
Abstract
Nonalcoholic fatty liver disease (NAFLD) is associated with increased oxidative stress that leads to hepatocyte and mitochondrial damage. In this study we investigated the mechanisms involved in the induction of oxidative stress and impairment of mitochondrial quality control and mitophagy in hepatocytes by the saturated fatty acid palmitate and Western diet feeding in mice and if their harmful effects could be reversed by the neurotrophic factor glial cell derived neurotrophic factor (GDNF). Western diet (WD)-feeding increased hepatic lipid peroxidation in control mice and, in vitro palmitate induced oxidative stress and impaired the mitophagic clearance of damaged mitochondria in hepatocytes. This was accompanied by reductions in hepatocyte sirtuin 3 (SIRT3) deacetylase activity, gene expression and protein levels as well as in superoxide dismutase enzyme activity. These reductions were reversed in the liver of Western diet fed GDNF transgenic mice and in hepatocytes exposed to palmitate in the presence of GDNF. We demonstrate an important role for Western diet and palmitate in inducing oxidative stress and impairing mitophagy in hepatocytes and an ability of GDNF to prevent this. These findings suggest that GDNF or its agonists may be a potential therapy for the prevention or treatment of NAFLD.
Similar content being viewed by others

Introduction
Nonalcoholic fatty liver disease and high-fat diet (HFD)-induced obesity in rodents are associated with mitochondrial dysfunction1,2,3. This has been suggested to be due to increased hepatic oxidative stress resulting in part from defective mitochondrial respiratory chain in the face of increased β-oxidation, and from increased peroxisomal fatty acid oxidation2,4. A key feature of the mitochondrial dysfunction is reduction in the activity of the mitochondria matrix-based NAD-dependent deacetylase sirtuin 3 (SIRT3)5,6,7,8. This is accompanied by hyperacetylation and inhibition of the activity of several mitochondrial proteins including enzymes involved in the removal of reactive oxygen species (ROS) such as the superoxide dismutases (SODs) and increased hepatic lipid peroxidation and DNA damage2,3,7,8,9,10,11. The overexpression of SIRT3, on the other hand, lowers ROS production and restores antioxidant activity in hepatocytes following exposure to the saturated fatty acid palmitic acid (PA)11.
Mitochondria constantly undergo a quality control process through fusion, fission and mitophagy (selective removal of mitochondria through autophagy) to maintain a healthy population of mitochondria12. In NAFLD and HFD models of obesity, dysregulated mitochondrial fission, fusion, and mitophagy have been suggested to contribute to the mitochondrial dysfunction observed in these conditions13,14,15. Thus, mechanisms to reduce oxidative stress and to improve mitochondria quality control could have therapeutic value in treating and preventing NAFLD.
We have previously shown that the transgenic overexpression in mice of GDNF, a neurotrophic factor that is also involved in enteric neuronal development, is protective against HFD-induced hepatic steatosis and that hepatocytes from HFD-fed GDNF transgenic (GDNF Tg) mice have higher mitochondrial β-oxidation rates than HFD-fed control (CNTRL) mice16,17,18. We have also shown that GDNF enhances autophagic flux in hepatocytes in response to exposure to PA and is protective against PA-induced lipotoxicity18. Whether GDNF protection against lipotoxicity and improvement of mitochondrial function involves the enhanced removal of toxic quantities of ROS and damaged mitochondria through mitophagy is not known. In this study we investigated whether GDNF can enhance the removal of ROS, and whether GDNF increases mitophagy in hepatocytes to eliminate damaged mitochondria. We show that GDNF is protective against Western diet induced hepatic oxidative damage and PA-induced increase in ROS and impairment of mitophagic flux. We also show that GDNF is protective against PA-induced reduction in hepatocyte SOD enzyme activity and SIRT3 deacetylase activity. We show that knockout of SIRT3 expression abolishes GDNF protection against palmitate-induced hepatocyte death. Finally, we show that GDNF enhances hepatocyte levels of key proteins involved in mitochondrial quality control. Our findings demonstrate an important ability of GDNF to regulate hepatocytes’ mitochondrial function to protect against palmitate-induced oxidative stress and impairment of mitophagy.
