1H-1,2,3-triazole substitution exhibited 72.6% inhibition (Fig. 5).

Percent inhibition of selected compounds against SARS CoV-2 spike protein RBD.
Molecular dynamic simulation
Protein stability analysis (RMSD)
To investigate the structural modifications that occur in proteins because of the binding of inhibitors (compounds) we used AMBER22 to simulate the interaction that would occur between Spike protein RBD and the compounds that were chosen. The topology and coordinate data were produced with the help of the LEap modules in AMBER22, which utilized the ff198SB forcefield. Based on good inhibition against spike protein RBD we select two compounds C3 and C6E for simulation to further confirm the docking interaction. The root-mean-square deviation (RMSD) was computed to gain an understanding of the degree to which the compounds diverged from their primary structure. The total average RMSD for APO was 2.7429 ± 0.0512 Å. A fluctuation of 2.8345 Å was observed at the start of the simulation at 33 ns and then 3.2441 Å at 107 ns, after that the protein was stable throughout the entire simulation. For the spike protein 6MOJ, the total RMSD value is 2.7429 ± 0.023 Å. The protein was stable through the entire simulation while a small rise was observed from 85 to 95 ns of 2.9248 Å. Similarly, the total average RMSD value for the ligand C3 was calculated at 1.6774 ± 0.012 Å. A small fluctuation of 1.9065 Å was observed at 127 ns then the entire simulation the ligand was stable. The RMSD value for ligand C6E was 1.5083 ± 0.0132 Å, however small variation was observed at 44 ns (1.5561 Å), 80 ns (1.6187 Å), and 98 ns (1.6205 Å) than stable at the end of the simulation (Fig. 6).

Root mean square deviation analysis of spike protein RBD domain in complex with human ACE2 receptor (6M0J), free state (APO) and selectd compounds (C3 and C6E) in 200 ns simulation time.
Residues fluctuations analysis (RMSF)
The root mean square fluctuation (RMSF) is a measurement performed to assess the extent of deviation of an atomic position relative to its average position within the framework of a protein structure. In this study, the RMSF analysis was performed on the Spike protein–protein complex with specific compounds (C3 and C6E) to investigate the fluctuations of these compounds concerning the protein. Increased RMSF values after compound attachment suggested a more flexible structure, which indicated well for subsequent interactions with ligand molecules. The average RMSF value for protein 6MOJ was calculated 1.0431 ± 0.012 Å. The protein was stable at the end of the simulation while some fluctuation was observed at 38 ns (3.4612 Å), 53 ns (2.6399 Å), and 187 ns (3.1878 Å). Similarly, for the APO form of protein, the average RMSF value was 0.9697 ± 0.065 Å. The residue fluctuation was observed at the start of the simulation at 35 ns (1.3634 Å) and 145 ns (2.3890 Å) for the APO form of spike protein RBD. The average RMSF value of inhibitor compound C3 was calculated as 0.93210 ± 065 Å. The compound was stable throughout the simulation while small residual fluctuation was detected at 38 ns of 2.4194 Å and 2.3456 Å at 146 ns. For the ligand C6E, a small residual increase was observed at 38 ns (2.1943 Å), 143 ns (1.9673 Å), and 187 ns (1.8934 Å) then stable at the end of the simulation with an average RMSF value of 0.9537 ± 065 Å (Fig. S1).
Radius of gyration (Rog) analysis
It is important in figuring out the radius of gyration because it gives a basic overview of an object's shape and helps forecast its behaviors under different conditions, considering things like stability, potential for energy transfer and preservation, and responsiveness to outside influences. The average Rog value for the APO form of protein is 18.4030 ± 0346 Å. A small increase was observed from the start of the simulation till 98 ns (18.5063 Å) then remained stable at the end of the simulation. Similarly, the average Rog value for spike protein 6MOJ was calculated 18.3886 ± 3.981 Å and during simulation small fluctuation was detected at 48 ns (18.4274 Å) and 83–89 ns (18.4467 Å). For the inhibitor compound C3 the total Rog value was calculated as 18.4347 ± 0321 Å. The compound was stable while showing an increase of 43 ns (18.4467 Å) to 120 n (18.5437 Å) at the start of the simulation. Similarly, the ligand C6E shows an average Rog value of 18.3742 ± 0321 Å. At the start of the simulation, an increase in Rog value was observed from 25 to 65 ns (18.5073 Å) then became stable at the end of the simulation (Fig. 7).

Radius of gyration analysis was used to calculate the compactness of the spike RBD domain in free (APO), activated state (6M0J) and selected inhibitors attached (C3 and C6E).
