
Abstract
Mycobacterium bovis (M. bovis) infection has been identified in black (Diceros bicornis) and white (Ceratotherium simum) rhinoceros populations in Kruger National Park, South Africa. However, it is unknown whether M. bovis infected rhinoceros, like humans and cattle, can shed mycobacteria in respiratory secretions. Limited studies have suggested that rhinoceros with subclinical M. bovis infection may present minimal risk for transmission. However, recent advances that have improved detection of Mycobacterium tuberculosis complex (MTBC) members in paucibacillary samples warranted further investigation of rhinoceros secretions. In this pilot study, nasal swab samples from 75 rhinoceros with defined infection status based on M. bovis antigen-specific interferon gamma release assay (IGRA) results were analysed by GeneXpert MTB/RIF Ultra, BACTEC MGIT and TiKa–MGIT culture. Following culture, speciation was done using targeted PCRs followed by Sanger sequencing for mycobacterial species identification, and a region of difference (RD) 4 PCR. Using these techniques, MTBC was detected in secretions from 14/64 IGRA positive rhinoceros, with viable M. bovis having been isolated in 11 cases, but not in any IGRA negative rhinoceros (n = 11). This finding suggests the possibility that MTBC/M. bovis-infected rhinoceros may be a source of infection for other susceptible animals sharing the environment.
Similar content being viewed by others


Introduction
Mammalian tuberculosis (TB) is a chronic progressive disease that is primarily caused by infection with Mycobacterium bovis (M. bovis). Between 2016 and 2019, M. bovis infection was discovered in black (Diceros bicornis) and white (Ceratotherium simum) rhinoceros in Kruger National Park (KNP), South Africa1,2,3. These findings were followed by a population-wide study in KNP (of 437 rhinoceros sampled between 2016–2020), which revealed that 15.4% (95% CI; 10.4–21.0%) of rhinoceros were infected with M. bovis, based on a combination of mycobacterial culture and antigen-specific interferon gamma release assay (IGRA) results4. The relatively high prevalence suggests that rhinoceros in KNP are regularly infected with M. bovis. In the cases in which infection was diagnosed postmortem, lesions and M. bovis isolation were primarily associated with the respiratory tract2,5,6,7,8. However, it is unknown whether infected rhinoceros, like humans and cattle, can shed mycobacteria in respiratory secretions9,10.
Although mycobacterial culture is the gold standard for diagnosing Mycobacterium bovis infection, it is widely recognized that antemortem detection can be insensitive, due to intermittent shedding, inability to obtain appropriate samples, and harsh decontamination processes, resulting in false negative diagnosis of infected individuals11,12. Therefore, diagnostic tests based on mycobacterial antigen-specific immunological responses, such as the delayed hypersensitivity response in the tuberculin skin test or in vitro cytokine release assays, are commonly used to identify M. bovis infected hosts11,13. Currently, the only available antemortem test that has been validated for detection of M. bovis infection in rhinoceros is the QuantiFERON TB Gold Plus Mabtech equine interferon gamma release assay (IGRA)1,14. Although tests based on host responses are useful for screening and surveillance, direct detection of M. bovis in secretions, such as respiratory samples, is crucial for understanding the epidemiology of TB and determining risk of spread from infected animals15,16,17,18.
Some studies have suggested that subclinically infected rhinoceros are unlikely to shed M. bovis, and, therefore, may present minimal risk for transmission6,19. Conventional mycobacterial culture methods, applied to bronchoalveolar lavage samples, recovered M. bovis in only 1 out of 60 samples collected over 20 months from three experimentally-infected white rhinoceros6. It is unknown whether the low recovery rate was due to the true absence of M. bovis in lavage samples, or low sensitivity of culture methods6. However, recent advances have improved detection of MTBC in paucibacillary samples and warrant further investigation of rhinoceros secretions4,8,20.
Applications of novel enhanced mycobacterial culture techniques and improved PCR-based speciation have led to increased M. bovis detection in wildlife13,16,18,21,22,23,24. Use of cationic D-enantiomer peptide supplementation and modified decontamination methods (TiKa-MGIT) have facilitated Mycobacterium tuberculosis complex (MTBC) isolation from paucibacillary tissue and respiratory samples21,22. Culture-independent direct detection, using Cepheid’s GeneXpert MTB/RIF Ultra qPCR assay (Ultra), supports same day MTBC DNA detection from a variety of animal specimens16,18,24,25,26,27. Development of PCRs priming highly conserved genomic region flanking areas within the rpoB and hsp65 genes with high genomic diversity provides new methods for detection and speciation of Mycobacteria spp23,24,28,29. The value of applying these techniques has already been demonstrated in studies of wildlife TB24. Therefore, these may also be valuable for detecting M. bovis, especially in antemortem samples, from suspected infected rhinoceros, providing key information for future epidemiological studies. In this study, the overall aims were 1) to determine whether M. bovis (DNA and viable bacilli) could be detected using novel direct detection approaches for nasal swabs from suspected M. bovis infected (IGRA positive) rhinoceros in KNP, and 2) to calculate the proportion of IGRA positive rhinoceros with M. bovis present in nasal secretions to elucidate the potential for mycobacterial shedding.