Results
Western diet and palmitate-induced oxidative damage and impairment of mitophagy in hepatocytes is reversed by GDNF
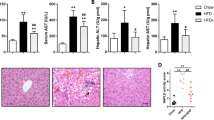
High levels of saturated fatty acids such as those in Western diets and high fat diets induce oxidative stress and damage in hepatocytes due to the intracellular buildup of ROS resulting from increased mitochondrial fatty acid β-oxidation3,9,19,20. We investigated the ability of Western diet feeding and PA to induce oxidative damage and to impair mitophagy in hepatocytes and if GDNF can protect against these harmful effects. Control mice fed a Western diet (WD) and provided with drinking water supplemented with fructose and glucose (FG) for 25 weeks had significantly (P < 0.001) higher hepatic levels of malondialdehyde (MDA) (an indicator of lipid peroxidation) indicative of increased hepatic oxidative damage when compared to control mice fed a regular rodent diet (RD) (Fig. 1A). In comparison, WD/FG-fed GDNF Tg mice had MDA levels that were significantly lower than those of WD/FG-fed control mice and only slightly higher than those of RD-fed control and GDNF Tg mice (Fig. 1A). We exposed RALA255-10G rat hepatocytes in vitro to PA and GDNF and assessed changes in intracellular ROS levels as well as the rate of mitophagic clearance of damaged mitochondria to determine if GDNF could prevent the accumulation of toxic levels of ROS and impairment of mitophagy resulting from the increased β-oxidation. We observed significant (P < 0.05) increase in ROS levels in hepatocytes exposed for 8 h to 0.2 mM PA and significant decrease in hepatocytes exposed to 0.2 mM PA in the presence of GDNF as well as in hepatocytes exposed to GDNF alone (Fig. 1B). The rate of mitophagic clearance was also significantly lower in hepatocytes exposed for 6 h to 0.2 mM PA when compared to hepatocytes exposed to Vehicle (Fig. 1C) indicative of impaired mitophagy. This decrease was, however, prevented in hepatocytes exposed to 0.2 mM PA in the presence of GDNF (Fig. 1C).

GDNF reverses Western diet- and palmitate-induced oxidative damage and impairment of mitophagy in hepatocytes. (A) Hepatic malondialdehyde (MDA) levels in control (CNTRL) and GDNF transgenic (GDNF Tg) mice fed regular diet (RD) or Western diet with fructose and glucose in drinking water (WD/FG) for 25 weeks. Plotted are means + SE. ***P < 0.001; *P < 0.05, relative to RD-fed control mice. N = 4 mice per group. (B) Intracellular ROS levels measured as CM-H2DCFDA fluorescence intensity in rat hepatocytes cultured for 24 h in the presence and absence of palmitate and GDNF. Plotted are means ± SE. **P < 0.01, *P < 0.05 relative to Vehicle-treated hepatocytes; N = 5. (C). Rat hepatocytes cultured for 6 h in the presence or absence of palmitate (PA), GDNF, and the lysosomal inhibitor chloroquine (CQ) and stained with MitoTracker Red CMX-ROS and plot of mitophagic flow expressed as % of Vehicle. Scale, 50 µm. Plotted are means ± SE. *P < 0.05; N = 2 repeats.
GDNF increases hepatocyte SOD activity and protects against PA and Western diet-induced loss of SIRT3
The first critical step in the detoxification of reactive superoxide radicals released during β-oxidation is catalyzed by two SOD enzymes, SOD1 which is localized mostly in the cytoplasm, but also in the mitochondrial intermembrane space, and SOD2 (manganese superoxide dismutase) which is localized in the mitochondrial matrix and mitochondrial inner membrane21,22,23,24,25. We assessed hepatocyte SOD activity to determine if GDNF’s ability to lower intracellular ROS levels was associated with increased SOD activity. Total SOD activity increased 40% in rat hepatocytes cultured for 24 h in medium supplemented with 0.15 mM PA and 100% in hepatocytes cultured in medium supplemented with 0.15 mM PA in the presence of GDNF (Fig. 2A) indicating an ability of GDNF to further enhance cellular antioxidant capacity to counter the oxidative stress induced by PA. The activity of the SOD enzymes is regulated through deacetylation of key lysine residues which is mediated by the NAD-dependent deacetylase SIRT326. We investigated the effects of GDNF on SIRT3 protein levels and deacetylase activity to determine if there was a correlation between the observed GDNF-induced increase in SOD activity and SIRT3 levels. SIRT3 activity levels were significantly (P < 0.05) decreased in rat hepatocytes exposed for 24 h to 0.2 mM PA while no changes were observed in hepatocytes exposed to PA in the presence of GDNF (Fig. 2B). We also observed significant reductions in SIRT3 gene expression and modest reductions in protein levels in primary human hepatocytes cultured in medium supplemented with 0.2 mM PA alone but significant increases in both gene expression and protein levels in hepatocytes cultured in medium with 0.2 mM PA in the presence of GDNF (Fig. 2C,D). We also assessed the effects of WD/FG on hepatic SIRT3 levels. Hepatic SIRT3 protein levels in WD/FG-fed control mice were significantly (P < 0.001) reduced when compared to RD-fed control mice in agreement with previously published data7,27. SIRT3 levels in the liver of WD/FG and RD-fed GDNF Tg mice were, however, unchanged relative to control mice (Fig. 2E).