Spike protein dominant motions
The principal component analysis (PCA) was used to calculate the dominant motions in the spike protein based on the Cα atoms in a 200 ns simulation trajectory. The total mobility observed in the protein was calculated by the eigenvalues, for which the eigenvectors were calculated from the covariance matrix. Ten eigenvectors for each system were calculated to explore the dominant motions that contribute to the significant structural changes during simulation (Fig. 8). The highest motions observed were in the reference-activated system 6M0J with a value of 32% in 200 ns simulation, followed by the C6E system (31%). The motions observed in the protein were in the protein–protein interaction interface, where the loop B side shows significant structural modification (Fig. 7). The internal sheets of the structure went through structural confirmation upon binding with the hACE2 protein. Meanwhile, the C6E system shows slight confirmation of the A loop position, and the B loop region shows most of the motions in the cartoon representation. Comparing the motions in the active state (6M0J) and inhibited system (C6E), they were opposite in direction, whereas the internal sheets in the C6E system do not show any structural modification (stays blue). The C3 system also shows a variation in the structural dynamic of 30% from its initial coordinates in the simulation run. The C3 system cartoon representation from the 200 ns simulation shows that the active site region of the protein depicts most of the structural confirmation of the C3 protein. The APO system (spike in free state) shows the lowest structural confirmation during the 200 ns simulation run (24%). The APO protein cartoon representation from the PCA analysis shows that the spike structure shows most of the motions in the overall loop’s regions of the protein. The PCA results from the 200 ns simulation of the spike protein's dynamics in free activated and inhibited states highlight key insights into structural mobility and interactions. The most significant movements were observed in the reference-activated system (6M0J) and the C6E system, particularly at the protein–protein interaction interfaces and loop regions. Notably, the directional opposition of movements between the active (6M0J) and inhibited (C6E) states emphasizes the insightful impact of ligand binding on the protein’s conformation. These results represent valuable perspectives of the selected compounds for therapeutic design to emphasize the importance of targeting areas of high mobility and conformational changes within the spike protein to disrupt its function effectively.

Principal component analysis (PCA) was conducted on four distinct systems which include the spike protein in its free state (APO) as the reference, the activated 6M0J system (comprising the spike RBD domain and ACE2 complex), and two systems with selected inhibitors attached (C3 and C-6E). The cumulative movements derived from the top ten eigenvectors for each of these systems are expressed as percentages in the 2D graph. The color-coded cartoon representation transitions from blue to red and shows the regions of the protein from their initial state to their altered state at the 200 ns simulation period. The residues region where the dominant motions were observed were highlighted in yellow.
The spike RBD domain underwent structural conformation changes in the free state, activated and inhibited during the simulation run. To observe these changes, the PC1 and PC2 values were plotted against each other. The transitions of the protein during the simulation run and where it stays the most, at which confirmation was shown in the Fig. 9. Three distinct confirmation clusters were observed in the APO system. Cluster A shows that the system stayed for 16% of the simulation time, from which the system moves to cluster B, where the system shows a diverse behavior for 38% of the simulation time. The system stabilizes in the cluster C region, where the simulation ends and the overall simulation time stayed in cluster C was 46%. The activated reference system 6M0J shows rapid conformational changes in the clusters. Cluster A shows that the simulation stayed at 25% and then shifted to cluster B with a stay of 18% simulation time. The system stabilizes in cluster C, where the simulation stays for the rest of the simulation period (57%). The behavior of the inhibitor-attached systems changed due to the inhibitor attachment with the protein active site. The C3 system shows two dominant clusters, where the system stayed in cluster A for 44% of the simulation time and then shifted to cluster B (56%) where the system stayed till the end of the simulation time. Similarly, the C6E system shows two distinct structural confirmations, where the system stays at cluster A for 36% and then stabilizes in cluster B (64%) for the rest of the simulation time. The structural conformation changes based on PC1 and PC2 of the spike RBD domain in the free state (APO), activated state (6M0J), and selected inhibitor attached states (C3 and C6E) revealed notable differences. In the APO system, a progression through three conformation clusters was observed, with a significant duration in the final cluster, while the activated 6M0J system showed quicker conformational transitions, ultimately stabilizing in its final cluster. In contrast, the inhibitor-attached systems (C3 and C6E) displayed distinct patterns from the reference Apo and 6M0J, predominantly occupying two major clusters each, reflecting the influence of inhibitor attachment on the protein’s conformational dynamics.

Flipping over the orientation of PC1 and PC2 components in the PCA analysis for the spike RBD domain in the free state (APO), activated state (6M0J), and systems with attached selected inhibitors (C3 and C6E) was conducted. Color coding was utilized to represent the density of frames over the simulation from blue, green, and yellow, where yellow means high density. The clusters within each system were labeled A, B, and C, and the simulation duration for which the system remained in each cluster was represented as a percentage.
