
Abstract
Loss of epithelial polarity and gain in invasiveness by carcinoma cells are critical events in the aggressive progression of cancers and depend on phenotypic transition programs such as the epithelial-to-mesenchymal transition (EMT). Many studies have reported the aberrant expression of voltage-gated sodium channels (NaV) in carcinomas and specifically the NaV1.5 isoform, encoded by the SCN5A gene, in breast cancer. NaV1.5 activity, through an entry of sodium ions, in breast cancer cells is associated with increased invasiveness, but its participation to the EMT has to be clarified. In this study, we show that reducing the expression of NaV1.5 in highly aggressive human MDA-MB-231 breast cancer cells reverted the mesenchymal phenotype, reduced cancer cell invasiveness and the expression of the EMT-promoting transcription factor SNAI1. The heterologous expression of NaV1.5 in weakly invasive MCF-7 breast cancer cells induced their expression of both SNAI1 and ZEB1 and increased their invasive capacities. In MCF-7 cells the stimulation with the EMT-activator signal TGF-β1 increased the expression of SCN5A. Moreover, the reduction of the salt-inducible kinase 1 (SIK1) expression promoted NaV1.5-dependent invasiveness and expression of EMT-associated transcription factor SNAI1. Altogether, these results indicated a prominent role of SIK1 in regulating NaV1.5-dependent EMT and invasiveness.
Similar content being viewed by others

Introduction
The development of secondary tumours, also called metastases, in organs distant from the primary solid tumour, represents the ultimate level of cancer aggressiveness, associated with more than 90% of patient deaths1. At the cancer cell level, critical steps in the metastatic cascade involve morphological and functional changes, such as the loss of cell-cell junctions, loss of apicobasal epithelial polarity, and therefore the loss of the vectorial transport of ions and molecules across the epithelium, towards the acquisition of an elongated, spindle-shaped, “mesenchymal” phenotype. These changes in cell morphologies are associated with the gain of invasive capacities2, allowing carcinoma cells to penetrate surrounding tissues and intravasate into the lymphatic or blood circulation. These steps are parts of the multistage epithelial-to-mesenchymal transition (EMT) which promotes cancer cell dissemination from the primary tumour, while the reverse process, called mesenchymal-to-epithelial transition (MET) is proposed to support the outgrowth of tumoral foci in distantly colonized organs3. EMT is a very plastic programme reminiscent from trans-differentiation processes physiologically participating to tissue constitution during embryogenesis and tissue repair. In these physiological contexts, EMT/MET are tightly and timely regulated, and participate to the organogenesis and at maintaining organ functions. In the tumour context, multiple environmental conditions can induce EMT, such as hypoxia or paracrine signals coming from both stromal and immune cells (e.g. transforming growth factor beta, TGF-β1). The majority of these signals converge and act through the induction of EMT–associated transcription factors such as zinc finger proteins from the SNAI family, zinc finger and homeodomain proteins from the ZEB family or basic-loop helix proteins of the TWIST family4. It is well characterized that microenvironmental conditions in the tumour might induce plasticity programs in cancer cells and contribute to the tumour progression5,6. This might be the case of the ionic composition and concentration of the extracellular content, which could participate in signalling pathways promoting cancer cell proliferation, dedifferentiation and invasiveness. As such, early studies questioned the relationships between the Na+ content, cell permeability to Na+ and the consequences on malignant cell proliferation, invasive capacities and the development of metastases7. Indeed, energy-dispersive X-ray microanalyses recorded higher concentrations of Na+ in tumour cells comparatively to non-cancer cells8. These initial measurements were confirmed later on, using non-invasive 23Na-magnetic resonance imaging and an increased concentration of Na+ in malignant breast tumours was identified compared to surrounding normal tissues9,10,11, suggesting that this parameter could represent an indicator of malignancy, as well as a predictive marker of breast cancer response to the chemotherapy treatment12,13. This increase in Na+ concentration is very likely to concern the intracellular compartment of cancer cells9, in which it could modulate a whole set of signalling pathways. Salt-inducible kinase 1 (SIK1) is a serine/threonine kinase of the AMP-activated protein kinase (AMPK) family, originally cloned from adrenocortical glands of rats receiving a high salt diet14, which takes part into a sodium-sensing intracellular network regulating sodium content15,16. In breast cancer, a reduced expression of SIK1 has been associated with metastatic progression and with a poor outcome17. Furthermore, silencing SIK1 was demonstrated to prevent p53-mediated anoikis and to promote cancer cell colony growth and metastases development18, suggesting that SIK1 could act as a tumour-suppressor19. Moreover, knocking down SIK1 increased the migration of gastric adenocarcinoma cells20. SIK1 expression was significantly lower in hepatocellular carcinoma, compared to normal liver biopsies, and its overexpression in cancer cells suppressed the expression of EMT markers, tumour growth and metastases in a xenograft tumour model21.
Voltage-gated sodium channels (NaV) are multimeric transmembrane complexes composed of one large pore-forming principal subunit (9 genes SCN1A-SCN5A, SCN8A-SCN11A encoding 9 proteins, NaV1.1–1.9)22,23 and one or two smaller transmembrane subunits considered as auxiliary (4 genes SCN1B to SCN4B, generating five subunits, β1, β1B, β2, β3 and β4, which all possess a single membrane-spanning domain with the exception of β1B)24. The activity of NaV generally gives rise to rapid and transient depolarizing Na+ currents (INa) that are responsible for the generation of action potentials in excitable cells25. For this reason, NaV have been considered as characteristic features of excitable cells. However, it has been shown that these channels are not only physiologically expressed in excitable cells, but also in normal immune and microglial cells, in which they participate to their activation and migration in response to environmental signals of threat26,27,28,29,30.
Multiple studies have also demonstrated the expression of NaV in non-excitable cancer cells, while they are not expressed in normal cells, suggesting that they could be associated with dedifferentiation of transformed cells and more generally with carcinogenesis and cancer progression31,32,33. In a wide range of cancer cell lines and primary cultures of carcinomas, NaV channels appeared to be fully functional at the plasma membrane, giving rise to recordable sodium currents, and to regulate cellular functions related to the invasive capacity31,34,35,36,37,38,39,40. Specifically, the NaV1.5 isoform (usually characterized as being the main cardiac isoform) which is the product of the SCN5A gene, was found to be highly overexpressed at both mRNA and protein levels in breast tumours, compared to normal tissues, and was correlated with cancer recurrence, metastases development and reduced patients survival41,42,43. In animal models of mammary cancer, the expression of NaV1.5 in breast cancer cells enhanced primary tumour growth and metastases development, and this was reduced in presence of pharmacological inhibitors of NaV44,45.
The activity of NaV1.5, resulting in the persistent entry of Na+ at the basal membrane potential (“window” current), was demonstrated in highly aggressive MDA-MB-231 human breast cancer cells, in which it was promoting extracellular matrix degradation and cancer cell invasiveness46,47. The activity of the channel is critical, since its inhibition using small molecules reduces extracellular matrix invasion48. In comparison, and while SCN5A was expressed at the mRNA level, no transient sodium current could be recorded in non-tumoural immortalized MCF-10A mammary cells, or even in weakly invasive and poorly dedifferentiated MCF-7 cancer cells42,47,49. Similar results were obtained in the context of non-small cell lung cancer cells, for which NaV activity was recorded in several cancer cell lines such as H460, H23 and Calu-1, but not in non-cancer lung epithelial cells BEAS-2B and NL-20. In lung cancer cells, NaV activity resulted in increases of intracellular sodium concentration and invasiveness35.