Materials and methods
Study population
Black (n = 39) and white (n = 472) rhinoceros in KNP were opportunistically sampled during immobilisations performed as part of management and veterinary activities between January 2020 and April 2022. Demographic characteristics were documented during immobilisation; these included species (black or white rhinoceros), sex (male or female) and age class, which was estimated by veterinary staff during capture and summarised as follows: calf (0–2 years); subadult (> 2 to 7 years); adult (> 7 years).
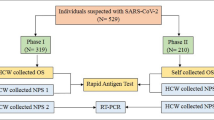
Routinely collected samples included heparinized whole blood and nasal swabs, which were processed as described below, and in Fig. 1. A subset of nasal swab samples was chosen based on rhinoceros interferon gamma release assay (IGRA) results. The IGRA was applied for measurement of antigen-specific interferon gamma (IFNg) release in whole blood that had been stimulated using the QuantiFERON Gold Plus (QFT) platform (Qiagen, Venlo, Limburg, Netherlands), along with an anti-equine interferon gamma ELISA (Mabtech Ab, Nacka Strand, Sweden)1,4. Specifically, antigen-specific IFNg concentrations in this study were determined by subtracting the concentration in the nil tube from the TB2 antigen tube, as recommended by the manufacturer. This study subset consisted of 75 total rhinoceros: 64 that were IGRA positive (suspect M. bovis infected) and 11 that were IGRA negative (suspect uninfected). The number of IGRA negative animals included in the study subset was determined by available resources for testing and randomly selected from all IGRA negative rhinoceros. A detailed explanation of the selection criteria for this subset is provided in Fig. 1.

Flow chart for identifying the study population and methods pipeline for detection of MTBC in nasal swabs from African rhinoceros. KNP, Kruger National Park; b, black rhinoceros; w, white rhinoceros; IGRA, interferon-gamma release assay; Ag, antigen; IFNg, interferon-gamma; cMGIT, conventional BACTEC MGIT Mycobacterial Growth Indicator Tube culture method; TiKa–MGIT, TiKa decontamination and growth supplement enhanced MGIT culture; NCBI BLASTn, National Centre for Biotechnology Information Basic Local Alignment Search Tool (nucleotide); MTBC, Mycobacterium tuberculosis complex; M. bovis, Mycobacterium bovis; RD4, (genetic) Region of Difference 4. ~A rhinoceros was classified as IGRA positive or negative, according to previously defined cutoff values1,4. It was considered IGRA negative if it had a TB Ag-specific IFNg response ≤ 21 pg/mL, a mitogen IFNg response ≥ 21 pg/mL, and a nil IFNg response ≤ 21 pg/mL. It was classified as IGRA positive if it had a TB Ag-specific IFNg response > 21 pg/ml. Individuals who could not be defined as IGRA positive or negative according to the described case definitions were considered inconclusive and excluded. $IGRA positive rhinoceros were included in the study subset for examination of nasal swabs based on the magnitude of their Ag-specific IFNg responses. Of the 93 IGRA positive individuals, the 64 with the highest Ag-specific IFNg responses (ultimately, all with Ag-specific [IFNg] ≥ 40 pg/ml) were included in the study subset. The remaining 29 IGRA positive individuals with lower Ag-specific IFNg (ultimately, all with Ag-specific [IFNg] < 40 pg/ml) were excluded from the study subset. *A small selection of IGRA negative rhinoceros were included for comparison.
Nasal swab sample collection and processing
A single nasal swab (FLOQswab, Copan Diagnostics, Murrieta, California, USA) was collected from each rhinoceros at the time of blood collection. The swab was immediately transferred into 1 ml of sterile saline and immediately frozen, then transported to Stellenbosch University. Nasal swabs were thawed and further processed for direct detection of MTBC, using the Ultra qPCR assay and two different mycobacterial culture methods followed by molecular identification of mycobacterial DNA, as shown in Fig. 1.