To further explore the direction and magnitude of the principal movements, porcupine plots were constructed for each system (Fig. 10). In the porcupine plot, the arrow length shows the magnitude of the motion caused in the residue positions, while the arrowhead shows the direction of the movement. The free state spike protein (APO) shows a close confirmation in the porcupine plot, where the active site residues move toward the active site region. In contrast, the magnitude and direction of the movements change in the active state of the 6M0J system. The active pocket’s right side moves towards the active site while affecting the helixes in the vicinity of the active pocket, while the left side remains stable with minor motion deviation. Comparing the APO and activated state 6M0J with the C3 inhibited state, the behavior of movement in the inhibited system was changed. The active pocket of the C3 inhibitor attached system moves outward, which retains an open conformation of the spike RBD. Similarly, the C6E follows the pattern of the C3 system, where the active pocket moves away from the center of the active site. The C3 system shows a higher magnitude of movement when compared with the C6E system. The outcomes from the porcupine plot results show distinct movement patterns of spike RBD protein in free, active, and inhibited states. In the free state spike RBD (APO), active site residues converged towards the center of the active site, which indicates a compact conformation, while the active state (6M0J) displayed divergent movement, having one side of the active pocket shifting towards the active site and affecting the helices in the active pocket vicinity. In contrast, inhibitor-attached systems (C3 and C6E) showed an outward movement of the active pocket, suggesting an open conformation, where the C3 system particularly exhibited a more noticeable movement magnitude than C6E, which highlights the variable impact of the selected inhibitors on the protein’s structure.

The porcupine plot, derived from the PCA analysis which includes the spike RBD in free state (APO), activated state (6M0J), and selected inhibitors attached systems (C3 and C6E). The arrow size indicates the extent of movement, and the arrow's orientation reveals the direction of the motion.
Hydrogen bond analysis
Hydrogen bonds are essential in maintaining the structure and function of biological molecules. In molecular dynamic simulations, they play a crucial role in determining the dynamics and stability of molecular systems. The SARS-CoV-2 spike protein interacting residues with the ACE2 receptor include ASN155, GLN166, GLY170, GLY164, GLN161, TYR117, THR168, GLY171, ASN155, TYR117, GLY114, LYS85, and TYR121. The bond lifetime between the ligands and these residues ranged from 11.00 to 81.20%. The ligand C3 makes hydrogen bond with residue GLY-170, GLY164, GLN161 and ASN169 which have a bond occupancy of 71.95%, 42.30%, 16.80% and 15.90%. The supporting residues GLN161 (5X), TYR117 (4X), GLN166 (4X), ARG71 (2X), TYR173, THR168, and TYR121 which have a bond occupancy > 10% make connection with ligand. Similarly, the ligand atom C6E makes hydrogen bonds with receptor residues THR138, GLN166 (2X), SER-62, THR138 (2X), TYR19 with a bond occupancy of 65.24%, 39.00%, 33.00%, 31.00%, 24.00%, 20.30% and 17.00% respectively. The supporting residues THR168, TYR19 (4X), GLN-66 (2X), THR138 (2X), ASN118 (5X), ARG14(2X), GLN161, GLY170 and GLU152(2X) which make hydrogen bond with receptor atom having bond occupancy > 10%. The investigation of hydrogen bond interactions revealed that the chosen chemicals establish strong bonds with the key residues present in the active pocket of Spike protein (Table S5).
Binding free energy calculation
The Molecular Mechanics and Generalized Born Solvent Accessible Surface Area (MM-GBSA) technique has been utilized to determine the binding free energies between various inhibitor chemicals and Spike protein (6MOJ). Because it can provide information about the strength of a protein–ligand interaction, the use of estimated binding free energy is important in the field of drug design. This in turn makes it easier to assess potential chemicals' binding affinities. In this study, we examined several types of energies, including van der Waals forces (ΔEVDW), electrostatic energy (ΔEEEL), polar solvation energy (ΔEGB), the nonpolar component of solvation energy (ΔESURF), and the total binding free energy (ΔGTOTAL) for inhibitors compounds with SARS CoV-2 Spike protein (RBD) (Table 3). The total binding free energy of protein 6MOJ is (ΔGTOTAL) − 64.42 ± 0.19 kcal/mol. The reduced surface area of protein is − 13.47 Å. The total (ΔEVDW) energy is − 92.32 ± 0.14 kcal/mol, total (ΔEEEL) − 627.31 ± 1.0 kcal/mol, and total (ΔEGB) is 668.68 ± 0.94 kcal/mol. The total binding free energy for compound C3 is (ΔGTOTAL) − 38.0 ± 0.08 kcal/mol with a reduced surface area of − 5.60 Å. The total (ΔEVDW) is − 41.20 ± 0.12 kcal/mol, (ΔEEEL) is − 9.90 ± 0.09 kcal/mol and (ΔEGB) 18.68 ± 0.09 kcal/mol. Similarly, for the ligand compound C6E, the total binding free energy (ΔGTOTAL) is − 41.98 ± 0.08 kcal/mol with a reduced surface area of − 6.55 Å. For the ligand C6E the (ΔEVDW) is − 37.75 ± 0.11 kcal/mol, (ΔEEEL) is − 15.85 ± 0.10 kcal/mol and (ΔEGB) is 18.17 ± 0.12 kcal/mol.