In breast cancer cells, the Na+ influx mediated through non-inactivated NaV1.5 channels was demonstrated to allosterically increase the activity of the Na+-H+ exchanger NHE1, thus promoting the efflux of H+ and further increasing the entry of Na+ into cancer cells, subsequently alkalinizing the intracellular pH and lowering the extracellular pH47,49,50. The acidification of the pericellular microenvironment was demonstrated to be favourable to the activity of extracellular proteases digesting the extracellular matrix, such as acidic cysteine cathepsins, thus allowing invasion of the extracellular matrix by cancer cells47,49,50,51. Furthermore, NaV1.5 activity was shown to sustain Src kinase activity, the polymerisation of actin and the acquisition by cancer cells of a spindle-shaped elongated morphology50. Altogether, these results suggest a critical role for NaV1.5 in the so-called “mesenchymal invasion”, in which cancer cells having a mesenchymal phenotype invade tissues thanks to their proteolytic capacity52. However, the participation of NaV channels in the EMT is still elusive.
This study was aimed to elucidate the role of NaV1.5 in the EMT and its potential regulation by SIK1. Here, we show that NaV1.5 expression promotes EMT in breast cancer cells and is upregulated by TGF-β1. Furthermore, knocking down SIK1 expression induces NaV1.5 expression and is correlated with the increase of cancer cell invasiveness.
Results
NaV1.5 activity in breast cancer cells promotes the acquisition of a mesenchymal phenotype and invasive capacities
Highly aggressive, triple-negative, MDA-MB-231 human breast cancer cells have been shown to be very invasive both in vitro and in vivo43,44,45,53. These cells, which endogenously express the SCN5A gene and display NaV1.5-dependent fast inward sodium currents41,47, show a typical spindle-shaped mesenchymal phenotype and multiple filopodia, as observed in scanning electron microscopy (Fig. 1a, left). However, when we stably knocked-down the expression of SCN5A, using a specific small-hairpin RNA (shNaV1.5 cells), thus leading to the absence of plasma membrane NaV1.5 currents, we noticed a dramatic change in the morphology of cancer cell that appeared to be less elongated (Fig. 1a, right). Indeed, we measured a significant increase of the cell circularity index in shNaV1.5 cells (median = 0.335, n = 40) compared to MDA-MB-231 cells expressing a null-target shRNA (shCTL cells, median = 0.679, n = 67, p = 0.003) (Fig. 1b), as well as a reduction in the number of filopodia per cell (median = 57 filopodia/shNaV1.5 cell, n = 39 versus 88.5 filopodia/shCTL cell, n = 24, p = 0.002) (Fig. 1c). Furthermore, the loss of SCN5A expression resulted in a 33%-reduction of MDA-MB-231 cell invasiveness through matrigel-coated inserts (Fig. 1d, p = 0.013). These results are in line with previously published data using tetrodotoxin (TTX) to block NaV1.5 activity, and demonstrating a rapid loss of mesenchymal phenotype50. Therefore, we assessed the expression level of EMT-inducing transcription factors in shNaV1.5 compared to more invasive shCTL breast cancer cells, and identified that SNAI1 expression was specifically and significantly reduced by 69.4% (p < 0.001), while the expression of other EMT-promoting transcription factors ZEB1, ETS1 and TWIST1 was not affected (Fig. 1e). Correlatively, the pharmacological inhibition of NaV1.5 using TTX (30 µM) in Nav1.5-expressing shCTL cells reduced SNAI1 expression by 39% (p = 0.033), while its activation using veratridine (50 µM) had opposite effects and induced its expression by 52% (P = 0.003). TTX and veratridine treatments had no effect either on SCN5A or on ZEB1 expression (Fig. 1f). These results support a role for both NaV1.5 expression and activity in maintaining a mesenchymal phenotype in aggressive cancer cells. We then questioned whether the experimental heterologous overexpression of NaV1.5 in weakly invasive and epithelial-type breast cancer cells, which do not express the protein endogenously, could promote EMT and increase invasiveness. Therefore, we transfected MCF-7 cells, known to express NaV1.5 mRNA and proteins that remain intracellular and do not show functionality at the plasma membrane42,49, with either a plasmid (pcDNA3.1- hNaV1.5-GFP) encoding for human NaV1.5 tagged with eGFP at its C-terminus or with a plasmid (pcDNA3.1-eGFP) encoding for only eGFP. Even though the transfection efficacy was quite low with both plasmid types (<30%), an increased amount of NaV1.5 proteins was measured in cells transfected with pcDNA-NaV1.5-GFP showing a green fluorescence emission (Fig. 2a). The epifluorescence imaging showed a strong GFP signal in the perinuclear area, which most probably corresponded to intracellular organelles involved into the membrane protein translation, maturation and addressing (i.e. endoplasmic reticulum, Golgi)54, as well as a plasma membrane labelling. The functionality of NaV1.5 at the plasma membrane of MCF-7 cells transfected with pcDNA-NaV1.5-GFP was verified by the recording of voltage-gated transient currents that were absent in cells transfected with pcDNA-eGFP (Fig. 2b). These transient currents were inhibited by 30 µM TTX confirming the activity of NaV channels (Fig. 2c). NaV1.5-mediated sodium currents recorded from transfected MCF-7 show typical current-voltage relationships, with a threshold of activation at −60 mV and a maximal peak current of –23.6 ± 5.4 pA/pF (mean ± sem, n = 7) at −10 mV (Fig. 2d). Fitting activation-voltage and inactivation-voltage relationships of sodium currents gave V1/2-activation and V1/2-inactivation voltages of −39.5 ± 0.6 mV and −68.6 ± 1.2 mV, respectively (Fig. 2e). A window of voltage was present between −60 and −35 mV and the mean membrane potential was −36.9 ± 6.9 mV (n = 7 cells), suggesting that a window current could allow a persistent entry of sodium into the cells. This heterologous expression of NaV1.5 in MCF-7 cells, albeit with a poor efficacy, resulted in a significant increase of invasiveness by 46.6% (p = 0.029) (Fig. 2f), as well as the increase of both SNAI1 (x4.5) and ZEB1 (x2.2) expression, that were prevented by the treatment with TTX (Fig. 2g).

SCN5A/NaV1.5 expression and activity promotes the acquisition of a mesenchymal phenotype in MDA-MB-231 breast cancer cells. (A), Representative scanning electron microscopy micrographs performed from MDA-MB-231 shCTL and shNaV1.5 cells. Scale bar, 10 µm. As shown as an illustration underneath the micrographs, MDA-MB-231 shCTL cells (left panel) show fast NaV1.5-related inward current, with a small persistent component, when recorded with a voltage step from −100 mV to −5 mV, while MDA-MB-231 shNaV1.5 cells (right panel), knocked-down for the expression of the SCN5A gene, do not. (B) A cell circularity index was calculated from SEM micrographs taken from shCTL and shNaV1.5 cells (n = 40 and 67 cells per condition, respectively). P = 0.003 (Mann-Whitney rank sum test). (C) Number of filipods per cell counted from SEM micrographs taken from shCTL and shNaV1.5 (n = 24 and 29 cells per condition, respectively). P = 0.002 (Mann-Whitney rank sum test). (D) Summary of cancer cell invasiveness results from 3 independent experiments, for MDA-MB-231 shCTL and shNaV1.5 cells. Results indicate the total number of cells counted per field taken at the x20 objective. Five fields were counted for each individual Matrigel-coated invasion insert, n = 15 inserts (5 independent experiments). P = 0.0013 (Mann-Whitney rank sum test). (E) mRNA expression levels of SCN5A, SNAI1, ZEB1, ETS1 and TWIST1 genes assessed by RT-qPCR in MDA-MB-231 shNaV1.5 cells and expressed as ratios to expression levels in MDA-MB-231 shCTL cells (n = 3–8 separate experiments). SCN5A and SNAI1 were significantly lower in shNaV1.5 compared to shCTL cells (p < 0.001, Mann-Whitney rank sum test). (F) mRNA expression levels of SCN5A, SNAI1 and ZEB1 genes assessed by RT-qPCR in MDA-MB-231 shCTL cells treated with either tetrodotoxin (TTX, 30 µM) or veratridine (Vera, 50 µM) and expressed as ratios to control condition (vehicle). (n = 5–8 separate experiments). TTX treatment significantly reduced the expression level of SNAI1 gene (P = 0.033, Mann-Whitney rank sum test) while Vera increased it (P = 0.003, Mann-Whitney rank sum test).

