
Abstract
Ex-vivo tumor tissue culture systems are used as models to test specific anti-cancer drugs. Their main advantage is that they are closely comparable with the in vivo tumor in their host organism. We previously reported that precision-cut organotypic tissue slices of pancreatic ductal adenocarcinoma (PDAC) can be successfully cultured ex-vivo for at least 4 days. In order to study how culturing might affect transcription patterns, we now performed genome-wide transcriptome profiling of both baseline (0 h) and explanted tumors at daily intervals (24, 48 and 72 h) after start of culturing. The total-RNA from five samples of surgically resected human PDAC tumors at baseline and at different time points in culture was sequenced. Differential gene expression analysis of the whole transcriptome, testing 58,713 genes and over 206,000 transcripts, found that only a small number of genes showed significant changes in expression between baseline and cultured samples. The cultured tumor slices showed upregulation of a median of 12, 10 and 15 genes and downregulation of a median of 15, 12 and 25 genes at 24, 48 and 72 h in culture, respectively. One sample had morphologically increasing loss of tissue viability (range 0–18%). The vascular endothelial growth factor A (VEGFA) was significantly upregulated during the entire culture period in this case. Pathway over-representation analysis suggested that VEGFA together with the PTGS2 gene were upregulated at the same time as HIF-1-triggered cell apoptosis via NF-ĸB and the AP-1 activating factor was induced. Indeed, increased areas of apoptotic lesions were visible in this sample after 24 hours of culture. In conclusion, genome-wide transcriptome analysis supports that ex-vivo cultured tissue slices of PDAC may be a representative model of the original tumor. Transcriptome analysis was found to be a valuable complement to morphology for evaluation of ex-vivo cultures of PDAC.
Similar content being viewed by others

Introduction
Pancreatic cancer is currently the fourth leading cause of cancer-related death in the West and predicted to rank second by 20301. Discrepancies in pancreatic cancer incidence between countries suggest that environmental and geographical factors may play a substantial role as risk factors for the disease2. Pancreatic cancer is usually diagnosed at advanced stages and characterized by aggressive tumor biology and pronounced resistance to treatment, which result in an overall 5-year survival rate of <7%3. PDAC tumors with distant metastases are generally not resected due to their dismal prognosis and surgical removal is indicated only for localized cancer. Risk factors are a family history of pancreatic cancer, smoking, obesity, chronic pancreatitis and genetic alterations, e.g. in BRCA1, BRCA2, PALB2 and ATM4.
Most pancreatic cancers have sporadic gene alterations, such as amplifications, deletions, translocations, inversions, frame-shifts, and substitutions. Mutations of multiple genes in pancreatic cancer occur in over 70% of all diagnosed cases. Most of the genetic alterations cause overexpression or activate signal transduction pathways5. As a result, translated oncoproteins or suppressor cellular regulatory mechanisms force cells into dysregulated growth6,7,8.
Histopathologically, most PDACs tumors are characterized by the presence of a prominent desmoplastic stroma with microenvironment that is composed of an admixture of acellular and cellular elements, including the extracellular matrix, mesenchymal cells (mainly cancer associated fibroblasts) and immune cells. The tumor microenvironment has pleiotropic tumor-promoting effects and contributes to an immune suppressive milieu, which can limit treatment efficacy9,10. Since the microenvironment in pancreatic cancer has unique features that directly impact the molecular signatures of the tumor cells, it is important to study the global molecular events that take place in the tumor tissues11,12.
Transcriptome sequencing uses massive parallel sequencing technologies to measure tissue RNA expression. Sequence data can be aligned to a reference genome to build full-length transcripts. Annotated transcripts are used to study differentially expressed genes while non-mapped reads can be analyzed to study fusion genes or viral transcripts. Transcriptome sequencing data can also be used to define novel transcripts leading to discovery of neoantigens13,14,15.