Conventional and modified mycobacterial cultures
Frozen nasal swab samples were thawed and 2 ml sterile phosphate (PO4) buffer was added and mixed in the BSL-3 laboratory. Each sample was split into three equal aliquots (Fig. 1). The first aliquot (1 ml supernatant) was stored at − 80 °C and was not used further for this study. The second aliquot (nasal swab tip and 1 ml supernatant) was decontaminated with N-acetyl L-cysteine sodium hydroxide (NALC-NaOH) and processed for culture using the BACTEC Mycobacteria Growth Indicator Tube (MGIT) 960 TB System (Becton Dickinson, Franklin Lakes, New Jersey, USA), as previously described12,22,23. The third 1 ml aliquot was processed for culture using a modified version of the conventional MGIT (cMGIT) system, TiKa-MGIT (TiKa Diagnostics, London, United Kingdom)22. Briefly, samples were transferred to 30 ml sterile tubes containing 10 ml TiKa-Kic decontamination agent (TiKa Diagnostics) and incubated overnight (for a minimum of 20 h) at 37ºC. Thereafter, samples were centrifuged at 3000 × g for 20 min and the supernatant was discarded. The cell pellets were resuspended in 1.6 ml PO4 buffer and thoroughly mixed before 1 ml of each sample was removed and kept aside for testing (post-TiKa decontamination) in the GeneXpert MTB/RIF Ultra qPCR assay (Cepheid, Sunnyvale, California, USA). The remaining 600 µl was used to inoculate MGIT tubes containing 800 µl BD BACTEC MGIT 960 Supplement Kit (Becton Dickinson) and 8.5 µl TiKa growth supplement B (TiKa Diagnostics). All culture samples were incubated in the BACTEC MGIT 960 TB System incubator at 37˚C for a minimum of 56 days. Samples with no growth after 56 days were regarded as culture negative, and no further downstream analysis was performed. One ml aliquots of culture growth positive samples were removed from the bottom of the MGIT tube, where bacterial growth had settled as seen by visible turbidity, boiled for 30 min at 99 °C and then removed from the BSL-3 facility for downstream testing.
Molecular detection of MTBC DNA using PCR and amplicon sequencing
Specialised PCRs, priming highly conserved genomic regions within the hsp65 and rpoB genes28,29, were performed to screen boiled culture aliquots to determine presence of Mycobacterium spp., as previously described23,24. Presence of amplified products with the correct target sizes (hsp65: ± 439 bp and rpoB: ± 764 bp) was confirmed using 1% agarose gel electrophoresis, followed by gel imaging using the ChemiDoc M.D. Universal Hood III Gel Documentation System (Bio-Rad Laboratories, Hercules, California, USA). Amplicons were sent to the Central Analytical Facility (CAF), Stellenbosch University, for Sanger sequencing. Sequence pairwise alignments were performed using A plasmid Editor (ApE; Version 3.1.3)30. Generated consensus sequences were analysed using the National Centre for Biotechnology Information (NCBI) nucleotide Basic Local Alignment Search Tool (BLASTn)31 to find sequence alignment matches in the NCBI database32.
Culture isolates, that produced consensus target sequences with ≥ 95% shared MTBC reference sequence identity, were selected for further investigation. Additional 1 ml aliquots of these cultures were boiled at 99˚C for 30 min and removed from the BSL-3 facility. Total DNA was extracted from each sample aliquot using the QIAGEN DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), as previously described. The hsp65 PCR was repeated, using extracted DNA instead of boiled culture as the template. Presence of the hsp65 amplicon was confirmed using 1% agarose gel electrophoresis, and amplicons were sent to CAF for Sanger sequencing. Sequence pairwise alignments and analysis using the NCBI database were completed as described above. Presence of MTBC in the extracted DNA samples was determined according to a threshold of 95% similarity between the query (sample DNA) amplicon sequence, and the MTBC reference target sequence. Extracted DNA samples identified to have MTBC present underwent an additional PCR targeting the genetic region of difference 4 (RD4) to confirm the presence of M. bovis, as previously described33. A 25 µl reaction contained 12.5 µl Q5 High-Fidelity 2X Master Mix (New England Biolabs), 0.5 µl of each 50 µM primer stock solution, 6 µl sterile, nuclease free water, and 5 µl extracted DNA. PCR cycling conditions were as follows: 1 cycle initial denaturation at 98˚C for 15 min, followed by 40 cycles of denaturation (98˚C for 30 s), annealing (62˚C for 1 min) and elongation (72˚C for 1 min). Final elongation took place at 72˚C for 2 min. Presence of the amplified products was confirmed by 1% agarose gel electrophoresis, followed by gel imaging using the ChemiDoc M.D. Universal Hood III Gel Documentation.