SCN5A/NaV1.5 heterologous expression in poorly aggressive MCF-7 cells increases invasive phenotype. (A) Representative micrographs showing MCF-7 cells transfected with NaV1.5-GFP in phase contrast (left) and in epifluorescence (right), 24 h after transfection. Scale bar, 20 µm. (B) Representative whole-cell current recordings obtained in MCF-7 cells transfected with either eGFP pcDNA3.1 (top recordings, black traces), or from MCF-7 cells transfected with NaV1.5-GFP pcDNA3.1 (bottom, blue traces), in response to depolarizing 30-ms pulses from −95 to +60 mV in 5-mV steps applied every 2 s from a holding potential of ─100 mV. Red dotted lines indicate baseline levels (zero current). (C) Representative whole-cell voltage-gated sodium currents recorded at −5 mV from a holding potential of −100 mV in absence (Control, PSS solution), presence of TTX (30 µM) and after washing out the toxin with PSS (Washout). (D) Current density-voltage (I-V) relationship for NaV1.5-GFP channels heterologously expressed in MCF-7 cells (n = 7). Peak sodium currents were averaged and plotted as a function of the depolarizing potential (Vm). (E) Conductance-voltage (blue circles) and steady-state inactivation-voltage (red triangles) relationships of peak Na+ currents recorded in MCF-7 cells heterologously expressing NaV1.5-GFP channels. Smooth lines are fits to Boltzmann functions and V1/2 values were calculated for each parameter. V1/2-activation: −39.5 ± 0.6 mV and V1/2-inactivation: −68.6 ± 1.2 mV. Data were obtained from five cells, and are presented as mean ± sem. (F) Invasiveness of MCF-7 cell transfected with NaV1.5-GFP expressed as a ratio to expression levels in cells transfected with eGFP. Data were obtained from 4 independent experiments, with 3 inserts per condition (P = 0.029, Mann-Whitney rank sum test). (G) mRNA expression levels of SNAI1 and ZEB1 genes assessed by RT-qPCR in MCF-7 cells transfected with NaV1.5-GFP and treated with tetrodotoxin (TTX, 30 µM) or not (CTL, vehicle) and expressed as ratios to expression levels in MCF-7 transfected with eGFP (n = 4 separate experiments, Mann-Whitney rank sum test). NS stands for “no statistical difference”.
The EMT inducer TGF-β1 stimulates SCN5A expression and NaV1.5-mediated invasiveness
TGF-β1 is a well-known signal coming from the mesenchymal stroma and inflammatory cells of the tumour, acting as a primary inducer of EMT and metastases development55, and we questioned whether it could potentiate SCN5A expression and mesenchymal phenotype. MDA-MB-231 cells treated with TGF-β1 (5 ng/mL) demonstrated an increase of SNAI1 expression, measured in qPCR, at 24 h (+25%, p = 0.028), at 48 h (+87%, p = 0.028) and at 72 h (+45%, p = 0.028) compared to control condition. However, TGF-β1 but did not induce SCN5A which is endogenously expressed in MDA-MB-231, nor ZEB1 (Fig. 3a). TGF-β1 significantly increased MDA-MB-231 invasiveness by 61%, and this effect was prevented by the use of TTX (Fig. 3b).

TGFβ1 treatment enhances SCN5A expression and invasive phenotype. (A) Effect of TGF-β1 (5 ng/mL) on the expression of SCN5A, SNAI1 and ZEB1 genes in MDA-MB-231 cells, 24 h, 48 h or 72 h after the treatment. Results were obtained by RT-qPCR from 4 independent experiments and expressed as a ratio to the control condition (vehicle). P values indicate significant differences compared with the control condition at the same time point (Mann-Whitney rank sum test), otherwise there is no statistical difference. (B) Effect of TGF-β1 (5 ng/mL for 24 h) on the invasiveness of MDA-MB-231 cells. Data were obtained from 3 independent paired experiments, with 3 inserts per condition, in the absence or presence of 30 µM TTX. p < 0.0001, when comparing TGF-β1 to CTL, and p < 0.001 when comparing TGF-β1 to TGF-β1 + TTX (Mann-Whitney rank sum test). (C) Effect of TGF-β1 (5 ng/mL) on the expression of SCN5A, SNAI1 and ZEB1 genes in MCF-7 cells, 24 h, 48 h or 72 h after the treatment. Results were obtained by RT-qPCR from 4 independent experiments and expressed as a ratio to the control condition (vehicle). P values indicate significant differences compared with the control condition at the same time point (Mann-Whitney rank sum test), otherwise there is no statistical difference. (E) Effect of TGF-β1 (5 ng/mL for 24 h) on the invasiveness MCF-7 cells, in presence or absence of 30 µM TTX. Data were obtained from 3 independent paired experiments, with 3 inserts per condition. p < 0.001, when comparing TGF-β1 to CTL, and P = 0.019 when comparing TGF-β1 to TGF-β1 + TTX (Mann-Whitney rank sum test).
Weakly invasive MCF-7 cells treated with TGF-β1 also displayed a +31.1% induction of SNAI1 at 72 h and a +30.0% induction of ZEB1 at 48 h. For these two genes, a tendency for an induction of expression by TGF-β1 was observed for the two other times considered (Fig. 3c). This treatment also induced the expression of SCN5A gene by 35.2% at 24 h (p = 0.029), 85.3% at 48 h (p = 0.029) and 53.3% at 72 h (p = 0.008) (Fig. 3c). This treatment resulted in a 40.8% increase of MCF-7 invasion (p < 0.001), which was prevented by the use of TTX (Fig. 3d). These results indicate that EMT-inducing conditions, such as the stimulation of cancer cells with TGF-β1, could promote SCN5A expression and NaV1.5-dependent increase in invasive capacities.
Loss of SIK1 expression correlates with breast cancer progression, stimulates SCN5A expression and NaV1.5-mediated invasiveness
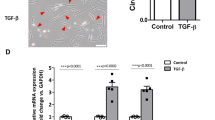
SIK1 has been identified as an important protagonist controlling sodium homeostasis15,16 and was recently proposed as acting as a tumour-suppressor19. An in silico study confirmed that SIK1 expression was reduced in breast primary and metastatic breast tumours, as compared with normal tissues (Fig. 4A). This lower expression of SIK1 gene in breast tumours, compared to normal mammary tissues, was observed at all tumour stages (Fig. 4b). Breast tumour biopsies obtained from the Tours University Hospital were analysed to assess SIK1 expression in the different breast tumour subtypes. While the statistical significance was not reached, a tendency was observed for a lower expression in triple-negative tumours, compared to luminal A, luminal B, or HER2 (Fig. 4c), a lower expression in grade III, compared to grades I and II (Fig. 4d), and a lower expression in HER2+ compared to pooled HER2- tumours (Fig. 4e).

The Salt-inducible Kinase type 1 (SIK1) is downregulated in breast cancer. (A) The expression level of SIK1 in normal breast (normal tissue, n = 114), primary breast tumours (n = 1097) and metastatic tumours (n = 7) is analysed from datasets coming from the “The Cancer Genome Atlas” (http://cancergenome.nih.gov) from the US National Cancer Institute. For each array, data were log2-transformed and centered to the median. P values indicate significant differences (Mann-Whitney rank sum test) as compared to normal tissue. (B) The expression level of SIK1 in normal breast (normal tissue, n = 114) and in different breast tumour grades: I (n = 125), IIA (n = 243), IIB (n = 115), IIIA (n = 85), IIIB (n = 10), IIIC (n = 31), IV (n = 4). P values indicate significant differences (Mann-Whitney rank sum test) as compared to normal tissue. (C–E) Expression level of SIK1 assessed by RT-qPCR from breast cancer biopsies obtained from the University Hospital of Tours and expressed as –deltaCt using HPRT1 as a reference gene, (C) depending on breast cancer subtypes: luminal A (Lum A, n = 15), luminal B (Lum B, n = 16), HER2-enriched (HER2, n = 10) and triple-negative breast tumour (TN, n = 11). (D) depending on tumour grade: I (n = 5), II (n = 17) and III (n = 28). (E) depending on the expression of HER2 negative (n = 35) or positive (n = 14).