GDNF increases hepatocyte SOD activity and protects against PA and Western diet-induced loss of SIRT3. (A) Total SOD activity levels in rat hepatocytes cultured for 24 h in the presence or absence of palmitate (PA) and GDNF. Plotted are means ± SE. ***P < 0.001, relative to vehicle; N = 3. (B) SIRT3 deacetylase activity levels in rat hepatocytes cultured for 24 h in the presence or absence of palmitate and GDNF. Plotted are means + SE. *P < 0.05, relative to Vehicle-treated hepatocytes; N = 3. Analysis of SIRT3 mRNA (C) and protein (D) levels in primary human hepatocytes cultured for 24 h in the presence and absence of palmitate (PA) and GDNF. The bands were cropped from the original blots presented in Supplementary Fig. S1. Protein loading was adjusted by analyzing for α-tubulin levels. Plotted are means + SE. ***P < 0.001; **P < 0.01; *P < 0.05, relative to Vehicle-treated hepatocytes; N = 4. (E) Western blot analysis of hepatic SIRT3 and GAPDH (loading control) protein levels in control (CNTRL) and GDNF transgenic (GDNF Tg) mice fed regular diet (RD) or Western diet with fructose and glucose in drinking water (WD/FG) for 25 weeks. The bands were cropped from the original blots presented in Supplementary Fig. S2. Plotted are means + SE. ***P < 0.001; *P < 0.05, relative to RD-fed control mice. N = 4 mice per group. NB.
GDNF reverses Western diet-induced suppression of hepatic PTEN-induced putative kinase 1 levels
We also investigated the effects of Western diet and PA on some key proteins involved in mitochondrial quality control. PTEN-induced putative kinase 1 (PINK1) is a mitochondrial protein that accumulates on the outer mitochondrial membrane marking them for removal through mitophagy. The liver of WD/FG-fed control mice had significantly (P < 0.001) reduced PINK1 (64 kDa) levels relative to RD-fed control mice while the liver of WD/FG-fed GDNF Tg mice showed no significant changes (Fig. 3A). In human and rat hepatocytes exposed for 24 h to 0.15 mM and 0.2 mM PA, the levels of the 64 kDa PINK1 protein were unchanged and significantly increased in hepatocytes exposed to palmitate in the presence of GDNF (Fig. 3B,C). The levels of OPA1 mitochondrial dynamin like GTPase, another mitochondrial membrane protein that is involved in mitochondrial fusion under certain stress conditions, were increased in rat hepatocytes after 24 h exposure to PA and increased further in hepatocytes exposed to PA in the presence of GDNF (Fig. 3D). These results demonstrate an important role for GDNF in regulating the levels of proteins involved in sensing mitochondrial stress and damage.

GDNF reverses Western diet-induced suppression of hepatic PTEN-induced putative kinase 1 (PINK1) levels. (A) Western blot analysis of hepatic PINK1 and GAPDH (loading control) protein levels in control (CNTRL) and GDNF transgenic (GDNF Tg) mice fed regular diet (RD) or Western diet with fructose and glucose in drinking water (WD/FG) for 25 weeks. The bands were cropped from the original blots presented in Supplementary Fig. S3. Plotted are means + SE. ***P < 0.001, relative to RD-fed control mice. N = 4 mice per group. (B). Western blot analysis of PINK1 and α-tubulin (loading control) protein levels in human hepatocytes exposed for 24 h to PA in the presence or absence of GDNF. The bands were cropped from the original blots presented in Supplementary Fig. S4. Plotted are means ± SE. ***P < 0.001; **P < 0.01, relative to vehicle and 0.2 mM PA + GDNF, respectively; N = 3. Western blot analysis of (C) PINK1 and β-actin (loading control) and (D) OPA1 and α-tubulin (loading control) protein levels in rat hepatocytes exposed to PA and GDNF for 24 h. The bands were cropped from the original blots presented in Supplementary Figs. S5 and S6. Plotted are means ± SE. ***P < 0.001, *P < 0.05, relative to Vehicle; N = 4.