Earlier, Misra and Moro et al., showed that pancreatic cancer cells, the tumor microenvironment and the interspersed remnants of nonneoplastic pancreas in 350 µm thick slices maintained their structural integrity, phenotypic characteristics and functional activity when kept in culture for at least 4 days16.
In the present study, we employed deep sequencing to characterize the transcriptome of five cases of PDAC that were selected based on the different degree of cell death in the tissues and tumor grade. We used freely available, open source bioinformatic tools to perform differential gene expression and pathway enrichment analysis. We hypothesized that tissue viability (and its loss) in ex-vivo cultured tissue slices would be reflected in the differences in transcription of genes involved in pathways related to cell death.
Results
We sequenced FFPE tissue from five tumor samples at baseline and their matched cultured tissue slices at 24, 48, and 72 h. In total 5 baseline tumor samples and 15 corresponding organotypic (OT) cultures were assessed.
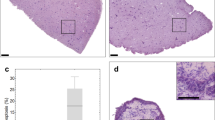
Two OT cultures were from well differentiated and three from moderately to poorly differentiated ductal adenocarcinomas. These were selected from the previously published series (Misra and Moro et al.) for being histomorphologically comparable (same tumor type and grouped by grade of differentiation) but showing different levels of tissue viability during culture16. In particular, four cultures (OT1, OT5, OT11 and OT12) showed no or minimal loss of viability (range 0–6%), while one (OT9) showed mild to moderate loss of tissue viability (range 0–18%) (Fig. 1. legend provided). The tumor cell outgrowth along the edge of the tissue slices ranged from 71–100% (of the perimeter) for the four cultures with high tissue viability and 55–76% for the one with viability loss. Patient gender and clinicopathological data as well as the percentage ranges of non-viable tissue for each OT culture are presented in Table 1.

Morphology-based analysis of tissue viability in the baseline (0 h) and cultured (24–2 h) tissue slices of PDAC samples OT5, OT9 and OT12. Non-viable tissue regions are indicated as red areas in the slice diagrams, their total percentages (with respect to the whole slice area) in red font and by red arrows in representative photomicrographs (hematoxylin-eosin staining, bar line: 100 μm).
Based on human assembly Homo sapiens GRCh38.95 annotations, 58,713 genes (coding and non-coding) and 206,487 gene transcripts were tested. Transcript isoform test detected no statistically significantly differentially expressed isoforms at any time point.
To analyze the differences in gene expression, each FFPE block prepared at time points 24 h, 48 h, and 72 h was compared to the baseline FFPE tumor tissue sample. Using TopHat2/Cuffdiff a limited number of differentially expressed genes were selected when allowing a false discovery rate of 0.05 (Table 2).
Over-representation pathway analysis
To evaluate if any of the differentially expressed genes (DEGs) were associated significantly with certain pathways, over-representation analysis was performed. Those genes that were expected to be in at least in one pathway are presented in Table 3 with symbols to show if they were up- or downregulated. Overall results from the top 3 significantly estimated pathways for all samples are presented in Table 4. For sample OT11 no differentially expressed genes were detected after 24 h of culture. Accordingly, none of the mapped entities (18 out of 20) after 48 h of culture for sample OT12 were over-represented in an enriched pathway.
Over-representation analysis for sample OT9, indicated that the gene sets PTGS2; IGFBP1; PMAIP1; and VEGFA could activate the therapy-induced HIF-1 survival signaling pathway followed by Quercetin, Nf-kB and AP-1 Induced Cell Apoptosis (48 h), the latter being in line with the histological detection of cell death/apoptosis (Fig. 1).
After 72 h, several mitochondrial transcripts were predominantly downregulated, suggesting a drop in oxidative phosphorylation, NADH dehydrogenase activity, and the respiratory electron transport system (Table 3).
The Cathepsin L gene transcript, CTSL was frequently upregulated in all cases. CTSL together with isoforms of MMP metalloproteinase and/or collagen formation factors were mapped in the Reactome database, mainly in pathways controlling the skeleton of the solid tissues (Tables 3 and 4).