The use of a specific siRNA to knock down the expression of SIK1 in MDA-MB-231 cells (inhibition by 63.5%, p = 0.029) increased the expression of SCN5A by +97.5% (p = 0.029) with no effect on SNAI1, 24 h after transfection (Fig. 5a). This resulted in a +122.2% increase of invasiveness in shCTL cells (p = 0.029). In comparison, knocking-down the expression of SIK1 in shNaV1.5 cells, which present a lower invasive capacity as compared to shCTL cells, had no stimulating effect on invasiveness (Fig. 5b). Because NaV1.5 was demonstrated to promote breast cancer cell invasiveness through a promotion of extracellular acidification involving NHE1 allosteric activation49,50, we monitored the H+ efflux at the introduction of NaCl in MDA-MB-231 cells previously acidified (NH4Cl pulse-wash protocol in sodium-free medium). We noticed that silencing SIK1 promoted H+ efflux in shCTL by +44.5% (Fig. 5c), but not in shNaV1.5 cells (Fig. 5d). In shCTL and shNaV1.5 cells, knocking-down SIK1 had no effect on NHE1 mRNA expression monitored by qPCR (data not shown). Correlatively, knocking-down the expression of SIK1 in MCF-7 cells also induced the expression of SCN5A by +52.8% (p = 0.026), of SNAI1 by +79.6% (p = 0.08) (Fig. 5e), and invasiveness by +70.1% (p < 0.01) (Fig. 5f). This increase in MCF-7 cell invasiveness was prevented by the use of 30 µM TTX (Fig. 5f).

Knocking down SIK1 expression increases NaV1.5-dependent cancer cell invasiveness. (A) Effect of silencing SIK1 (siSIK1) in MDA-MB-231 cells on the mRNA expression of SIK1, SCN5A and SNAI1 genes. Results were obtained from 4 independent RT-qPCR experiments, 24 h after siRNA transfection, and are expressed relatively to the results obtained when transfecting control irrelevant siRNA (siCTL). P values indicate significant differences (Mann-Whitney rank sum test) as compared to siCTL condition. (B) Effect of silencing SIK1 (siSIK1) on the invasiveness of MDA-MB-231 shCTL and shNaV1.5 cells. Results are coming from 4 independent experiments and are expressed relatively to the effect of the siCTL condition in shCTL cells. P values indicate significant differences (Mann-Whitney rank sum test) as compared to siCTL condition in shCTL cells. NS stands for “no statistical difference” between siCTL and siSIK1 conditions in shNaV1.5 cells. (C) Relative H+ efflux measurements induced by reintroduction of 130 mM NaCl in MDA-MB-231 shCTL cells transfected with either siCTL or siSIK1, after a NH4Cl wash pulse-induced intracellular acidification in a Na+-free solution. Results are coming from 5 independent experiments and are expressed relatively to the efflux measured in the siCTL condition. P value indicates a significant difference (Mann-Whitney rank sum test) when compared to the siCTL condition. (D) Relative H+ efflux measurements induced by reintroduction of 130 mM NaCl in MDA-MB-231 shNaV1.5 cells transfected with either siCTL or siSIK1, as in C. Results are coming from 5 independent experiments and are expressed relatively to the efflux measured in the siCTL condition. NS indicates “no statistical difference” when compared to the siCTL condition. (E) Effect of silencing SIK1 (siSIK1) in MCF-7 cells on the mRNA expression of SIK1, SCN5A and SNAI1 genes. Results were obtained from 7 independent RT-qPCR experiments, 24 h after siRNA transfection, and are expressed relatively to the results obtained when transfecting control irrelevant siRNA (siCTL). P values indicate significant differences (Mann-Whitney rank sum test) as compared to the siCTL condition. (F) Effect of silencing SIK1 (siSIK1) on the invasiveness of MCF-7 cells, in the presence of 30 µM tetrodotoxin (TTX) or not (CTL, vehicle). Results are coming from 7 independent experiments and are expressed relatively to the effect of the CTL condition in siCTL cells. P values indicate significant differences (Mann-Whitney rank sum test). NS stands for “no statistical difference” between CTL and TTX conditions in siCTL cells.
Taken together, these results indicate that the loss of SIK1 participate in promoting breast cancer cell invasiveness through the induction of SCN5A expression and NaV1.5 activity.
Discussion
Increasing evidence indicates that pore-forming α32,56 and auxiliary β subunits53,57,58,59,60 of voltage-gated sodium channels are key contributors of oncogenic properties and metastatic progression. Particularly, pore-forming NaV α have been demonstrated to be overexpressed in a wide range of biopsies and cancer cells originating from different types of carcinomas, including breast cancer42,46, prostate cancer39,61, lung cancer35,62, cervical cancer36,38,40, ovarian cancer63, gastric cancer64 and colon cancer37,65. In all these cancer types, different isoforms of pore-forming NaV are expressed at the mRNA level, probably also at the protein level, but generally only one is functional at the plasma membrane of cancer cells giving rise to recordable sodium currents, with the only exception so far of the non-small-cell cancer cell line Calu-1 in which several NaVα might simultaneously be functional35. In all cases, NaV expression has been shown to increase the invasive potency and the extracellular matrix degradation capacity. Several studies identified that functional NaV expressed at the plasma membrane of breast, colon and cervical cancer cells were neonatal splice variants40,42,66,67,68, and not the adult splice variant isoform that might be restricted to non-cancer excitable cells. Whether this is the case for other types of cancers still has to be investigated.
The use of pharmacological inhibitors such as TTX47, the anticonvulsant phenytoin45, the antianginal ranolazine44,69, the local anaesthetics lidocaine or ropivacaine67, or even newly developed small NaV inhibitory molecules48, has been shown to reduce cell invasiveness, albeit not completely abrogating it, and to modify cancer cell morphology to a less aggressive phenotype. By comparison, the use of pharmacological NaV inhibitors in weakly invasive cancer cells not showing any plasma membrane sodium current, yet expressing NaV mRNA and protein, such as MCF-7 breast46,49 or A-549 non-small cell cancer cells35 had no effect on cell invasiveness. Taken together all of these results strongly suggest that the functionality of NaV at the plasma membrane, i.e. the sodium influx, independently on NaV molecular nature, is critical for the gain of aggressiveness in cancer cells. However, this does not exclude the possibility that NaV expression in intracellular compartments might also be involved in cancer cell migration, invasion, endocytic recycling or phagocytic activity, as is the case for microglial and immune cells26,29. Nevertheless, in almost all of these previously published studies, researchers inhibited the expression or activity of NaV in aggressive, endogenously NaV-expressing cancer cells. In this study, we demonstrated that the heterologous expression of NaV in weakly invasive cancer cells is sufficient to increase cell invasiveness. Furthermore, the adult splice variant of the channel that we heterologously overexpressed seems to promote cancer cell invasiveness similarly to the neonatal one, further supporting the idea that NaV activity and not the NaV protein per se, as a structural molecule, is important to cancer progression. These results are in line with recently published data obtained with the overexpression of the adult NaV1.6 isoform in C33A cervical cancer cells40. The reason why some NaV isoforms are specifically expressed and correlated to some specific cancers, NaV1.5 in breast and colon cancers, NaV1.6 in cervical cancer, NaV1.7 in prostate cancer, remains to be determined.