Knockdown of SIRT3 expression abolishes GDNF’s protection against palmitate-induced hepatocyte cell death
Since we observed a strong ability of GDNF to enhance SIRT3 protein levels and deacetylase activity, using the RALA255-10G rat hepatocyte cell line we investigated if this conferred any survival advantage. We observed significant (P < 0.001) increases in SIRT3 protein levels in non-transfected hepatocytes cultured for 24 h in medium supplemented with 0.2 mM PA and further increases in hepatocytes cultured in medium supplemented with PA in the presence of GDNF or GDNF alone (Fig. 4A). This observation, although opposite to the previous observations in primary human hepatocytes, is not unique as increased hepatic SIRT3 gene expression and protein levels have been reported following 1 week of HFD feeding7. We then examined if reducing hepatocyte SIRT3 levels using siRNA can impair the ability of GDNF to protect against PA-induced cell death. GDNF was protective against PA-induced cell death in rat hepatocytes transfected with non-coding control siRNA as evidenced by the lower cleaved caspase-3 levels relative to hepatocytes cultured in the presence of PA alone (Fig. 4B). This protection was, however, lost in hepatocytes transfected with SIRT3 siRNA (Fig. 4B) thus demonstrating the important role of SIRT3 in mediating GDNF’s protection against PA-induced hepatocyte cell death. The proposed mechanism how GDNF acts to reduce oxidative stress and enhance mitophagic flux and survival in hepatocytes is summarized in Fig. 4C.

Knocked down of SIRT3 expression abolishes GDNF protection against palmitate-induced hepatocyte cell death. (A) Western blot analysis of SIRT3 and β-actin (loading control) protein levels in rat hepatocytes exposed to PA and GDNF for 24 h. The bands were cropped from the original blots presented in Supplementary Fig. S7. Plotted are means ± SE. ***P < 0.001, relative to Vehicle; N = 4. (B) Western blot analysis of SIRT3, cleaved caspase-3 and α-tubulin (loading control) levels in control siRNA and SIRT3 siRNA-transfected rat hepatocytes after 24 h exposure to palmitate (PA) and GDNF. The bands were cropped from the original blots presented in Supplementary Fig. S8. Plotted are means + SE. ***P < 0.001, relative to Vehicle-treated hepatocytes; N = 3. (C) Summary of proposed mechanism of GDNF action to protect hepatocytes against oxidative stress resulting from increased β-oxidation involving enhancement of superoxide dismutase (SOD) activity and SIRT3 levels as well as enhancement of resistance against fatty acid-induced suppression of mitophagy. To counter the increased mitochondrial generation of reactive oxygen species (ROS) GDNF increase SIRT3 levels and deacetylase activity which results in enhanced activity of mitochondrial SOD enzymes and their catalysis of the breakdown of ROS. GDNF also enhances mitochondrial cycling and removal of excess and damaged mitochondria through mitophagy.
Discussion
In this study we explored the role of a Western diet and the saturated fatty acid palmitate in inducting oxidative damage in hepatocytes and the ability of the neurotrophic factor GDNF to enhance mitochondrial quality control and prevent oxidative damage.
In NAFLD, hepatocytes respond to increased cellular free fatty acid (FFA) levels arising from FFA uptake, adipose tissue lipolysis and de novo lipogenesis by increasing the rate of mitochondrial β-oxidation which in turn results in increased intracellular ROS levels28,29. Increased oxidative stress with decreased SIRT3 protein and activity levels have been reported during NAFLD and in HFD rodent models of obesity2,3,7,9,11,26,30. This has been attributed in part to a defect in the mitochondrial respiratory chain resulting uncontrolled intracellular ROS levels4. The importance of SIRT3 in protecting hepatocytes against oxidative stress has been demonstrated in studies in which knockout of SIRT3 has been shown to result in the hyperacetylation of mitochondrial proteins including manganese superoxide dismutase and complex I of the electron transport chain6,7,26,31 while the transgenic overexpression of SIRT3 prevents oxidative stress by increasing antioxidant enzyme levels and reducing ROS levels11. SIRT3 acts to reduce ROS levels by deacetylating and activating SOD226.