The long noncoding RNAs MALAT1 and NEAT1
High levels of transcripts of long non-coding lncRNAs, MALAT1 and NEAT1 (located on the long arm of chromosome 11) have been described in various cancer types including pancreatic cancer17,18. We found that both transcripts were highly expressed (based on FPKM values) in all examined samples (data not shown) but there were no overall expression changes of these transcripts during the incubations.
Analysis of biologically relevant fusion transcripts
No possible oncogenic fusions were detected in sample OT5 at any time-point.
FusionCatcher detected “likely” fusion of CDKL5-NEAT1 in specimen OT9 after 48 h but this fusion did not contain sequences from exonic regions of the involved genes.
Materials and methods
Preparation of precision-cut tissue slices
This study was approved by the Regional Ethical Review Board, Stockholm (IRB diary number 2012/1657-31/4). In brief, as previously described (Misra and Moro et al.), fresh samples of pancreatic cancer tissue were cut into 350 µm thick slices using a vibrating blade microtome. The slicing process yielded between 12 and 17 slices per tissue sample. The first slice (baseline tissue slice, time point 0 h) was immediately fixed in formalin and embedded in paraffin. This was used as baseline for morphological (tissue viability, grade of tumor differentiation) assessments in the cultured tissue slices. The subsequent eight slices were placed on inserts and cultured at 37 °C in a humidified incubator in 41% O2 for up to 96 h. Every 24 h, duplicate tissue slices were harvested, formalin fixed and embedded in paraffin. The tissue slices were subsequently sectioned and processed for histology. Loss of tissue viability was determined on hematoxylin-eosin stained sections according to the percentage of necrotic and apoptotic areas with respect to the entire surface of the tissue slice. Among the duplicate slices for 24, 48 and 72 h, the one with higher tumor tissue content was selected for transcriptome analysis.
Isolation of total-RNA and library preparation
RNA was extracted and treated with DNase from the formalin-fixed paraffin embedded (FFPE) 2×10 µm curls using the Maxwell RSC FFPE RNA kit (Promega, Madison, USA). Extracted RNA was quantified by Qubit 4.0 and RNA HS Assay Kit (Thermofisher Scientific, Waltham, USA). A maximum of fifty nanogram of extracted RNA was used to prepare cDNA libraries skipping the RNA fragmentation step. Whole transcriptome sequencing libraries were prepared using the Takara Smarter total-RNA Seq kit V2 Pico Input Mammalian (Takara Bio Inc, Kusato, Japan). Briefly, cDNA was prepared by random hexamer priming while preserving strand of origin information. During a first PCR amplification, full-length Illumina adapters, including barcodes were added. The ribosomal cDNA sequences (originating from rRNA) were cleaved in the presence of RNAse H and the mammalian-specific R-Probes. The remaining fragments were enriched via a second round of PCR amplification using primers universal to all libraries. The final library contained sequences allowing clustering on any Illumina flow cell. All libraries including a non-template control were quantified on Bioanalyzer (Agilent Technologies, Santa Clara, USA). To equalize sequencing input amount, each library was quantified by Qubit 4.0 HS Assay Kit (Thermofisher Scientific, Waltham, USA).
NextSeq 500 sequencing, bioinformatic analysis and statistical evaluation
In total twenty normalized libraries from five PDAC were sequenced on a NextSeq. 500 Illumina system (Illumina, San Diego, USA). Paired-end cycle sequencing 2 × 75 was run on High Output V2 Kit which in total generated a median of 40 million raw paired-end reads/sample. Indices, included in the Illumina adapters, were used to demultiplex and assign raw sequence reads. Datasets were analyzed using the Chipster virtual bioinformatic interface at CSC Finland19 to process and analyze RNA data for gene expression. All sequences were quality-checked by FASTQC20, adapters were preprocessed and trimmed if necessary. To estimate the stability of the sequencing reactions insertion/deletion rate values were observed after FASTQ alignment by STAR using Homo sapiens genome version GRCh38.95 as ref. 21 (data not shown).