GeneXpert MTB/RIF ultra qPCR assay
The Ultra qPCR assay (Cepheid) was used for direct detection of MTBC DNA in the remaining 1 ml sample aliquots kept aside after overnight TiKa decontamination (Fig. 1). One ml of GeneXpert sample reagent was added to the 1 ml sample aliquot. The mixture was incubated for 15 min at room temperature, followed by vortexing for 10 s, and incubated for another 5 min before a final vortex for 10 s. The total volume (2 ml) of lysed sample was transferred to the Ultra cartridge sample chamber, loaded into the GeneXpert instrument and PCR performed, according to manufacturer’s guidelines18,25. Possible result outputs included MTB not detected, MTB detected high/medium/low/very low/trace. Detection of any MTB (trace amounts or greater) was interpreted as an Ultra positive result.
Data analyses
The proportion of IGRA positive individuals was determined for the entire study population. Demographic characteristics of the rhinoceros subset selected for further investigation were summarized. Frequency distributions of MTBC detected in nasal swabs from IGRA positive and negative groups were determined across each of the three different detection methods (Ultra qPCR, cMGIT culture/PCR, TiKa-MGIT culture/PCR). In addition, parallel interpretation was used to estimate the proportion of IGRA positive, as well as IGRA negative, rhinoceros with MTBC detected in nasal swabs using any of the three methods; 95% confidence intervals were calculated using the Agresti-Coull method34.Test agreement of these three methods within the IGRA positive group was evaluated using a Cohen’s kappa statistic, and qualitatively interpreted35. All statistical calculations were performed in R (version 4.3.0, R Core Team).
Ethics
All procedures including immobilisation of animals and blood and swab collection were undertaken by South African Veterinary Council—registered wildlife veterinarians for management or other veterinary procedures unrelated to this study. Procedures were carried out according to the SANParks Standard Operating Procedures for the Capture, Transportation and Maintenance in Holding Facilities of Wildlife.Ethical approval for this project was granted by the Stellenbosch University Animal Care and Use Committee (ACU-2020-0966, ACU-2020-19019, ACU-2021-19019, ACU-2022-19019) and the Stellenbosch University Biological and Environmental Safety Research Ethics Committee (SU-BEE-202122561; SU-BES-202322561). Section 20 approval was issued by the South African Department of Agriculture, Land Reform and Rural Development (DALRRD; 12/11/1/7/2, 12/11/1/7/2A (JD)).
An approved Biomaterial Transfer Agreement (BMTA 005/22; 011/19) was obtained from South African National Parks (SANParks) which includes evaluation by their Animal Care and Use Committee. A Threatened or Protected Species (TOPS) permit was obtained through the South African Department of Environmental Affairs (DEA Standing Permit S02556; S65805 and DEA Registration Certificate 29416; 02256).
All procedures involving potentially infectious material (e.g. mycobacterial culture) were performed in a Biosafety Level 3 (BSL3) facility that is certified for compliance under the Directorate Animal Health (DAH) of DALRRD. All methods were performed in accordance with the relevant guidelines and regulations as described by Stellenbosch University’s Animal Care and Use Committee and SANParks. ARRIVE guidelines for reporting animal research have been followed (https://arriveguidelines.org/).
Results
The M. bovis infection status, based on IGRA results, was determined for 492 of the 511 (472 white, 39 black) sampled rhinoceros. A total of 93 out of 492 individuals (19%; 95% CI: 16–23%) were IGRA positive, which included 87 white rhinoceros and 6 black rhinoceros. Demographic characteristics of the rhinoceros included in the study subset (64 IGRA positive and 11 IGRA negative) are outlined in Table 1. A total of 14 of the 64 (22%; 95% CI: 13–34%) study rhinoceros that tested IGRA positive had MTBC detected in their nasal swab by at least one of the methods (parallel interpretation), with viable M. bovis isolated in 11/64 (17%; 95% CI: 10–28%) cases. Individual test results are shown in Table 2. In summary, 4/64 (6%) swabs were MTBC positive by Ultra, 5/64 (8%) were M. bovis positive by cMGIT, and 9/64 (14%) were M. bovis positive by TiKa-MGIT. None of the swab samples had MTBC detected using all three methods; however, four samples had MTBC detected by two methods—specifically, three swabs were M. bovis positive by cMGIT/PCR and TiKa-MGIT/PCR, and one swab was MTBC positive by Ultra and TiKa-MGIT/PCR (with confirmation of M. bovis in the latter). None of the nasal swabs from IGRA negative rhinoceros had MTBC detected by any of the applied methods. Importantly, none of the rpoB target amplicon sequences from any of the samples had ≥ 95% similarity to any analogous MTBC sequence in the NCBI BLASTn database; therefore, identification of possible MTBC presence in the samples was determined solely based on a ≥ 95% similarity of the sample hsp65 target amplicon sequences to the MTBC reference hsp65 sequence (NCBI sequence ID: CP074075.1: 528,755 to 529,195) in the NCBI BLASTn database. In the IGRA positive group (n = 64), there was fair test agreement between MTBC detection by cMGIT and TiKa-MGIT culture methods (κ = 0.3648, p = 0.04), and no test agreement between cMGIT culture and Ultra (κ = − 0.0746, p = 0.003) and Tika culture and Ultra (κ = 0.0737, p = 0.6), respectively.