Mitophagy has been demonstrated to play an important role in protecting against the initial insult resulting from oxidative stress and changes in the levels of proteins involved in sensing mitochondrial damage such as PINK1 and in driving mitochondrial fusion and fission have been studied in HFD-fed mice. In HFD-fed mice, changes in hepatic PINK1 and OPA1 levels involving slight increases in PINK1 levels and decreases in OPA1 levels have been reported13. In addition, increases in the hepatic levels of other proteins involved in mitochondrial fission and fusion including dynamin related protein 1 and mitofusin 1 and reductions in the levels of protein involved in mitophagy have also been reported in HFD-fed mice15.
In this study we observed an important role of GDNF in preventing hepatic lipid peroxidation and oxidative stress in hepatocytes by reducing reactive oxygen species levels and enhancing mitophagic flux and the expression of proteins involved in mitochondrial quality control. These effects of GDNF occurred in the background of enhanced SOD and SIRT3 activity suggesting that GDNF possibly acts through SIRT3 to protect against oxidative stress. Indeed, when we knocked out SIRT3 expression we observed an inability of GDNF to protect against PA-induced lipotoxicity. In our study hepatic PINK1 levels were highly elevated in RD and WD/FG-fed GDNF transgenic mice. In addition, PINK1 and OPA1 protein levels were modestly increased, respectively, in human and rat hepatocytes exposed to palmitate alone and highly elevated in hepatocytes exposed to PA in the presence of GDNF. Moreover, hepatocytes exposed to GDNF alone also had highly elevated PINK1 and OPA1 levels.
Our study, thus, demonstrates an important role of GDNF in regulating hepatocyte ROS and SIRT3 levels, mitochondrial cycling and mitophagy and in protecting against WD- and palmitate-induced oxidative stress and cell death. Hence, GDNF agonists may be potential therapy for the prevention or treatment of NAFLD.
Materials and methods
Animals
This study followed ARRIVE guidelines on the use of experimental animals. Animal studies were conducted in 5–6 weeks old female CF-1 (control) mice and GDNF transgenic (GDNF Tg) littermates. GDNF transgenic mice are on a CF-1 background and overexpress GDNF in cells expressing glial fibrillary acidic protein (GFAP) which is expressed in several tissues including the liver16,18. The mice were maintained on a 12 h light–dark cycle in a temperature-controlled barrier facility with free access to food and water. Four control (CF-1) mice and 4 GDNF Tg mice were assigned to a regular rodent diet (RD) (2018SX; Teklad Global 18% Protein Extruded Rodent Diet, Harlan Laboratories, Madison, WI; 6.2% fat by weight/18% kcal from fat) with regular drinking water and maintained on the diet for 25 weeks. Another 4 CF-1 mice and 4 GDNF Tg mice were assigned to a Western Diet (WD) (TD.120528, Harlan; 21.2% fat by weight/42% kcal from fat, with increased Sucrose, and 1.25% cholesterol) along with drinking water supplemented with a high sugar solution (FG) [23.1 g/L d-fructose (Sigma-Aldrich, cat. #1286504 USP) and 18.9 g/L d-glucose (Sigma-Aldrich, cat. #1181302 USP)]32. These mice were also maintained on the diet for 25 weeks. The complete composition of the diets is shown in Table 1. All animal studies were approved by the Atlanta Veteran Affairs Medical Center Institutional Animal Care and Use Committee and conducted according to the recommended guidelines.
Cell culture
RALA255-10G rat hepatocytes were cultured in Gibco Dulbecco’s modified Eagle’s medium (DMEM) high glucose, HEPES, without pyruvate (#12430054, Life Technologies Corp, Grand Island, NY, USA) as previously described33. Human hepatocytes (H1000.H15B+, Lot No. HC3-37, Sekisui XenoTech, Kansas City, KS, USA) were cultured according to the vendors instructions. Cell culture media were replaced with fresh media every 48 h.
The rat and human hepatocytes were each assigned to 4 treatment groups: Vehicle, Palmitate, GDNF, and PA plus GDNF. Stock (6 mM) palmitate (Sigma-Aldrich, St. Louis, MO, USA) conjugated to fatty acid-free bovine serum albumin (BSA) (Sigma-Aldrich) was prepared as previously described34 and used at final concentration of 0.1–0.3 mM. Recombinant human and mouse GDNF (Shenandoah Biotechnology, Warwick, PA, USA) were used at a final concentration of 100 ng/mL.