For comparative analysis of gene expression, TopHat2 alignment tool for paired-end reads followed by the Cufflinks algorithm package version 2.1.1 for assembly and Cuffdiff for differential expression22. Multiple GTF files were merged using Cuffmerge and sorted by chromosome and start position.
GTF and the corresponding BAM files were compared to the reference baseline sample using the Cuffdiff tool to study the DEGS of the potentially novel transfrags where at least one splice junction was shared with a reference transcript. To calculate the DEGS, Cuffdiff differences in FPKM (Fragment Per Kilobase Million) values were considered significant when p-values were equal or less than 0.05.
Hypergeometric test for ConsensusPathDB
To predict the possible molecular functional and gene-pathway interactions, Hypergeometric distribution probability test (over-representation analysis) was used. This pathway analysis is described by the ConsensusPathDB (CPDB) created by Herwig et al.23. This test searches gene lists for over-represented pathways in the CPDB database at: http://cpdb.molgen.mpg.de/. The p-values of each pathway test reflect the significance of the observed overlap between the input gene list and the module’s members as compared to random expectations. CPDB is an open source database provided by the Max Planck Institute for Molecular Genetics and contains pathway information from 32 publicly available databases, including BioCarta, HumanCyc, Reactome, KEGG and WikiPathways.
Detection of fusion genes and viral/phage transcripts
To analyze the RNA sequencing data for detection of somatic fusion transcripts, FusionCatcher_v1.10 (https://github.com/ndaniel/fusioncatcher), using human genome database v95 was installed and used on a local Linux server24. The command line option was used to enable FusionCatcher to search for fusions in raw paired-end FASTQ files, skipping the alignment tool BLAT and adding -V to the command line to engage and store unaligned reads to be able to BLAST for possible viral transcripts. Only fusion transcripts not labeled as “banned” in the output file (because of their known presence in healthy individuals) or involving intronic sequences and not associated with a protein product were considered as a novel finding.
Ethics declaration
This study was approved by the Regional Ethical Review Board, Stockholm (IRB diary number 2012/1657-31/4). Written informed consent was obtained from all patients prior to surgery. All study methods were performed in accordance with the relevant guidelines and regulations.
Discussion
Sequencing of the total transcriptome from surgically resected human PDAC at four time points in culture in vitro revealed that limited number of genes (median 10 to 25) exhibited altered expression after ex-vivo culture. Among those, the pancreas-specific transcripts (PNLIPRP1: pancreatic lipase related protein 1 and CPA1: carboxypeptidase A1 and CPA2) were suppressed.
In this study modern deep sequencing with short reads was used. In this way a very deep transcriptional mapping could be performed, even in specimens that may contain fragmented ribonucleic acids. The fact that we found similar expression levels in the explanted tumor for >200,000 transcripts indicates that the analysis was both deep and comprehensive.
Tumor dissection to enrich for certain cell types was not performed, hence it was not possible to study the contribution or representation of the cellular compartment expression in diverse pathways. Rather, we studied the overall expression from all cellular components within the tissue including the tumor, stroma, infiltrating immune cells and nonneoplastic pancreatic tissue. In this way, it was possible to capture a genome-wide image of globally expressed genes, reflecting the molecular events that take place in the tumor tissue and its surrounding microenvironment.