Discussion
The widespread infectivity of the COVID-19 pandemic has made it a serious public health concern73. Human lung epithelial cells are infected when the SARS-CoV-2 virus attaches its spike protein to the human ACE2 receptor on the cell surface74. After binding to the hACE2 receptor the SARS-CoV-2 and other coronaviruses, allow their RNA to penetrate the target cell75. The estimated binding affinity between SARS-CoV-2 and hACE2 is ~ 16 nM which is as much as twenty times higher than that of SARS-CoV76. To prevent virus entry into human cells, scientists are working to create drug compounds that interfere with the spike protein’s ability to connect to hACE277,78. Several investigations towards blocking SARS CoV-2 entry into the host cell have focused on the spike protein79,80,81,82,83. Also, vaccines have been developed as an effective agents but still possess some modest side effects84,85,86. Over time, viruses can undergo mutations that lead to the formation of new versions that can either fully or partially prevent immunization and develop resilience to current medications and vaccines. The creation of novel inhibitors may offer substitute therapeutic approaches that are successful in combating newly developing variations. Designing inhibitors against the spike protein could aim for a broad-spectrum effect, targeting multiple variants or even different coronaviruses. Since drugs developed to treat SARS-CoV-2 could provide an effective first line of defense against future coronaviruses, their development is more rational and preferable77. Similarly, in-silico methods are becoming increasingly significant in the pharmaceutical industry87. In-silico medication design affects the overall drug development schedule by facilitating the quick identification and discovery of innovative therapeutic medicines88,89,90,91. Herein, we employed a combination of computational methods like structure-based virtual screening and 2D-similarity searching and an enzyme inhibition bioassay to uncover novel potential inhibitors of SARS-CoV-2 spike protein.
In this study, the crystal structure of the spike protein receptor binding domain (RBD) was taken together with the human hACE2 receptor.The natural products and their derived compounds were targeted against spike protein through virtual screening. Among the top 10% natural compounds, the acetyl 11-keto-β-boswellic acid (AKBA) showed good binding and docking score which was further confirmed by in-vitro inhibition bioassay where AKBA showed > 80% inhibition of spike protein RBD. Based on in-vitro results of AKBA, we searched for more AKBA-like molecules in our in-house database through 2D-similarity searching. Later, we obtained 19 compounds that have 85% similarity with AKBA with docking scores of − 5.55 to − 4.73 kcal/mol. The docking analysis helps strengthen the protein–ligand bond, suggesting a strong binding affinity between ligands and spike protein RBD. The pharmacokinetics of these compounds showed their appropriate drug-like properties with no toxicity and allergenicity. The compounds have good bioavailability cannot penetrate the Blood–brain barrier and cannot elicit an immune response especially A3, A4, C3, C6A, C6B, C6C, C6E, C6H, C6I, and C6J which can not violate Lipinski’s rule having good pharmacokinetics properties. Based on good docking score, binding position, and pharmacokinetics properties, these compounds were further subjected to in vitro inhibition assay to confirm their drug candidacy. Fortunately, nine new inhibitors were identified against the SARS CoV-2 spike protein. Among them, C3, C-6C, C6E and C6D, exhibited higher anti spike protein capability with 91.00%, 90.00%, 87.00%, and 86.20%, inhibition. Acetyl 11-keto-β-boswellic acid has been recognized as a potential natural product for the treatment of various viral illnesses26,92. Boswellia plays a crucial role in various diseases, for instance, its serrata gum resin is already reported in inflammation, chikungunya, and vesicular stomatitis and β-boswellic acids are also reported against HSV-1, HIV, and herpes virus93,94,95,96. In the recent COVID-19 pandemic, boswellic acids with glycyrrhizin (GR) combination displayed successful actions against COVID-1997,98. Due to high inhibition potential against spike protein RBD the top compounds C3 and C6E docking interaction were confirmed by molecular dynamic simulation. The results of binding free energy calculations demonstrated that compounds C3 and C6E displayed the most elevated energy values, with values of ΔGTOTAL − 38.0 ± 0.08 kcal/mol and − 41.98 ± 0.08 kcal/mol respectively. Based on the medicinal implications of AKBA, and good inhibitory sresults in our current findings, these newly identified spike protein inhibitors appeal further investigation to be used as possible drug candidates to tackle this severe infection (Supplementary Information).