In MDA-MB-231 breast cancer cells, NaV1.5 activity was shown to promote invadopodial activity and the proteolytic degradation of the extracellular matrix47,49,50. Furthermore, NaV1.5 activity was shown to sustain the polymerisation of actin, the generation of F-actin stress fibres and the acquisition by cancer cells of a fibroblast-like morphology50. These results suggested a role for NaV1.5 in “mesenchymal invasion”. However, the participation of NaV activity in the epithelial-to-mesenchymal dedifferentiation was unclear. In this study, we demonstrated for the first time that both the expression and the activity of NaV1.5 sustain the expression of the EMT-promoting transcription factor SNAI1, the acquisition of a mesenchymal phenotype and of enhanced invasive capacities. In weakly invasive and epithelial-phenotype MCF-7 cells, the overexpression and activity of NaV1.5 increased the expression of SNAI1 but also induced ZEB1. Our results using both pharmacological tools and shRNA in cells endogenously expressing NaV1.5, and in cells heterologously expressing NaV1.5, are in apparent contradiction with data published by Nelson and collaborators45. In this previously published study, MDA-MB-231 cells stably knocked-down for the expression of NaV1.5 (shRNA transduction) did not show any significant reduction in the protein expression from SNAI1, although a tendency for a small reduction could be observed on the western blot shown, and no effect on vimentin or SLUG expression. Only a reduction in CD44 was identified. A possible explanation for this discrepancy could be the selection, in their study, of a specific clone expressing the transduced shRNA.
In weakly aggressive MCF-7 breast cancer cell line, TGF-β1 stimulation promoted NaV1.5 expression, EMT transcription factors expression and mesenchymal invasion. Considering the fact that the treatment with TGF-β1 increased SCN5A expression, and that TGF-β1-induced increase in MCF7 cancer cell invasiveness was prevented by the use TTX, it is most likely that NaV1.5 channels expressed and functional (giving rise to sodium currents) at the plasma membrane of cancer cells are responsible for this effect. Indeed, electrophysiological experiments performed in MCF7 treated with TGF-β1 (for 72 h) showed in several, but not all cells, the apparition of transient inward currents (Suppl. Fig. 1) which are absent in non-treated cells (Fig. 2b) and could correspond NaV1.5-mediated sodium currents. However, we cannot exclude the possibility that intracellular NaV1.5 channels might also be involved in the regulation of cancer cell invasiveness.
It is of interest to compare these results, obtained in human breast cancer cells, to data acquired with cardiac cells expressing NaV1.5. While TGF-β1 might be important for the embryogenic development of the heart, its overexpression has also been associated with cardiac remodelling and heart diseases70. Indeed, a regulation of cardiac NaV1.5 by TGF-β1 seems to be associated with cardiac fibrosis and conduction troubles. In neonatal rat cardiac myocytes, TGF-β1 treatment reduced NaV1.5 activity with a 30%-decreased peak Na+ current, which could be attributed to a reduced protein expression of the channel, combined with a leftward shift of the inactivation-voltage relationship71. Scn5a haplo-insufficient mice (Scn5a+/-), thus showing a loss-of-function of NaV1.5, are characterized by impaired sinoatrial node automatic activity and slowed sinoatrial conduction. Interestingly, it is also associated with increased collagen and fibroblast levels, high levels of TGF-β1 and vimentin. Loss of NaV1.5 was associated with a TGF-β1-mediated cardiac fibrosis72. More recently, Derangeon and collaborators demonstrated, in a model of progressive cardiac conduction disease, that the ventricular fibrosis that develops with aging in Scn5a+/- mice is secondary to the activation of the TGF-β1 signalling pathway73. Cardiac fibrosis could result from an increased collagen expression and maturation from cardiac fibroblasts, or from a decrease in the collagen fibres turnover, which is normally regulated by proteolytic enzymes. It is of particular interest to notice that a loss-of-function of NaV1.5 in cardiac cells promotes collagen deposit and fibrosis, while its gain of function in cancer cells induces extracellular matrix degradation and invasion. The fact that in one case TGF-β1 reduces NaV1.5 expression and activity in cardiac cells, but increases them in cancer cells might be representative of a complex feedback regulation that might be lost during carcinogenesis.
The secondary objective of this study was to determine whether NaV expression in cancer cells could be a part of a general dysregulation of the SIK signalling pathway, known to control sodium homeostasis. A reduced expression of SIK1 has been identified in breast cancer and has been associated with metastatic progression and with a poor outcome17,18. In other cancer types, the loss of SIK1 expression promoted the expression of EMT markers, as well as invasive capacities both in vitro and in vivo20,21. Indeed, SIK proteins have been demonstrated to participate in gene regulation74.
In the current study, we confirmed a reduction of SIK1 expression in breast tumours, and further identified that reducing SIK1 expression in breast cancer cells induced SCN5A expression and NaV1.5-dependent invasiveness. The loss of SIK1 expression in epithelial-like phenotype MCF-7 cells promoted the expression of SNAI1, but did not significantly modulate its expression in MDA-MB-231 cancer cells already largely engaged in a mesenchymal phenotype.
Taken together, these results suggest that loss of SIK1 in cancer cells might be a critical step in tumour progression by inducing NaV1.5 expression and activity, triggering the epithelial-to-mesenchymal transition, and the acquisition of pro-metastatic capacities that might be associated with a complete dysregulation of Na+ homeostasis in the cancer.
Methods
In Silico RNA expression
The expression level of SIK1 gene in breast cancer tissues was performed using data available from “The Cancer Genome Atlas” for breast invasive carcinoma (TCGA BRCA) by using Cancer Genomics Browser (http://xena.ucsc.edu/).
Chemicals and drugs
Tetrodotoxin was purchased from Latoxan (France). All other drugs and chemicals were purchased from Sigma-Aldrich (France).
Breast tumour biopsies
Breast cancer biopsies, coming from surgical interventions performed by the gynaecological surgery department of the University-Hospital of Tours, were frozen in liquid nitrogen and conserved at the tumour collection (N°DC2008-308). All experiments and methods were performed in accordance with relevant guidelines and regulations, and approved by the “Comité de Protection des Personnes” (CPP) of Tours hospital. Informed consent was obtained from all subjects.
Cell lines and culture
Cell lines were purchased from the American Type Culture Collection (LGC Promochem, France). Cells were grown at 37 °C in 5% CO2 incubator, in a humidified atmosphere. MCF-7 and MDA-MB-231 breast cancer cells were cultured in DMEM supplemented with 5% foetal calf serum (FCS). MDA-MB-231 shCTL and shNaV1.5 breast cancer cells were generated as previously described44,53, from MDA-MB-231 cells stably expressing the luciferase gene. Briefly these two cell lines were obtained by transduction with a lentiviral vector encoding a short hairpin RNA (shRNA) specifically targeting human SCN5A transcripts (shNaV1.5 cell line) or a null-target shRNA (shCTL cell line). The sequence encoding shSCN5A, inhibiting the expression of NaV1.5 protein, was obtained by DNA polymerase fill-in of two partially complementary primers: 5′-GGATCCCCAAGGCACAAGTGCGTGCGCAATTCAAGAGA-3′ and 5′-AAGCTTAAAAAAAGGCACAAGTGCGTGCGCAATCTCTTGAA-3′. We constructed a lentiviral vector expressing a null-target shRNA (pLenti-shCTL), using the following primers: 5′-GGATCCCCGCCGACCAATTCACGGCCGTTCAAGAGACG-3′ and 5′-AAGCTTAAAAAGCCGACCAATTCACGGCCGTCTCTTGAACG-3′.
Tests assessing for mycoplasma contamination were performed once a week, every week (Lonza, MycoAlert™ Mycoplasma Detection Kit).
RNA extraction, reverse transcription, and real-time PCR
Total RNA extraction was performed (RNAgents® Total RNA Isolation System, Promega, France) and RNA yield and purity were determined by spectrophotometry. Only samples with an A260/A280 ratio above 1.6 were kept for reverse-transcription. To do so, RT kits Ready-to-go® You-prime First-Strand Beads (Amersham Biosciences, UK) and random hexamers pd(N)6 5′-Phosphate (0.2 µg, Amersham Biosciences) were used. Samples were incubated at 37 °C for 60 min. Real time PCR experiments were performed as previously described47. Results obtained from cell lines are expressed as the relative gene expression using the comparative 2−ΔΔCt method75 with PPIA as a reference gene. Results coming from breast cancer biopsies, for which there are no associated non-cancer tissues and no experimental conditions, were expressed as – ΔCt using HPRT1 as a reference gene. Primers sequences can be found in Table 1.