Mycobacteria other than MTBC were present in the nasal swab samples, based on DNA amplification of conserved regions of hsp65 and rpoB in initial PCRs. Although Sanger sequencing and sequence alignment matches in the NCBI database identified NTM species, no further characterization was performed in this study.
Discussion
Mycobacterium tuberculosis complex organisms were detected in nasal swabs using the applied direct detection methods from 14/64 (22%, 95% CI: 13–34%) of the IGRA positive rhinoceros tested, with viable M. bovis isolated in 11/64 (17%; 95% CI: 10–28%) cases. Similarly, MTBC has been directly detected in nasal samples from various species with confirmed infection. For example, in a small group (n = 12) of African buffaloes (Syncerus caffer) with culture-confirmed M. bovis infection, MTBC DNA was detected in 5 of 12 (41%) nasal swabs16. In a separate study of a human population with culture-confirmed M. tuberculosis infection (n = 80), M. tuberculosis DNA was detected in oral swabs from 29 (36.3%) individuals36. In a KNP population of African wild dogs (Lycaon pictus) with immunological sensitisation to M. bovis (determined using an IGRA) (n = 136), M. bovis was only recovered by conventional mycobacterial culture in 4 oronasal samples17. However, to our knowledge, this is the first report of M. bovis detection in nasal (swab) samples from African rhinoceros. These findings demonstrate that MTBC/M. bovis can be sporadically detected in nasal cavities of infected African rhinoceros.
The presence of M. bovis in nasal secretions of rhinoceros could be indicative of several possible scenarios. Since these respiratory samples were obtained from rhinoceros that were free-ranging in an M. bovis endemic area and share the environment with infected hosts37,38, the presence of M. bovis in these samples may result from environmental contamination or exposure. Alternatively, M. bovis may be present in respiratory secretions of a truly infected rhinoceros that is shedding. In this study, MTBC DNA and/or viable M. bovis were only detected in nasal secretions from study individuals with immunological evidence of infection (IGRA positive), and not from any IGRA negative individuals, which supports the hypothesis that at least some of the IGRA positive individuals were truly infected and may be shedding mycobacteria. This raises concern that M. bovis infected rhinoceros may be a source of exposure for other susceptible animals sharing the environment15,17,39. Inclusion of a larger sample size of IGRA negative rhinoceros in future studies could better inform this hypothesis.
This study applied multiple techniques for direct detection of MTBC, including the culture-independent GeneXpert MTB/RIF Ultra qPCR assay, followed by two mycobacterial culture methods, including cMGIT culture and TiKa-MGIT culture with subsequent application of genus PCRs and Sanger sequencing to identify presence of MTBC DNA. The cMGIT culture and TiKa-MGIT cultures only agreed 36% of the time (p = 0.04); this reflects the substantially higher recovery of MTBC by TiKa-MGIT culture (n = 9 positive) compared to cMGIT culture (n = 5 positive). This is concordant with findings from other comparative studies thus far, which support the use of TiKa agents to enhance culture recovery from paucibacillary specimens, irrespective of sample type22,24.
Both culture methods identified more MTBC positive samples than the Ultra, and there was no agreement between the Ultra and the two culture methods, respectively. In Ultra positive, culture negative samples (Table 2), this could reflect the presence of only non-viable bacilli in some of the samples, which can be detected using Ultra, but not culture. Conversely, M. bovis was isolated using culture from several samples that tested Ultra negative (Table 2). This may reflect the ability of culture (and to a greater extent, TiKa-MGIT culture) to select for and enhance viable M. bovis to a level that is detectable by molecular methods, even in the presence of other environmental microorganisms (including non-tuberculous mycobacteria (NTMs)) that may otherwise obscure the presence of M. bovis and confound culture independent detection using Ultra22. Alternative explanations for the culture positive, but Ultra negative results include the presence of PCR inhibitors, or low numbers of bacilli, below the limit of detection for the Ultra, but sufficient to grow in culture. A previous study has shown that the Ultra’s limit of detection for M. tuberculosis was 2 colony forming units (CFU) per ml, versus 30 CFU/ml for M. bovis18.