Conclusions
An international health problem exists because of the SARS-CoV-2-caused COVID-19 pandemic. Scientists’ current efforts are directed toward the development of therapeutics for this devastating pandemic. Because the virus’ spike protein interacts with the host cell's hACE2 receptor, this interaction has been identified as a potential site for therapeutic development. After discovering that acetyl 11-keto-boswellic acid (AKBA) is a potential compound for spike protein through virtual screening and inhibitory tests, we scanned the remaining AKBA derivatives in our in-house database using 2D-similarity searching. Subsequently, 19 compounds were chosen based on their homology to AKBA (> 85%) and docked with the spike protein’s receptor binding domain (RBD). Eleven compounds (A2-A3, A7, C3, C6B, C6C, C6D, C6E, C6G, C6J, and C6K) showed considerable inhibitory activity with good percent inhibition (range: > 72–90). Our in-silico results were subsequently corroborated by in vitro bioassay. Particularly impressive anti-SARS CoV-2 spike protein activities were shown by C3 (90.00%), C6E (91.00%), C-6C (87.00%), and C6D (86.20%). The docking interaction of highly inhibitory potential potent compounds have further confirm by MD simulation which give us a good binding energy.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Yuki, K., Fujiogi, M. & Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 215, 108427 (2020).
Hafeez, A., Ahmad, S., Siddqui, S. A., Ahmad, M. & Mishra, S. A review of COVID-19 (Coronavirus Disease-2019) diagnosis, treatments and prevention. Ejmo 4, 116–125 (2020).
Çalıca-Utku, A. et al. Main symptoms in patients presenting in the COVID-19 period. Scottish Med. J. 65, 127–132 (2020).
Bowleg, L. We’re not all in this together: On COVID-19, intersectionality, and structural inequality. Am. Public Health Assoc. 110, 917–917 (2020).
Babaahmadi, V., Amid, H., Naeimirad, M. & Ramakrishna, S. Biodegradable and multifunctional surgical face masks: A brief review on demands during COVID-19 pandemic, recent developments, and future perspectives. Sci. Total Env. 798, 149233 (2021).
Ibrahim, I. M., Abdelmalek, D. H., Elshahat, M. E. & Elfiky, A. A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 80, 554–562 (2020).
Banerjee, A. K., Begum, F. & Ray, U. Mutation hot spots in Spike protein of COVID-19. Preprints 2020, 2020040281 (2020).
Smith, M. & Smith, J. C. Repurposing therapeutics for COVID-19: Supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. J. Med. Virol. 2020, 859 (2020).
Liu, Z. et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. 92, 595–601 (2020).
Tian, F. et al. Mutation N501Y in RBD of spike protein strengthens the interaction between COVID-19 and its receptor ACE2. BioRxiv 2021, 431117 (2021).
Koley, T. et al. Structural analysis of COVID-19 spike protein in recognizing the ACE2 receptor of different mammalian species and its susceptibility to viral infection. 3 Biotech 11, 1–16 (2021).
Hussain, M. et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 92, 1580–1586 (2020).
Khattab, E. S., Ragab, A., Abol-Ftouh, M. A. & Elhenawy, A. A. Therapeutic strategies for Covid-19 based on molecular docking and dynamic studies to the ACE-2 receptors, Furin, and viral spike proteins. J. Biomol. Struct. Dyn. 40, 13291–13309 (2022).
Odolczyk, N., Marzec, E., Winiewska-Szajewska, M., Poznański, J. & Zielenkiewicz, P. Native structure-based peptides as potential protein–protein interaction inhibitors of SARS-CoV-2 spike protein and human ACE2 receptor. Molecules 26, 2157 (2021).
Ullah, A. et al. Sirtuin 1 inhibition: A promising avenue to suppress cancer progression through small inhibitors design. J. Biomol. Struct. Dyn. 2023, 1–17 (2023).
Rosa, S. G. V. & Santos, W. C. Clinical trials on drug repositioning for COVID-19 treatment. Rev. Panam. Salud Públ. 44, e40 (2020).
Chang, C.-K. et al. Targeting protein-protein interaction interfaces in COVID-19 drug discovery. Comput. Struct. Biotechnol. J. 19, 2246–2255 (2021).
Riad, A. et al. Prevalence of COVID-19 vaccine side effects among healthcare workers in the Czech Republic. J. Clin. Med. 10, 1428 (2021).
Beatty, A. L. et al. Analysis of COVID-19 vaccine type and adverse effects following vaccination. JAMA Netw. Open 4, e2140364–e2140364 (2021).
Morfin, F. & Thouvenot, D. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 26, 29–37 (2003).
Van Dorp, L. et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 83, 104351 (2020).
Li, S.-Y. et al. Identification of natural compounds with antiviral activities against SARS-associated coronavirus. Antiviral Res. 67, 18–23 (2005).
Smith, C. J. & Perfetti, T. A. A comparison of the persistence, toxicity, and exposure to high-volume natural plant-derived and synthetic pesticides. Toxicol. Res. Appl. 4, 2397847320940561 (2020).
Henkel, T., Brunne, R. M., Müller, H. & Reichel, F. Statistical investigation into the structural complementarity of natural products and synthetic compounds. Angew. Chem. Int. Ed. 38, 643–647 (1999).
Jantan, I., Ahmad, W. & Bukhari, S. N. A. Plant-derived immunomodulators: An insight on their preclinical evaluation and clinical trials. Front. Plant Sci. 6, 655 (2015).