Transfection of cells with plasmid and small interfering RNA
MDA-MB-231 and MCF-7 human breast cancer cells were transfected with 20 nM small interfering RNA (siRNA, Tebu-Bio, France) targeting the expression of SIK1 (siSIK1, sc-91428) or scrambled siRNA (siCTL, siRNA-A sc-37007). SiRNA Transfections were performed with Lipofectamine RNAi max (Invitrogen, France). For some experiments, MCF-7 cells were transfected with a pcDNA3.1 plasmid encoding for human NaV1.5-GFP (generous gift of Dr. Pascale Guicheney, Inserm UMR_S 956, UPMC, Paris), or alternatively by a pcDNA3.1 plasmid encoding for enhanced green fluorescent protein (eGFP). Cells, at a 80–90% confluence state, were transfected with 1 µg cDNA using Lipofectamine 2000 (Invitrogen, France). Transfection efficiency was verified by qPCR using an iCycler® system (BioRad, USA), and by western blotting.
Electrophysiology
Whole-cell currents were recorded, as already described46, under the voltage-clamp mode of the patch-clamp technique, at room temperature, using an Axopatch 200B patch clamp amplifier (Axon Instrument, USA). Patch pipettes were pulled from borosilicate glass (TW150-3, World Precision Instruments, France) to a resistance of 3–5 MΩ using. Analogue signals were filtered at 5 kHz, and sampled at 10 kHz using a 1440 A Digidata converter. Cell capacitance and series resistance were electronically compensated by about 60%. The P/2 sub-pulse correction of cell leakage and capacitance was used to study Na+ current (INa). Sodium currents were recorded by depolarizing the cells from a holding potential (HP) of –100 mV to a maximal test pulse of –30 mV for 30 ms every 500 ms. Sodium current-voltage (INa-V) relationships were determined using the following protocol: the membrane was depolarized from a HP of –100 mV to potentials from –80 to +60 mV, with 5-mV increments, for 50 ms and at a frequency of 2 Hz. Availability-voltage relationships were obtained by applying 50 ms prepulses using the INa-V curve procedure, followed by a depolarizing pulse to –5 mV for 50 ms. Currents were normalized to the amplitude of the test current without a prepulse. Currents amplitudes were normalized to cell capacitance and expressed as current density (pA/pF). The Physiological Saline Solution (PSS) had the following composition (in mM): NaCl 140, KCl 4, MgCl2 1, CaCl2 2, D-Glucose 11.1, and HEPES 10, adjusted to pH 7.4 with NaOH (1 M). The intrapipette solution had the following composition (in mM): KCl 130, NaCl 15, CaCl2 0.37, MgCl2 1, Mg-ATP 1, EGTA 1, HEPES 10, adjusted to pH 7.2 with KOH (1 M).
Intracellular pH (pHi) measurements
Cells were incubated for 30 min at 37 °C in Hank’s medium containing 2 µM BCECF-AM (2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein; excitation 503/440 nm; emission 530 nm), then washed twice with PSS. H+ efflux was measured after an NH4Cl pulse-wash intracellular acidification protocol in absence of external sodium, followed by 130 mM NaCl extracellular introduction allowing re-alkalinisation, as previously described49,76.
Cell viability
Cancer cells were seeded in a 24-well plate at the density of 4 × 104 cells per well. Media were changed every day, and after 5 days growing, the number of viable cells was assessed by the tetrazolium salt assay as previously described47 and normalised to the appropriate control condition.
Cancer cell invasiveness assessment
Cancer cell invasiveness was using culture inserts with 8-µm pore size filters covered with Matrigel™ (Becton Dickinson, France), as already described77. The upper chamber of the insert was seeded with 6 × 104 cells in their conventional growing medium (containing 5% FCS). The lower compartment was filled with DMEM supplemented with 10% FCS, thus creating a chemoattractant gradient. Cells were let to invade for 24 h at the 37 °C and 5%-CO2 incubator. Cells that had invaded and were adherent to the lower side of the insert were stained with DAPI. Cells were manually counted on the whole area of the insert membrane. Assays were performed in triplicate in each separate experiment.
Epifluorescence imaging
Cells were cultured for 24–48 h on glass coverslips, then were then washed twice in PBS, before being fixed with 3.7% ice-cold paraformaldehyde in PBS. Cell permeabilization was obtained using a solution containing 50 mM NH4Cl, 1% BSA and 0.02% saponin. Saturation of epitopes was fulfilled by incubating 2 h with a solution containing 3% BSA and 3% Normal Goat Serum (NGS). Epifluorescence microscopy was performed with a Nikon TI-S microscope. Images were analysed using both NIS-BR software (Nikon, France) and ImageJ© software 1.38I (http://rsbweb.nih.gov/ij). Fluorescent probes and conjugated antibodies were purchased from Fisher Scientific (France).
Scanning electron microscopy
Cells were fixed by incubation for 24 h in 4% paraformaldehyde, 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2). Cells were then washed twice in phosphate-buffered saline (PBS), and post-fixed by incubation with 2% osmium tetroxide for 1 h. Samples were dehydrated by the incubation in a series of ethanol solutions, then dried in hexamethyldisilazane (HMDS, Sigma, USA). Cells were coated with 40 Å platinum, using a GATAN PECS 682 apparatus (Pleasanton, USA), and were analysed by scanning electron microscopy (Zeiss Ultra plus FEG-SEM, Germany). A circularity index was calculated from pictures as being 4π·Area/Perimeter2. An index value of “1.0” indicates a perfect circle, while a value approaching “0” corresponds to an increasingly elongated shape.
Statistical analyses
Data were displayed as box plots indicating the first quartile, the median, and the fourth quartile, whiskers indicating the minimal and maximal values, and square dots indicating the means. Non-parametric statistical tests were performed (Mann-Whitney rank sum test), using SigmaStat 3.0 software (Systat software Inc.) and P values are indicated on figures. NS stands for “not statistically different”.
References
Parkin, D. M., Bray, F., Ferlay, J. & Pisani, P. Global cancer statistics, 2002. CA Cancer J Clin 55, 74–108 (2005).
Friedl, P. & Alexander, S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 (2011).
Diepenbruck, M. & Christofori, G. Epithelial-mesenchymal transition (EMT) and metastasis: yes, no, maybe? Curr Opin Cell Biol 43, 7–13, https://doi.org/10.1016/j.ceb.2016.06.002 (2016).
Nieto, M. A., Huang, R. Y., Jackson, R. A. & Thiery, J. P. Emt: 2016. Cell 166, 21–45, https://doi.org/10.1016/j.cell.2016.06.028 (2016).
Gupta, G. P. & Massague, J. Cancer metastasis: building a framework. Cell 127, 679–695 (2006).
Hanahan, D. & Coussens, L. M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322, https://doi.org/10.1016/j.ccr.2012.02.022 (2012).
Cone, C. D. Jr. The role of the surface electrical transmembrane potential in normal and malignant mitogenesis. Ann N Y Acad Sci 238, 420–435 (1974).
Cameron, I. L., Smith, N. K., Pool, T. B. & Sparks, R. L. Intracellular concentration of sodium and other elements as related to mitogenesis and oncogenesis in vivo. Cancer Res 40, 1493–1500 (1980).
Ouwerkerk, R. et al. Elevated tissue sodium concentration in malignant breast lesions detected with non-invasive 23Na MRI. Breast Cancer Res Treat 106, 151–160, https://doi.org/10.1007/s10549-006-9485-4 (2007).
Jacobs, M. A. et al. Multiparametric and multinuclear magnetic resonance imaging of human breast cancer: current applications. Technology in cancer research & treatment 3, 543–550, https://doi.org/10.1177/153303460400300603 (2004).
Zaric, O. et al. Quantitative Sodium MR Imaging at 7 T: Initial Results and Comparison with Diffusion-weighted Imaging in Patients with Breast Tumors. Radiology 280, 39–48, https://doi.org/10.1148/radiol.2016151304 (2016).