Assessment of oxidative stress
Intracellular reactive oxygen species (ROS) levels were assessed in RALA255-10G rat hepatocytes cultured for 8 h in medium supplemented with or without palmitate (PA) and GDNF. The hepatocytes were stained with the general reactive oxygen species probe CM-H2DCFDA (Molecular Probes, Eugene, OR, USA) according to the manufacturer’s instructions and fluorescence measured on a SpectraMax iD3 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA). Lipid peroxidation was assessed in mice liver tissues using a Lipid Peroxidation (MDA) Assay Kit (#MAK085, Sigma-Aldrich).
Assessment of antioxidant and SIRT3 activity levels
Total cellular superoxide dismutase activity was assessed in RALA255-10G rat hepatocytes cultured for 24 h in medium supplemented with or without GDNF and palmitate using the Superoxide Dismutase (SOD) Colorimetric Activity Kit (# EIASODC, Life Technologies Corp. Frederick, MD, USA). SIRT3 deacetylase activity was also assessed in RALA255-10G rat hepatocytes cultured for 24 h in medium supplemented with or without GDNF and palmitate using a fluorometric SIRT3 Activity Assay Kit (# ab156067, Abcam, Waltham, MA, USA).
Assessment of mitophagic flux
Mitophagic flow was assessed by flow cytometry using a modification of a previously published protocol35. RALA255-10G rat hepatocytes were cultured for 6 h with or without palmitate (0.2 mM), GDNF (100 ng/mL), and with or without the lysosomal inhibitor chloroquine (CQ) (30 µM) (Cell Signaling Technology, Danvers, MA, USA). MitoTracker Red CMX-ROS (#M7512, Molecular Probes, Eugene, OR, USA) was then added to the cells at a final concentration of 25 nM in culture medium and the cells cultured for a further 15 min. The cells were washed once in PBS, stained for 30 min with LIVE/DEAD Fixable Near-IR Stain (#L34976, Life Technologies Corp., Carlsbad, CA, USA) and fixed with 3.7% formaldehyde in complete culture medium at 37 °C for 15 min. After washing 3 times with PBS with 1% bovine serum albumin, the cells were analyzed by flow cytometry at the Emory Pediatric and Winship Flow Cytometry Core (Emory University, Atlanta, GA, USA). Mitophagic flow was calculated from the difference in the number of MitoTracker Red CMX-ROS-positive hepatocytes between cells cultured in the presence of the inhibitor (CQ) and those cultured without CQ for each treatment.
Western blotting
Western blotting was performed as previously described17 using rabbit primary antibodies to SIRT3 (#5490, Cell Signaling Technology, Danvers, MA, USA), OPA1 (#80471, Cell Signaling Technology), cleaved caspase-3 (Asp175) (Cell Signaling Technology) and PINK1 (Sigma-Aldrich) diluted 1:1000. Mouse monoclonal primary antibodies to α-tubulin (DM1A) (Cell Signaling Technology) and β-actin (A5441, clone AC-15) (Sigma-Aldrich) were diluted, respectively, 1:1000 and 1:5000 before use. Horseradish peroxidase conjugated anti-mouse and anti-rabbit IgG (Cell Signaling Technology) secondary antibodies were used at 1:2,000 dilution. A semi quantitative measurement of band intensity was performed using the Carestream Molecular Imaging Software (Carestream Molecular Imaging, New Haven, CT, USA) and Fiji36.
Gene expression analysis
Total RNA was isolated using the RNeasy Mini kits (Qiagen GmbH, Hilden, Germany) and first-strand cDNA synthesized using SuperScript VILO (Invitrogen Life Technologies, Grand Island, NY, USA). Real-time PCR reactions were set up using Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) human SIRT3 upstream (5′-ATCGATGGGCTTGAGAGAGTGTC-3′) and downstream (5′-AACCCTGTCTGCCATCACGT-3′) primers and human GAPDH upstream (5′-AGCCTCAAGATCATCAGCAATGCC-3′) and downstream (5′-TGTGGTCATGAGTCCTTCCACGA-3′) primers. The primers were designed such that at least one primer in the pair spanned an intron to prevent it from priming on to genomic DNA. The inability of these primers to amplify genomic DNA was confirmed by PCR. Thermal cycling was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems).
siRNA transfection
Rat hepatocytes were transfected with SMARTpool, ON-TARGETplus Rat Sirt3 siRNA (Catalog # L-084761-03-0005; Dharmacon, Cambridge, United Kingdom) or ON-TARGETplus Non-targeting Control Pool (Catalog # D-001810-10-05) using Lipofectamine RNAiMax (Invitrogen Life Technologies) according to recommended procedure.