While both rpoB and hsp65 PCRs were initially applied post-culture in this study, evidence suggested that the rpoB target selected was less specific to MTBC. Since the rpoB primers targeted highly conserved genomic regions in Mycobacterium spp., both NTMs and MTBC DNA would have been amplified in the PCR. However, low levels of MTBC may have been obscured in the Sanger sequencing alignment matches if there was a high abundance of NTMs with rpoB target sequences29. The hsp65 target appeared to be more robust and better suited for detection of MTBC, with MTBC only detected in culture isolates based on this marker in combination with Sanger sequencing and a NCBI BLASTn database search, followed by species confirmation of M. bovis using the RD4 PCR. However, since the RD4 PCR was developed for use with tissues (higher mycobacterial load) compared to paucibacillary respiratory samples33, it is recommended that future studies explore the combinational use of different specialized PCRs for MTBC detection, rather than a single target, to increase confidence in results.
A limitation of this study was that rhinoceros were selected based on IGRA results. The presence of false IGRA positive rhinoceros, due to cross-reactive host immune response to NTMs, could not be ruled out. Previous studies in African buffaloes have reported the high diversity of NTMs present in respiratory samples23. A study investigating M. bovis shedding in African wild dogs in KNP described a low percentage of M. bovis positive cultures (3.1–3.5%) of respiratory samples from IGRA positive individuals17, like findings in the current study. However, Parsons et al. (2017) described a waning in QFT IGRA responses in three M. bovis experimentally infected white rhinoceros over time, with only a low IFNg in vitro response to purified protein derivative of M. avium19. This suggests that a positive QFT IGRA response in rhinoceros is more likely due to infection with MTBC rather than NTM.
Since only a single nasal swab was obtained from each study individual, there was a limited sample volume available for examination using direct detection methods. Furthermore, examination of a single swab sample per individual provided only a single time point representation of the possible shedding status. Sampling of a rhinoceros cohort at multiple time points may provide a better indication of the potential for mycobacterial shedding by rhinoceros over the course of M. bovis infection.
An additional limitation was the small number of IGRA negative (suspect uninfected) individuals included in this study for comparison. These samples were chosen randomly from the larger IGRA negative cohort, and the number was based on available resources for testing the samples. To increase confidence that M. bovis detection in rhinoceros nasal samples is indicative of true infection and possible shedding, a larger subset of IGRA negative individuals should be examined.
Finally, nasal swabs are much easier and cheaper to obtain from a large number of rhinoceros than bronchoalveolar/tracheal lavage samples. However, the nasal swab sample type is more likely to have environmental contamination. This could include contamination with environmental MTBC, which may introduce a false positive result that is suggestive of shedding when this is not the case. Alternatively, it may be contamination by NTMs or other environmental microorganisms that can obscure the presence of MTBC in the sample, resulting in misclassification of the sample as MTBC negative. Examination of respiratory lavage samples using the same techniques applied in this study, albeit only feasible in a smaller study population, may increase confidence that any M. bovis detected is truly indicative of shedding, and should minimise misclassification of M. bovis positive respiratory samples as negative.
Conclusion
This study showed that viable M. bovis may be detected in nasal secretions from rhinoceros with immunological evidence of infection. The KNP rhinoceros population may have an important role in persistence and transmission of M. bovis in the KNP system. Larger scale studies of antemortem respiratory samples of KNP rhinoceros, employing direct detection approaches with additional MTBC markers, and sequencing techniques with higher depth of coverage, may be warranted. In addition, comparison of M. bovis whole genome sequences isolated from rhinoceros and other reservoir hosts, such as African buffaloes, would provide valuable insight into the epidemiology of TB in the KNP system. Future research would enable better characterisation of possible shedding patterns, presence of other MTBC members, contribution of the KNP rhinoceros population to persistence of M. bovis in the ecosystem, and the possible transmission risk associated with the translocation of KNP rhinoceros for management and conservation purposes.
Data availability
Summarised data are included in the manuscript. The reference DNA sequence dataset analysed during the current study is available in the NCBI BLASTn database [https://blast.ncbi.nlm.nih.gov/Blast.cgi]. Rhinoceros-specific data are highly sensitive due to the ongoing crisis of poaching for rhinoceros horn and data restrictions apply.
References
Chileshe, J. et al. An interferon-gamma release assay for the diagnosis of the Mycobacterium bovis infection in white rhinoceros (Ceratotherium simum). Vet. Immunol. Immunopathol. 217, 109931 (2019).