Gomaa, A. A., Mohamed, H. S., Abd-Ellatief, R. B. & Gomaa, M. A. Boswellic acids/Boswellia serrata extract as a potential COVID-19 therapeutic agent in the elderly. Inflammopharmacology 29, 1033–1048 (2021).
Jamshidi, Z. et al. Effects of Boswellia species on viral infections with particular attention to SARS-CoV-2. Inflammopharmacology 30, 1541–1553 (2022).
von Rhein, C. et al. Curcumin and Boswellia serrata gum resin extract inhibit chikungunya and vesicular stomatitis virus infections in vitro. Antiviral Res. 125, 51–57 (2016).
Khan, A. et al. Identification of novel prolyl oligopeptidase inhibitors from resin of Boswellia papyrifera (Del.) Hochst. and their mechanism: Virtual and biochemical studies. Int. J. Biol. Macromol. 213, 751–767 (2022).
Cuaz-Pérolin, C. et al. Antiinflammatory and antiatherogenic effects of the NF-κB inhibitor acetyl-11-keto-β-boswellic acid in LPS-challenged ApoE−/− mice. Arterioscler. Thromb. Vasc. Biol. 28, 272–277 (2008).
Gong, Y. et al. The biological activity of 3-O-acetyl-11-keto-β-boswellic acid in nervous system diseases. Neuro Mol. Med. 24, 374–384 (2022).
Ali, A. & Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 10, 1–12 (2020).
Pearse, W. D., Lee, B. R., Stemkovski, M., Primack, R. B. & Lee, S. Consistent, linear phenological shifts across a century of observations in South Korea. BioRxiv 2022, 510037 (2022).
Hussain, N. et al. Structural insights into the organization and channel properties of human Pannexin isoforms 1 and 3. BioRxiv 2022, 507385 (2022).
Papageorgiou, L. et al. ADRA2B and HTR1A: An updated study of the biogenic amine receptors reveals novel conserved motifs which play key role in Mental Disorders. BioRxiv 2022, 508280 (2022).
Waqas, M. et al. Multi-fold computational analysis to discover novel putative inhibitors of isethionate sulfite-lyase (isla) from bilophila wadsworthia: Combating colorectal cancer and inflammatory bowel diseases. Cancers 15, 901 (2023).
Scarpino, A., Ferenczy, G. R. G. & Keserű, G. R. M. Comparative evaluation of covalent docking tools. J. Chem. Inf. Model. 58, 1441–1458 (2018).
Belinskaya, T., Pattabiraman, N., Choi, M. & Saxena, A. Differences in amino acid residues in the binding pockets dictate substrate specificities of mouse senescence marker protein-30, human paraoxonase1, and squid diisopropylfluorophosphatase. Biochim. Biophys. Acta BBA Proteins Proteom. 1824, 701–710 (2012).
Idrus, S., Tambunan, U. S. F. & Zubaidi, A. A. Designing cyclopentapeptide inhibitor as potential antiviral drug for dengue virus ns5 methyltransferase. Bioinformation 8, 348 (2012).
Jereva, D., Pencheva, T., Tsakovska, I., Alov, P. & Pajeva, I. In Advanced Computing in Industrial Mathematics: 13th Annual Meeting of the Bulgarian Section of SIAM, December 18–20, 2018, Sofia, Bulgaria, Revised Selected Papers 198–208 (Springer, 2018).
Eberini, I. et al. In silico identification of new ligands for GPR17: A promising therapeutic target for neurodegenerative diseases. J. Comput.-Aided Mol. Design 25, 743–752 (2011).
Kortagere, S., Chekmarev, D., Welsh, W. J. & Ekins, S. Hybrid scoring and classification approaches to predict human pregnane X receptor activators. Pharmaceut. Res. 26, 1001–1011 (2009).
Cereto-Massagué, A. et al. Molecular fingerprint similarity search in virtual screening. Methods 71, 58–63 (2015).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Tripathi, P., Ghosh, S. & Talapatra, S. N. Bioavailability prediction of phytochemicals present in Calotropis procera (Aiton) R. Br. by using Swiss-ADME tool. World Sci. News 2019, 147–163 (2019).
Banerjee, P., Eckert, A. O., Schrey, A. K. & Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 46, W257–W263 (2018).
Liu, Q.-M. et al. Deep-sea-derived butyrolactone I suppresses ovalbumin-induced anaphylaxis by regulating mast cell function in a murine model. J. Agric. Food Chem. 66, 5581–5592 (2018).
Erkes, D. A. & Selvan, S. R. Hapten-induced contact hypersensitivity, autoimmune reactions, and tumor regression: Plausibility of mediating antitumor immunity. J. Immunol. Res. 2014, 28 (2014).
Müller, W. E. et al. The biomaterial polyphosphate blocks stoichiometric binding of the SARS-CoV-2 S-protein to the cellular ACE2 receptor. Biomater. Sci. 8, 6603–6610 (2020).