Statistical analysis
Statistical analyses were conducted using the GraphPad Prism software version 3.00 for Windows (GraphPad Software, San Diego, CA). Data were tested for normality and subjected to unpaired t test or one-way ANOVA with Tukey posttest.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information file).
Abbreviations
- SIRT3:
-
Sirtuin 3
- PINK1:
-
PTEN-induced putative kinase 1
- GDNF:
-
Glial cell derived neurotrophic factor
- NAFLD:
-
Nonalcoholic fatty liver disease
- ROS:
-
Reactive oxygen species
- PA:
-
Palmitate
- OPA1:
-
OPA1 mitochondrial dynamin like GTPase
- HFD:
-
High-fat diet
- CM-H2DCFDA:
-
6-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester
- CNTRL:
-
CF-1 control mice
- EM:
-
Emission
- EX:
-
Extinction
- FA:
-
Fatty acid
- FG:
-
Fructose/glucose
- GDNF Tg:
-
GDNF transgenic mice
- RD:
-
Regular diet
- TG:
-
Triglyceride
- WD:
-
Western diet
References
Begriche, K., Massart, J., Robin, M. A., Bonnet, F. & Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58, 1497–1507. https://doi.org/10.1002/hep.26226 (2013).
Simões, I. C. M. et al. The alterations of mitochondrial function during NAFLD progression—an independent effect of mitochondrial ROS production. Int. J. Mol. Sci. 22, 6848. https://doi.org/10.3390/ijms22136848 (2021).
Koliaki, C. et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 21, 739–746. https://doi.org/10.1016/j.cmet.2015.04.004 (2015).
Pérez-Carreras, M. et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 38, 999–1007. https://doi.org/10.1053/jhep.2003.50398 (2003).
Hirschey, M. D. et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. https://doi.org/10.1038/nature08778 (2010).
Lombard, D. B. et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814. https://doi.org/10.1128/mcb.01636-07 (2007).
Hirschey, M. D. et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190. https://doi.org/10.1016/j.molcel.2011.07.019 (2011).
Kendrick, A. A. et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 433, 505–514. https://doi.org/10.1042/bj20100791 (2011).
Satapati, S. et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 125, 4447–4462. https://doi.org/10.1172/jci82204 (2015).
Sanyal, A. J. et al. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120, 1183–1192. https://doi.org/10.1053/gast.2001.23256 (2001).
Li, R. et al. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 18, 229–243. https://doi.org/10.1016/j.redox.2018.07.011 (2018).
Hill, B. G. et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 393, 1485–1512. https://doi.org/10.1515/hsz-2012-0198 (2012).
Wang, L. et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 61, 486–496. https://doi.org/10.1002/hep.27420 (2015).
Galloway, C. A., Lee, H., Brookes, P. S. & Yoon, Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G632-641. https://doi.org/10.1152/ajpgi.00182.2014 (2014).
Jacobi, D. et al. Hepatic Bmal1 regulates rhythmic mitochondrial dynamics and promotes metabolic fitness. Cell Metab. 22, 709–720. https://doi.org/10.1016/j.cmet.2015.08.006 (2015).
Mwangi, S. M. et al. Glial cell line-derived neurotrophic factor protects against high-fat diet-induced obesity. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G515–G525. https://doi.org/10.1152/ajpgi.00364.2013 (2014).
Mwangi, S. M. et al. Glial cell line-derived neurotrophic factor protects against high-fat diet-induced hepatic steatosis by suppressing hepatic PPAR-γ expression. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G103–G116. https://doi.org/10.1152/ajpgi.00196.2015 (2016).
Mwangi, S. M. et al. Glial cell line-derived neurotrophic factor enhances autophagic flux in mouse and rat hepatocytes and protects against palmitate lipotoxicity. Hepatology 69, 2455–2470. https://doi.org/10.1002/hep.30541 (2019).
Nakamura, S. et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 284, 14809–14818. https://doi.org/10.1074/jbc.M901488200 (2009).