Jacobs, M. A. et al. Multiparametric magnetic resonance imaging, spectroscopy and multinuclear ((2)(3)Na) imaging monitoring of preoperative chemotherapy for locally advanced breast cancer. Academic radiology 17, 1477–1485, https://doi.org/10.1016/j.acra.2010.07.009 (2010).
Jacobs, M. A. et al. Monitoring of neoadjuvant chemotherapy using multiparametric, (2)(3)Na sodium MR, and multimodality (PET/CT/MRI) imaging in locally advanced breast cancer. Breast Cancer Res Treat 128, 119–126, https://doi.org/10.1007/s10549-011-1442-1 (2011).
Wang, Z., Takemori, H., Halder, S. K., Nonaka, Y. & Okamoto, M. Cloning of a novel kinase (SIK) of the SNF1/AMPK family from high salt diet-treated rat adrenal. FEBS Lett 453, 135–139 (1999).
Sjostrom, M. et al. SIK1 is part of a cell sodium-sensing network that regulates active sodium transport through a calcium-dependent process. Proc Natl Acad Sci USA 104, 16922–16927, https://doi.org/10.1073/pnas.0706838104 (2007).
Ferrandi, M. et al. SIK1 localizes with nephrin in glomerular podocytes and its polymorphism predicts kidney injury. Hum Mol Genet 23, 4371–4382, https://doi.org/10.1093/hmg/ddu154 (2014).
Wang, Y. et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 365, 671–679, https://doi.org/10.1016/S0140-6736(05)17947-1 (2005).
Cheng, H. et al. SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci Signal 2, ra35, https://doi.org/10.1126/scisignal.2000369 (2009).
Shaw, R. J. Tumor suppression by LKB1: SIK-ness prevents metastasis. Sci Signal 2, pe55, https://doi.org/10.1126/scisignal.286pe55 (2009).
Selvik, L. K. et al. Salt-inducible kinase 1 (SIK1) is induced by gastrin and inhibits migration of gastric adenocarcinoma cells. PLoS One 9, e112485, https://doi.org/10.1371/journal.pone.0112485 (2014).
Qu, C. et al. Salt-inducible Kinase (SIK1) regulates HCC progression and WNT/beta-catenin activation. Journal of hepatology 64, 1076–1089, https://doi.org/10.1016/j.jhep.2016.01.005 (2016).
Goldin, A. L. et al. Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368 (2000).
Catterall, W. A. Voltage-Gated Sodium Channels at 60:Structure, Function, and Pathophysiology. J Physiol (2012).
O’Malley, H. A. & Isom, L. L. Sodium channel beta subunits: emerging targets in channelopathies. Annu Rev Physiol 77, 481–504, https://doi.org/10.1146/annurev-physiol-021014-071846 (2015).
Hodgkin, A. L. & Huxley, A. F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117, 500–544 (1952).
Black, J. A., Liu, S. & Waxman, S. G. Sodium channel activity modulates multiple functions in microglia. Glia 57, 1072–1081, https://doi.org/10.1002/glia.20830 (2009).
Persson, A. K. et al. Contribution of sodium channels to lamellipodial protrusion and Rac1 and ERK1/2 activation in ATP-stimulated microglia. Glia 62, 2080–2095, https://doi.org/10.1002/glia.22728 (2014).
Craner, M. J. et al. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 49, 220–229, https://doi.org/10.1002/glia.20112 (2005).
Black, J. A., Newcombe, J. & Waxman, S. G. Nav1.5 sodium channels in macrophages in multiple sclerosis lesions. Multiple sclerosis 19, 532–542, https://doi.org/10.1177/1352458512460417 (2013).
Lo, W. L., Donermeyer, D. L. & Allen, P. M. A voltage-gated sodium channel is essential for the positive selection of CD4(+) T cells. Nature immunology 13, 880–887, https://doi.org/10.1038/ni.2379 (2012).
Besson, P. et al. How do voltage-gated sodium channels enhance migration and invasiveness in cancer cells? Biochim Biophys Acta 1848, 2493–2501, https://doi.org/10.1016/j.bbamem.2015.04.013 (2015).
Roger, S., Gillet, L., Le Guennec, J. Y. & Besson, P. Voltage-gated sodium channels and cancer: is excitability their primary role? Front Pharmacol 6, 152, https://doi.org/10.3389/fphar.2015.00152 (2015).
Roger, S., Potier, M., Vandier, C., Besson, P. & Le Guennec, J. Y. Voltage-gated sodium channels: new targets in cancer therapy? Curr Pharm Des 12, 3681–3695 (2006).
Brackenbury, W. J. Voltage-gated sodium channels and metastatic disease. Channels (Austin) 6 (2012).
Roger, S. et al. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int J Biochem Cell Biol 39, 774–786 (2007).
Hernandez-Plata, E. et al. Overexpression of NaV 1.6 channels is associated with the invasion capacity of human cervical cancer. Int J Cancer 130, 2013–2023 (2012).
House, C. D. et al. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res 70, 6957–6967 (2010).
Diaz, D. et al. Functional expression of voltage-gated sodium channels in primary cultures of human cervical cancer. J Cell Physiol 210, 469–478 (2007).
Laniado, M. E. et al. Expression and functional analysis of voltage-activated Na+ channels in human prostate cancer cell lines and their contribution to invasion in vitro. Am J Pathol 150, 1213–1221 (1997).
Lopez-Charcas, O. et al. The invasiveness of human cervical cancer associated to the function of NaV1.6 channels is mediated by MMP-2 activity. Scientific reports 8, 12995, https://doi.org/10.1038/s41598-018-31364-y (2018).
Yang, M. et al. Therapeutic potential for phenytoin: targeting Na(v)1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res Treat (2012).
Fraser, S. P. et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin Cancer Res 11, 5381–5389 (2005).
Nelson, M., Yang, M., Millican-Slater, R. & Brackenbury, W. J. Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget 6, 32914–32929, https://doi.org/10.18632/oncotarget.5441 (2015).
Driffort, V. et al. Ranolazine inhibits NaV1.5-mediated breast cancer cell invasiveness and lung colonization. Mol Cancer 13, 264, https://doi.org/10.1186/1476-4598-13-264 (2014).
Nelson, M., Yang, M., Dowle, A. A., Thomas, J. R. & Brackenbury, W. J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol Cancer 14, 13, https://doi.org/10.1186/s12943-014-0277-x (2015).
Roger, S., Besson, P. & Le Guennec, J. Y. Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim Biophys Acta 1616, 107–111 (2003).
Gillet, L. et al. Voltage-gated Sodium Channel Activity Promotes Cysteine Cathepsin-dependent Invasiveness and Colony Growth of Human Cancer Cells. J Biol Chem 284, 8680–8691 (2009).
Dutta, S. et al. Discovery and evaluation of nNav1.5 sodium channel blockers with potent cell invasion inhibitory activity in breast cancer cells. Bioorganic & medicinal chemistry 26, 2428–2436, https://doi.org/10.1016/j.bmc.2018.04.003 (2018).
Brisson, L. et al. Na(V)1.5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H(+) efflux in caveolae. Oncogene 30, 2070–2076 (2011).
Brisson, L. et al. NaV1.5 Na+ channels allosterically regulate the NHE-1 exchanger and promote the activity of breast cancer cell invadopodia. J Cell Sci 126, 4835–4842 (2013).
Brisson, L., Reshkin, S. J., Gore, J. & Roger, S. pH regulators in invadosomal functioning: Proton delivery for matrix tasting. Eur J Cell Biol 91, 847–860 (2012).
Linder, S., Wiesner, C. & Himmel, M. Degrading devices: invadosomes in proteolytic cell invasion. Annu Rev Cell Dev Biol 27, 185–211 (2011).
Bon, E. et al. SCN4B acts as a metastasis-suppressor gene preventing hyperactivation of cell migration in breast cancer. Nature communications 7, 13648, https://doi.org/10.1038/ncomms13648 (2016).