Davies EJ. et al. also studied precision-cut slices prepared from human and animal models of different cancer types25. They investigated tissue viability and proliferation at different oxygen levels and analyzed the expression of a large set of RNA molecules that are involved in different cellular mechanisms, such as apoptosis, DNA damage and repair. They concluded that ischaemia during transportation from the surgical theatre and mechanical slicing (baseline tissue slice, timepoint 0 h) had little impact on stress gene expression, but that cultivation induced changes in gene and protein expression. They observed that atmospheric oxygen concentration is suitable for tissue slice culture and that the slice thickness has no impact on O2 diffusion and its availability to the tissue. Consistent with that, a previous study from our group showed no significant differences in tissue viability, proliferation, or metabolic activity between matched tissue slices cultured in atmospheric (21% O2) or hyperoxic (41% O2) conditions16. Davies EJ. et al. also showed overexpression of HIF-1α by immunohistochemistry in association with hypoxia and stress of the tissue slice compartments. In line with this observation, VEGFA and the HIF-1α signaling network were upregulated in the cultured slices of sample OT9 that exhibited increased amounts of apoptotic cell death. The vascular growth factor A, encoded by the VEGFA gene, is a key factor in angiogenesis and increased vascular permeability26. It can be speculated that upregulation of VEGFA was induced in hypoxic conditions because “HIF-1-induced” cell apoptosis via NF-ĸB and the AP-1 activating factor was suggested as a possible pathway.
Altogether, these findings support the role and value of assessing HIF-1α activity for the detection of cellular stress in the cultured tissue slices.
In the current study, the analysis of the total cellular transcriptome assessed the impact of the new environment on the cultured pancreatic cancer tissue. Ex-vivo model systems are influenced largely by culture conditions such as varying oxygen tension and absence of vital cellular stimulatory cues.
Future studies linking transcriptome data and morphological phenotypes of cancer tissue could be of value to shed light on the molecular origins of intratumoral heterogeneity.
Conclusions
Genome-wide transcriptome sequencing revealed transcriptional changes only in a limited number of genes during tissue culture ex vivo. Changes were mainly seen in genes involved in biological processes whose molecular function can affect the stabilization or upregulation of cellular compartments. Consistent with histomorphological analysis, upregulation of pathways related to cell death/apoptosis were observed in a cultured tumor with decreased tissue viability, while they were not upregulated in the tumor cultures with minimal tissue loss.
The present analysis supports the transcriptional stability of the tissue slices during ex-vivo culture and its ability to reflect changes related to cell death. This is of special relevance in order to successfully address and correctly assess biological processes and drug responses in slice cultures. In addition, these findings support the use of transcriptomic analysis as a valuable tool for evaluation of ex-vivo cultures of PDAC.
References
Rahib, L. et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 74, 2913–2921, https://doi.org/10.1158/0008-5472.CAN-14-0155 (2014).
Boffetta, P., Castaing, M. & Brennan, P. A geographic correlation study of the incidence of pancreatic and other cancers in Whites. Eur. J. Epidemiol. 21, 39–46, https://doi.org/10.1007/s10654-005-4639-y (2006).
Neoptolemos, J. P. et al. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 15, 333–348, https://doi.org/10.1038/s41575-018-0005-x (2018).
Zhan, W., Shelton, C. A., Greer, P. J., Brand, R. E. & Whitcomb, D. C. Germline Variants and Risk for Pancreatic Cancer: A Systematic Review and Emerging Concepts. Pancreas 47, 924–936, https://doi.org/10.1097/MPA.0000000000001136 (2018).
Pihlak, R., Weaver, J. M. J., Valle, J. W. & McNamara, M. G. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers (Basel) 10, https://doi.org/10.3390/cancers10010017 (2018).
Redston, M. S. et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 54, 3025–3033 (1994).
Smit, V. T. et al. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 16, 7773–7782, https://doi.org/10.1093/nar/16.16.7773 (1988).
Caldas, C. et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 8, 27–32, https://doi.org/10.1038/ng0994-27 (1994).
Hessmann, E. et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 67, 497–507, https://doi.org/10.1136/gutjnl-2016-311954 (2018).
Xiao, Q. et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 76, 5395–5404, https://doi.org/10.1158/0008-5472.CAN-15-3264 (2016).
Cozzitorto, C. & Spagnoli, F. M. Pancreas organogenesis: The interplay between surrounding microenvironment(s) and epithelium-intrinsic factors. Curr. Top. Dev. Biol. 132, 221–256, https://doi.org/10.1016/bs.ctdb.2018.12.005 (2019).