Miller, M. A. et al. Conservation of white rhinoceroses threatened by bovine tuberculosis, South Africa, 2016–2017. Emerg. Infect. Dis. 24, 3 (2018).
Miller, M. A., Michel, A., van Helden, P. & Buss, P. Tuberculosis in rhinoceros: An underrecognized threat?. Transbound. Emerg. Dis. 64, 1071–1078 (2017).
Dwyer, R. et al. Epidemiology of Mycobacterium bovis infection in free-ranging rhinoceros in Kruger National Park, South Africa. Proc. Natl. Acad. Sci. USA 119, e2120656119 (2022).
Miller, M. A., Buss, P. E., van Helden, P. D. & Parsons, S. D. C. Mycobacterium bovis in a free-ranging black rhinoceros, Kruger National Park, South Africa, 2016. Emerg. Infect. Dis. 23, 557–558 (2017).
Michel, A. L. et al. Experimental Mycobacterium bovis infection in three white rhinoceroses (Ceratotherium simum): Susceptibility, clinical and anatomical pathology. PLoS ONE 12, e0179943 (2017).
Valandikar, S. C. & Raju, R. Pulmonary TB in black rhino in Mysore zoo. Zoos Print J. 11, 16–17 (1996).
Stetter, M. et al. Epizootic of Mycobacterium bovis in a zoologic park. J. Am. Vet. Med. Assoc. 207, 1618–1621 (1995).
Zhang, H., Liu, M., Fan, W., Sun, S. & Fan, X. The impact of Mycobacterium tuberculosis complex in the environment on one health approach. Front. Public Health 10, 994745 (2022).
Lombard, J. E. et al. Human-to-cattle Mycobacterium tuberculosis complex transmission in the United States. Front. Vet. Sci. 8, 691192 (2021).
De La Rua-Domenech, R. et al. Ante mortem diagnosis of tuberculosis in cattle: a review of the tuberculin tests, γ-interferon assay and other ancillary diagnostic techniques. Res. Vet. Sci. 81, 190–210 (2006).
Goosen, W. J. et al. Agreement between assays of cell-mediated immunity utilizing Mycobacterium bovis-specific antigens for the diagnosis of tuberculosis in African buffaloes (Syncerus caffer). Vet. Immunol. Immunopathol. 160, 133–138 (2014).
Bernitz, N. et al. Review of diagnostic tests for detection of Mycobacterium bovis infection in South African wildlife. Front. Vet. Sci. 8, 588697 (2021).
Buss, P. et al. SANParks rhinoceros tuberculosis management plan. 78 (2017).
Santos, N., Almeida, V., Gortázar, C. & Correia-Neves, M. Patterns of Mycobacterium tuberculosis-complex excretion and characterization of super-shedders in naturally-infected wild boar and red deer. Vet. Res. 46 (2015).
Clarke, C., Cooper, D. V., Miller, M. A. & Goosen, W. J. Detection of Mycobacterium tuberculosis complex DNA in oronasal swabs from infected African buffaloes (Syncerus caffer). Sci. Rep. 12, 1834 (2022).
Meiring, C. et al. Shedding of Mycobacterium bovis in respiratory secretions of free-ranging wild dogs (Lycaon pictus): implications for intraspecies transmission. Transbound. Emerg. Dis. 68, 2581–2588 (2021).
Goosen, W. J. et al. The Xpert MTB/RIF Ultra assay detects Mycobacterium tuberculosis complex DNA in white rhinoceros (Ceratotherium simum) and African elephants (Loxodonta africana). Sci. Rep. 10, 14482 (2020).
Parsons, S. D. C. et al. The kinetics of the humoral and interferon-gamma immune responses to experimental Mycobacterium bovis infection in the white rhinoceros (Ceratotherium simum). Front. Immunol. 8, 1831 (2017).
Dwyer, R. A., Witte, C., Buss, P., Goosen, W. J. & Miller, M. Epidemiology of tuberculosis in multi-host wildlife systems: implications for black (Diceros bicornis) and white (Ceratotherium simum) rhinoceros. Front. Vet. Sci. 7, 580476 (2020).
Bull, T. J. et al. Improved culture medium (TiKa) for Mycobacterium avium subspecies paratuberculosis (MAP) matches qPCR sensitivity and reveals significant proportions of non-viable MAP in lymphoid tissue of vaccinated MAP challenged animals. Front. Microbiol. 7, 2112–2112 (2017).
Goosen, W. J. et al. Improved detection of Mycobacterium tuberculosis and M. bovis in African wildlife samples using cationic peptide decontamination and mycobacterial culture supplementation. J. Vet. Diagn. Investig. 104063872110441 (2021). https://doi.org/10.1177/10406387211044192.