Mercier, A., Bois, P. & Chatelier, A. Sodium Channel Trafficking. Handbook of experimental pharmacology 246, 125–145, https://doi.org/10.1007/164_2017_47 (2018).
Das, V., Bhattacharya, S., Chikkaputtaiah, C., Hazra, S. & Pal, M. The basics of epithelial-mesenchymal transition (EMT): A study from a structure, dynamics, and functional perspective. J Cell Physiol, https://doi.org/10.1002/jcp.28160 (2019).
Patel, F. & Brackenbury, W. J. Dual roles of voltage-gated sodium channels in development and cancer. The International journal of developmental biology 59, 357–366, https://doi.org/10.1387/ijdb.150171wb (2015).
Adachi, K. et al. Identification of SCN3B as a novel p53-inducible proapoptotic gene. Oncogene 23, 7791–7798 (2004).
Sanchez-Sandoval, A. L. & Gomora, J. C. Contribution of voltage-gated sodium channel beta-subunits to cervical cancer cells metastatic behavior. Cancer cell international 19, 35, https://doi.org/10.1186/s12935-019-0757-6 (2019).
Nelson, M., Millican-Slater, R., Forrest, L. C. & Brackenbury, W. J. The sodium channel beta1 subunit mediates outgrowth of neurite-like processes on breast cancer cells and promotes tumour growth and metastasis. Int J Cancer 135, 2338-2351 (2014).
Gong, Y. et al. Preserved SCN4B expression is an independent indicator of favorable recurrence-free survival in classical papillary thyroid cancer. PLoS One 13, e0197007, https://doi.org/10.1371/journal.pone.0197007 (2018).
Grimes, J. A. et al. Differential expression of voltage-activated Na+ currents in two prostatic tumour cell lines: contribution to invasiveness in vitro. FEBS Lett 369, 290–294 (1995).
Onganer, P. U. & Djamgoz, M. B. Small-cell Lung Cancer (Human): Potentiation of Endocytic Membrane Activity by Voltage-gated Na(+) Channel Expression in Vitro. J Membr Biol 204, 67–75 (2005).
Gao, R., Shen, Y., Cai, J., Lei, M. & Wang, Z. Expression of voltage-gated sodium channel alpha subunit in human ovarian cancer. Oncol Rep 23, 1293–1299 (2010).
Xia, J. et al. Voltage-gated sodium channel Nav 1.7 promotes gastric cancer progression through MACC1-mediated upregulation of NHE1. Int J Cancer 139, 2553–2569, https://doi.org/10.1002/ijc.30381 (2016).
House, C. D. et al. Voltage-gated Na+ Channel Activity Increases Colon Cancer Transcriptional Activity and Invasion Via Persistent MAPK Signaling. Scientific reports 5, 11541, https://doi.org/10.1038/srep11541 (2015).
Guzel, R. M., Ogmen, K., Ilieva, K. M., Fraser, S. P. & Djamgoz, M. B. A. Colorectal cancer invasiveness in vitro: Predominant contribution of neonatal Nav1.5 under normoxia and hypoxia. J Cell Physiol 234, 6582–6593, https://doi.org/10.1002/jcp.27399 (2019).
Baptista-Hon, D. T. et al. Potent inhibition by ropivacaine of metastatic colon cancer SW620 cell invasion and NaV1.5 channel function. Br J Anaesth 113(Suppl 1), i39–i48, https://doi.org/10.1093/bja/aeu104 (2014).
Brackenbury, W. J., Chioni, A. M., Diss, J. K. & Djamgoz, M. B. The neonatal splice variant of Nav1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res Treat 101, 149–160 (2007).
Bugan, I. et al. Anti-metastatic effect of ranolazine in an in vivo rat model of prostate cancer, and expression of voltage-gated sodium channel protein in human prostate. Prostate Cancer Prostatic Dis, https://doi.org/10.1038/s41391-019-0128-3 (2019).
Bujak, M. & Frangogiannis, N. G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74, 184–195, https://doi.org/10.1016/j.cardiores.2006.10.002 (2007).
Ramos-Mondragon, R., Vega, A. V. & Avila, G. Long-term modulation of Na+ and K+ channels by TGF-beta1 in neonatal rat cardiac myocytes. Pflugers Arch 461, 235–247, https://doi.org/10.1007/s00424-010-0912-3 (2011).
Hao, X. et al. TGF-beta1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circulation. Arrhythmia and electrophysiology 4, 397–406, https://doi.org/10.1161/CIRCEP.110.960807 (2011).
Derangeon, M. et al. Transforming growth factor beta receptor inhibition prevents ventricular fibrosis in a mouse model of progressive cardiac conduction disease. Cardiovasc Res 113, 464–474, https://doi.org/10.1093/cvr/cvx026 (2017).
Katoh, Y. et al. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J 273, 2730–2748, https://doi.org/10.1111/j.1742-4658.2006.05291.x (2006).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, https://doi.org/10.1006/meth.2001.1262 (2001).
Gore, J., Besson, P., Hoinard, C. & Bougnoux, P. Na(+)-H+ antiporter activity in relation to membrane fatty acid composition and cell proliferation. Am J Physiol 266, C110–120 (1994).
Jelassi, B. et al. P2X(7) receptor activation enhances SK3 channels- and cystein cathepsin-dependent cancer cells invasiveness. Oncogene 30, 2108–2122 (2011).
Acknowledgements
This work was supported by the University of Tours, the “Ligue Nationale Contre le Cancer – Interrégion Grand-Ouest”, the Région Centre-Val de Loire (grant “NaVMetarget”), the “Association CANCEN”. S.R. was recipient of a prize “Prix Ruban Rose Avenir 2017” from the Charity “le Cancer du sein: parlons-en!”. We thank Mrs Catherine Le Roy and Mrs Carole Desplanches for secretary and administrative assistance, and Mrs Isabelle Domingo for cell culture assistance. We thank Mr Julien Gaillard for the assistance with scanning electron microscopy.
Author information
Authors and Affiliations
Contributions
All authors contributed extensively to the work presented in this study. F.G. performed and analysed SEM experiments, performed siRNA silencing, assessed H+ effluxes. O.L.-C. performed all electrophysiological and epifluorescence experiments. F.G. and O.L.-C. performed cell culture, molecular and cellular biology experiments, assessed cell viability and invasiveness. L.P. participated to molecular biology experiments. C.G. performed RT-qPCR from human breast cancer biopsies. L.O. resected and selected tumours and patients. M.-L.J. maintained and annotated tumour biobank. E.B., S.R., S.C. and P.B. analysed in vivo data. E.B., V.D., E.P. and A.M. participated to lentiviral particles production and the generation of small hairpin RNA or overexpressing cancer cell lines. S. Chadet and S.R. performed in silico expression analyses. S. Chevalier, D. M. contributed in discussion and correction of the manuscript. S.R. and P.B. obtained research grants, directed the research, designed the study, analysed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gradek, F., Lopez-Charcas, O., Chadet, S. et al. Sodium Channel Nav1.5 Controls Epithelial-to-Mesenchymal Transition and Invasiveness in Breast Cancer Cells Through its Regulation by the Salt-Inducible Kinase-1. Sci Rep 9, 18652 (2019). https://doi.org/10.1038/s41598-019-55197-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55197-5
This article is cited by
-
Inactivation of epithelial sodium ion channel molecules serves as effective diagnostic biomarkers in clear cell renal cell carcinoma
Genes & Genomics (2023)
-
Genome-wide differential DNA methylation analysis of MDA-MB-231 breast cancer cells treated with curcumin derivatives, ST08 and ST09
BMC Genomics (2022)
-
Voltage imaging reveals the dynamic electrical signatures of human breast cancer cells
Communications Biology (2022)
-
Epithelial-mesenchymal transition process during embryo implantation
Cell and Tissue Research (2022)
-
Novel mutation of SIK1 gene causing a mild form of pediatric epilepsy in a Chinese patient
Metabolic Brain Disease (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