Begriche, K., Igoudjil, A., Pessayre, D. & Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 6, 1–28. https://doi.org/10.1016/j.mito.2005.10.004 (2006).
Forrester, S. J., Kikuchi, D. S., Hernandes, M. S., Xu, Q. & Griendling, K. K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 122, 877–902. https://doi.org/10.1161/circresaha.117.311401 (2018).
Weydert, C. J. & Cullen, J. J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 5, 51–66. https://doi.org/10.1038/nprot.2009.197 (2010).
Wang, Y., Branicky, R., Noë, A. & Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 217, 1915–1928. https://doi.org/10.1083/jcb.201708007 (2018).
Okado-Matsumoto, A. & Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 276, 38388–38393. https://doi.org/10.1074/jbc.M105395200 (2001).
Kira, Y., Sato, E. F. & Inoue, M. Association of Cu, Zn-type superoxide dismutase with mitochondria and peroxisomes. Arch. Biochem. Biophys. 399, 96–102. https://doi.org/10.1006/abbi.2001.2738 (2002).
Qiu, X., Brown, K., Hirschey, M. D., Verdin, E. & Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667. https://doi.org/10.1016/j.cmet.2010.11.015 (2010).
Bao, J. et al. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic. Biol. Med. 49, 1230–1237. https://doi.org/10.1016/j.freeradbiomed.2010.07.009 (2010).
Sunny, N. E., Parks, E. J., Browning, J. D. & Burgess, S. C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 14, 804–810. https://doi.org/10.1016/j.cmet.2011.11.004 (2011).
Lambert, J. E., Ramos-Roman, M. A., Browning, J. D. & Parks, E. J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146, 726–735. https://doi.org/10.1053/j.gastro.2013.11.049 (2014).
Zhang, T. et al. SIRT3 promotes lipophagy and chaperon-mediated autophagy to protect hepatocytes against lipotoxicity. Cell Death Differ. 27, 329–344. https://doi.org/10.1038/s41418-019-0356-z (2020).
Ahn, B. H. et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 14447–14452. https://doi.org/10.1073/pnas.0803790105 (2008).
Tsuchida, T. et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 69, 385–395. https://doi.org/10.1016/j.jhep.2018.03.011 (2018).
Wang, Y., Schattenberg, J. M., Rigoli, R. M., Storz, P. & Czaja, M. J. Hepatocyte resistance to oxidative stress is dependent on protein kinase C-mediated down-regulation of c-Jun/AP-1. J. Biol. Chem. 279, 31089–31097. https://doi.org/10.1074/jbc.M404170200 (2004).
Egnatchik, R. A., Leamy, A. K., Jacobson, D. A., Shiota, M. & Young, J. D. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol. Metab. 3, 544–553. https://doi.org/10.1016/j.molmet.2014.05.004 (2014).
Mauro-Lizcano, M. et al. New method to assess mitophagy flux by flow cytometry. Autophagy 11, 833–843. https://doi.org/10.1080/15548627.2015.1034403 (2015).
Schindelin, J. et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. https://doi.org/10.1038/nmeth.2019 (2012).
Acknowledgements
This work is supported by U.S. National Institutes of Health grants R01DK080684 (Shanthi Srinivasan), R01DK044234 (Mark J. Czaja and Shanthi Srinivasan), and R01DK047987 (Paul A. Dawson), Department of Veterans Affairs Research and Development Merit Review Awards I01 BX000136 (Shanthi Srinivasan), I01 BX004263 (Mike Hart), and BX002526 (Didier Merlin), and Department of Veterans Affairs Senior Research Career Scientist Award BX004476 (Didier Merlin). We thank Aaron Rae from the Emory Pediatrics' and Winship Flow Cytometry Core for experimental design and planning for flow cytometry and for performing flow cytometry data acquisition and analysis.
Author information
Authors and Affiliations
Contributions
S.M.M., M.J.C., C.M.H., and S.S. performed study concept and design, interpretation of data and drafting and review of the manuscript. S.M.M., G.L., and A.B. performed acquisition of data, analysis and interpretation of data, and review of the manuscript. D.M., Y.C.J. and P.A.D. performed development of methodology and review of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mwangi, S.M., Li, G., Balasubramaniam, A. et al. Glial cell derived neurotrophic factor prevents western diet and palmitate-induced hepatocyte oxidative damage and death through SIRT3. Sci Rep 12, 15838 (2022). https://doi.org/10.1038/s41598-022-20101-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20101-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