Sapan, C. V., Lundblad, R. L. & Price, N. C. Colorimetric protein assay techniques. Biotechnol. Appl. Biochem. 29, 99–108 (1999).
Pukac, L. A., Carter, J. E., Morrison, K. S. & Karnovsky, M. J. Enhancement of diaminobenzidine colorimetric signal in immunoblotting. Biotechniques 23, 385–388 (1997).
Salomon-Ferrer, R., Gotz, A. W., Poole, D., Le Grand, S. & Walker, R. C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013).
He, X., Man, V. H., Yang, W., Lee, T.-S. & Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 153, 11 (2020).
Bergonzo, C. & Cheatham, T. E. III. Improved force field parameters lead to a better description of RNA structure. J. Chem. Theory Comput. 11, 3969–3972 (2015).
Pearlman, D. A. et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91, 1–41 (1995).
Skjevik, Å. A., Madej, B. D., Walker, R. C. & Teigen, K. LIPID11: A modular framework for lipid simulations using amber. J. Phys. Chem. B 116, 11124–11136 (2012).
Ullah, A. et al. Bioinformatics and immunoinformatics approach to develop potent multi-peptide vaccine for coxsackievirus B3 capable of eliciting cellular and humoral immune response. Int. J. Biol. Macromol. 239, 124320 (2023).
Amarnath-Jonniya, N., Sk, M. F. & Kar, P. Elucidating specificity of an allosteric inhibitor WNK476 among With-No-Lysine kinase isoforms using molecular dynamic simulations. Chem. Biol. Drug Design 98, 405–420 (2021).
Smith, M. D., Rao, J. S., Segelken, E. & Cruz, L. Force-field induced bias in the structure of Aβ21–30: A comparison of OPLS, AMBER, CHARMM, and GROMOS force fields. J. Chem. Inf. Model. 55, 2587–2595 (2015).
Haddad, Y., Adam, V. & Heger, Z. Rotamer dynamics: Analysis of rotamers in molecular dynamics simulations of proteins. Biophys. J. 116, 2062–2072 (2019).
Xu, L., Sun, H., Li, Y., Wang, J. & Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 117, 8408–8421 (2013).
Zhang, M.-Q. & Wilkinson, B. Drug discovery beyond the ‘rule-of-five’. Curr. Opin. Biotechnol. 18, 478–488 (2007).
Ivanović, V., Rančić, M., Arsić, B. & Pavlović, A. Lipinski’s rule of five, famous extensions and famous exceptions. Popul. Sci. Article 3, 171–177 (2020).
Avdeef, A. & Kansy, M. “Flexible-acceptor” general solubility equation for beyond rule of 5 drugs. Mol. Pharmaceut. 17, 3930–3940 (2020).
Roskoski, R. Rule of five violations among the FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2023, 106774 (2023).
Bhattacharyya, J. et al. A paclitaxel-loaded recombinant polypeptide nanoparticle outperforms Abraxane in multiple murine cancer models. Nat. Commun. 6, 7939 (2015).
Forrest, M. L., Won, C.-Y., Malick, A. W. & Kwon, G. S. In vitro release of the mTOR inhibitor rapamycin from poly (ethylene glycol)-b-poly (ε-caprolactone) micelles. J. Controll. Release 110, 370–377 (2006).
Makino, K. et al. Efficient intracellular delivery of rifampicin to alveolar macrophages using rifampicin-loaded PLGA microspheres: Effects of molecular weight and composition of PLGA on release of rifampicin. Colloids Surfaces B: Biointerf. 36, 35–42 (2004).
Anandhavalli, M., Ghose, M. & Gauthaman, K. Association rule mining in genomics. Int. J. Comput. Theory Eng. 2, 269 (2010).
Veber, D. F. et al. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623 (2002).
Salam, S. S., Chetia, P. & Kardong, D. In silico docking, ADMET and QSAR study of few antimalarial phytoconstituents as inhibitors of plasmepsin II of P. falciparum against malaria. Curr. Drug Therapy 15, 264–273 (2020).
Muegge, I., Heald, S. L. & Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 44, 1841–1846 (2001).
Niknam, Z. et al. Potential therapeutic options for COVID-19: An update on current evidence. Eur. J. Med. Res. 27, 1–15 (2022).
Mir, J. M. & Maurya, R. C. Nitric oxide as a therapeutic option for COVID-19 treatment: A concise perspective. New J. Chem. 45, 1774–1784 (2021).
Jia, H., Neptune, E. & Cui, H. Targeting ACE2 for COVID-19 therapy: Opportunities and challenges. Am. J. Respir. Cell Molecular Biology 64, 416–425 (2021).
Han, P. et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 185, 630–640 (2022).
Jean, S.-S. & Hsueh, P.-R. Old and re-purposed drugs for the treatment of COVID-19. Expert Rev. Anti-infect. Therapy 18, 843–847 (2020).