Ahn, D. H. & Ramanathan, R. K. Targeting the stroma in pancreatic cancer. Chin. Clin. Oncol. 6, 65, https://doi.org/10.21037/cco.2017.11.02 (2017).
Triulzi, T. et al. Whole-transcriptome analysis links trastuzumab sensitivity of breast tumors to both HER2 dependence and immune cell infiltration. Oncotarget 6, 28173–28182, https://doi.org/10.18632/oncotarget.4405 (2015).
Zhang, W., Liu, S., Zhan, H., Yan, Z. & Zhang, G. Transcriptome sequencing identifies key pathways and genes involved in gastric adenocarcinoma. Mol. Med. Rep. 18, 3673–3682, https://doi.org/10.3892/mmr.2018.9370 (2018).
Tian, P. & Liang, C. Transcriptome profiling of cancer tissues in Chinese patients with gastric cancer by high-throughput sequencing. Oncol. Lett. 15, 2057–2064, https://doi.org/10.3892/ol.2017.7548 (2018).
Misra, S. et al. Ex vivo organotypic culture system of precision-cut slices of human pancreatic ductal adenocarcinoma. Sci. Rep. 9, 2133, https://doi.org/10.1038/s41598-019-38603-w (2019).
Huang, B. et al. Long non-coding RNA NEAT1 facilitates pancreatic cancer progression through negative modulation of miR-506-3p. Biochem. Biophys. Res. Commun. 482, 828–834, https://doi.org/10.1016/j.bbrc.2016.11.120 (2017).
Arun, G. & Spector, D. L. MALAT1 long non-coding RNA and breast cancer. RNA Biol. 16, 860–863, https://doi.org/10.1080/15476286.2019.1592072 (2019).
Kallio, M. A. et al. Chipster: user-friendly analysis software for microarray and other high-throughput data. BMC Genomics 12, 507, https://doi.org/10.1186/1471-2164-12-507 (2011).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. Available online at, http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578, https://doi.org/10.1038/nprot.2012.016 (2012).
Kamburov, A., Wierling, C., Lehrach, H. & Herwig, R. ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res. 37, D623–628, https://doi.org/10.1093/nar/gkn698 (2009).
Nicorici, D. et al. FusionCatcher – a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv, https://doi.org/10.1101/011650 (Nov. 2014).
Davies, E. J. et al. Capturing complex tumour biology in vitro: histological and molecular characterisation of precision cut slices. Sci. Rep. 5, 17187, https://doi.org/10.1038/srep17187 (2015).
Claesson-Welsh, L. & Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 273, 114–127, https://doi.org/10.1111/joim.12019 (2013).
Acknowledgements
This study was supported by funding from Cancerfonden (M.B.), The Jochnick Foundation (M.B.), Cancer och Allergifonden (M.B.), Stockholm County Council (M.B., J.D.) and Radiumhemmets Forskningsfonder (M.B.). Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Contributions
J.D., M.B., M.G. and C.F.M. participated in the study design; S.P.E. performed tissue embedding, sectioning, and H&E staining. M.G, E.H. and S.P.E. prepared RNA and NGS libraries; C.F.M. and C.S.V. examined the tumor tissue material; M.G. performed expression data analyses. M.G. and C.F.M. wrote the manuscript. All authors critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghaderi, M., Fernández Moro, C., Pouso Elduayen, S. et al. Genome-wide transcriptome profiling of ex-vivo precision-cut slices from human pancreatic ductal adenocarcinoma. Sci Rep 10, 9070 (2020). https://doi.org/10.1038/s41598-020-65911-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65911-3
This article is cited by
-
Culture of vibrating microtome tissue slices as a 3D model in biomedical research
Journal of Biological Engineering (2023)
-
Comparing P53 expression and genome-wide transcriptome profiling to Comet assay in lymphocytes from melanoma patients and healthy controls
Scientific Reports (2023)
-
Ex vivo culture of intact human patient derived pancreatic tumour tissue
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