Clarke, C. et al. Identification and characterisation of nontuberculous mycobacteria in African buffaloes (Syncerus caffer), South Africa. Microorganisms 10, 1861 (2022).
Goosen, W. J. et al. Culture-independent PCR detection and differentiation of Mycobacteria spp. in antemortem respiratory samples from African elephants (Loxodonta africana) and rhinoceros (Ceratotherium simum, Diceros bicornis) in South Africa. Pathogens 11, 709 (2022).
Clarke, C. et al. Novel molecular transport medium used in combination with Xpert MTB/RIF ultra provides rapid detection of Mycobacterium bovis in African buffaloes. Sci. Rep. 11, 7061 (2021).
Hlokwe, T. M. & Mogano, R. M. Utility of Xpert MTB/RIF Ultra assay in the rapid diagnosis of bovine tuberculosis in wildlife and livestock animals from South Africa. Prev. Vet. Med. 177, 104980 (2020).
Kerr, T. J. et al. Novel techniques for detection of Mycobacterium bovis infection in a cheetah. Emerg. Infect. Dis. 26, 630–631 (2020).
Telenti, A. et al. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 31, 175–178 (1993).
Adékambi, T., Colson, P. & Drancourt, M. rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J. Clin. Microbiol. 41, 5699–5708 (2003).
Davis, M. W. & Jorgensen, E. M. ApE, A Plasmid Editor: A freely available DNA manipulation and visualization program. Front. Bioinform. 2, 818619 (2022).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Sayers, E. W. et al. Database resources of the national center for biotechnology information. Nucl. Acids Res. 50, D20–D26 (2022).
Warren, R. M. et al. Differentiation of Mycobacterium tuberculosis complex by PCR amplification of genomic regions of difference. Int. J. Tuberc. Lung. Dis. 6 (2006).
Whitlock, M. C. & Schluter, D. The Analysis of Biological Data. (WH Freeman & Company, 2020).
Cohen, J. A coefficient of agreement for nominal scales. Educ. Psychol. Meas. 20, 37–46 (1960).
Molina-Moya, B. et al. Molecular detection of Mycobacterium tuberculosis in oral mucosa from patients with presumptive tuberculosis. J. Clin. Med. 9, 4124 (2020).
De Garine-Wichatitsky, M. et al. A review of bovine tuberculosis at the wildlife–livestock–human interface in sub-Saharan Africa. Epidemiol. Infect. 141, 1342–1356 (2013).
Michel, A. L. et al. Wildlife tuberculosis in South African conservation areas: Implications and challenges. Vet. Microbiol. 112, 91–100 (2006).
Barasona, J. A., Torres, M. J., Aznar, J., Gortázar, C. & Vicente, J. DNA detection reveals Mycobacterium tuberculosis complex shedding routes in its wildlife reservoir the Eurasian wild boar. Transbound. Emerg. Dis. 64, 906–915 (2017).
Acknowledgements
We thank the Veterinary Wildlife Services staff for assistance with collecting samples from Kruger National Park rhinoceros and providing laboratory space. We also thank Cepheid, TiKa Diagnostics Ltd, and the South African Medical Research Council, for their partnerships and ongoing support of our research.
Funding
This work was supported by the South African government through the South African Medical Research Council (SAMRC), Stellenbosch University Faculty of Medicine and Health Sciences, National Research Foundation South African Research Chair Initiative (SARChI grant 86949), Wellcome Foundation (grant #222941/Z/21/Z) National Geographic Society (NGS-61089C-19), Wildlife Disease Association crowd funding campaign through Experiment.com, and American Association of Zoo Veterinarians Wild Animal Health Fund (S005651 and S007355). The content is the sole responsibility of the authors and does not necessarily represent the official views of the funders.
Author information
Authors and Affiliations
Contributions
Conceptualization, R.D., R.W., W.J.G., C.W., P.B., M.M.; methodology, R.D, R.W., W.J.G, C.W, M.M.; software, R.D; formal analysis, R.D., C.W; investigation, R.D.; resources, P.B., W.J.G., M.M.; data curation, R.D; writing—original draft preparation, R.D.; writing—review and editing, R.D., R.W., P.B., C.W., M.M., W.J.G; visualization, R.D., C.W.; supervision, C.W., M.M., W.J.G.; project administration, W.J.G, M.M.; funding acquisition, W.J.G., M.M. All authors have revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dwyer, R., Witte, C., Buss, P. et al. Antemortem detection of Mycobacterium bovis in nasal swabs from African rhinoceros. Sci Rep 14, 357 (2024). https://doi.org/10.1038/s41598-023-50236-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-50236-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