Olaleye, O. A., Kaur, M. & Onyenaka, C. C. Ambroxol hydrochloride inhibits the interaction between severe acute respiratory syndrome coronavirus 2 spike protein’s receptor binding domain and recombinant human ACE2. BioRxiv 2020, 295691 (2020).
Ho, T.-Y., Wu, S.-L., Chen, J.-C., Li, C.-C. & Hsiang, C.-Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antiviral Res. 74, 92–101 (2007).
Wang, C. et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 11, 2251 (2020).
Du, L. et al. The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 7, 226–236 (2009).
Le, T. T. et al. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 19, 305–306 (2020).
Jeyanathan, M. et al. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 20, 615–632 (2020).
Raus, K., Mortier, E. & Eeckloo, K. Ethical reflections on Covid-19 vaccines. Acta Clin. Belg. 77, 600–605 (2022).
Țăran, A.-M., Mustea, L., Vătavu, S., Lobonț, O.-R. & Luca, M.-M. Challenges and drawbacks of the EU medical system generated by the COVID-19 pandemic in the field of health systems’ digitalization. Int. J. Env. Res. Public Health 19, 4950 (2022).
Sprent, J. & King, C. COVID-19 vaccine side effects: The positives about feeling bad. Sci. Immunol. 6, eabj9256 (2021).
Halim, S. A., Waqas, M., Khan, A. & Al-Harrasi, A. In silico prediction of novel inhibitors of SARS-CoV-2 main protease through structure-based virtual screening and molecular dynamic simulation. Pharmaceuticals 14, 896 (2021).
Rao, V. S. & Srinivas, K. Modern drug discovery process: An in silico approach. J. Bioinform. Seq. Anal. 2, 89–94 (2011).
Terstappen, G. C. & Reggiani, A. In silico research in drug discovery. Trends Pharmacol. Sci. 22, 23–26 (2001).
Singla, R. K. In silico drug design & medicinal chemistry. Curr. Top. Med. Chem. 15, 971 (2015).
Shekhar, C. In silico pharmacology: Computer-aided methods could transform drug development. Chem. Biol. 15, 413–414 (2008).
Ali, S. et al. Natural products can be used in therapeutic management of COVID-19: Probable mechanistic insights. Biomed. Pharmacother. 147, 112658 (2022).
Vakilian, S. et al. An engineered microfluidic blood-brain barrier model to evaluate the anti-metastatic activity of β-boswellic acid. Biotechnol. J. 16, 2100044 (2021).
Qurishi, Y., Hamid, A., Zargar, M., Singh, S. K. & Saxena, A. K. Potential role of natural molecules in health and disease: Importance of boswellic acid. J. Med. Plants Res. 4, 2778–2785 (2010).
Sethi, V., Garg, M., Herve, M. & Mobasheri, A. Potential complementary and/or synergistic effects of curcumin and boswellic acids for management of osteoarthritis. Therapeut. Adv. Musculoskel. Dis. 14, 1124545 (2022).
Fatima, S. W., Alam, S. & Khare, S. K. Molecular and structural insights of β-boswellic acid and glycyrrhizic acid as potent SARS-CoV-2 Envelope protein inhibitors. Phytomed. Plus 2, 100241 (2022).
Trivedi, V. L. et al. Anticancer properties of boswellic acids: Mechanism of action as anti-cancerous agent. Front. Pharmacol. 14, 1187181 (2023).
Caliebe, R. H., Scior, T. & Ammon, H. P. Binding of boswellic acids to functional proteins of the SARS-CoV-2 virus: Bioinformatic studies. Arch. Der. Pharm. 354, 2100160 (2021).
Acknowledgements
The project was supported by grant from The Oman Research Council (TRC) through the funded project (BFP/RGP/HSS/23/037). The authors extend their appreciation to "The Deputyship for Research & Innovation, Ministry of Education, in Saudi Arabia" for funding this research (IFKSUOR3-567-6).
Funding
The authors extend their appreciation to "The Deputyship for Research & Innovation, Ministry of Education, in Saudi Arabia" for funding this research (IFKSUOR3-567-6). The project was supported by grant from The Oman Research Council (TRC) through the funded project (BFP/RGP/HSS/23/037).
Author information
Authors and Affiliations
Contributions
A.K. and A.A.H. conceived and designed the study. A.U., M.W., S.U. and S.A.H. performed all computational analysis and in-vitro testing. B.A., F.S.A., N.M.E. and G.E.B. analyzed the data. A.U., M.W. and S.A.H. wrote the manuscript with input and comments from all co-authors. S.K.A. and R.C. help in the revising of manuscript. S.K.A., R.C., A.K. and A.A.H. edited the final version of manuscript. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ullah, A., Ullah, S., Halim, S.A. et al. Identification of new pharmacophore against SARS-CoV-2 spike protein by multi-fold computational and biochemical techniques. Sci Rep 14, 3590 (2024). https://doi.org/10.1038/s41598-024-53911-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-53911-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
