
Abstract
Autophagy is active during cellular remodeling including muscle differentiation. Muscle differentiation is dysregulated in type 2 diabetes and we therefore hypothesize that muscle precursor cells from people with type 2 diabetes (T2DM) have a dysregulation of their autophagy leading to impaired myogenesis. Muscle precursor cells were isolated from people with T2DM or healthy controls and differentiated in vitro. Autophagy marker levels were assessed by immunoblotting. Differentially expressed autophagy-related genes between healthy and T2DM groups were identified based on a previously published RNA-sequencing data-set, which we verified by RT-qPCR. siRNA was used to assess the function of differentially expressed autophagy genes. Basal autophagy increases during human muscle differentiation, while T2DM muscle cells have reduced levels of autophagy marker ATG7 and show a blunted response to starvation. Moreover, we demonstrate that the 3 non-canonical autophagy genes DRAM1, VAMP8 and TP53INP1 as differentially expressed between healthy and T2DM groups during myoblast differentiation, and that T53INP1 knock-down alters expression of both pro-and anti-apoptotic genes. In vitro differentiated T2DM muscle cells show differential expression of autophagy-related genes. These genes do not regulate myogenic transcription factors but may rather be involved in p53-associated myoblast apoptosis during early myogenesis.
Similar content being viewed by others


Introduction
Skeletal muscle function depends on the continuous repair and maintenance of muscle fibers in a complex process relying on satellite cells: a population of muscle precursor cells residing beneath the basal lamina, with the capacity to expand and progress through the myogenic program upon activation1,2. Throughout differentiation of these muscle precursor cells a considerable reprogramming of gene expression occurs; likewise, a substantial remodeling of cellular structures takes place3,4.
A candidate mechanism behind this remodeling is autophagy; a major cellular degradation pathway in which intracellular organelles can be broken down and recycled, thus allowing for cellular clean-up and homeostasis5. Adaptation to cellular stress is a key function of autophagy, and autophagy has concomitantly been shown to protect against metabolic diseases such as insulin resistance and diabetes6. Beyond its roles as a stress and starvation response, autophagy is also involved in cell survival, differentiation and development7,8, and importantly, recent murine-derived cell-based studies have shown that autophagy is required for in vitro myogenesis4,9. Moreover, it is becoming increasingly clear that autophagy is required for maintenance of skeletal muscle tissue. This is illustrated by several murine knock-out model-studies of autophagy-associated proteins showing that autophagy is critical for maintaining skeletal muscle mass, as well as muscle metabolism and function10,11,12,13, thus underlining the importance of functional autophagy in skeletal muscle.
Skeletal muscle tissue is a main target of insulin-mediated glucose uptake, and skeletal muscle insulin resistance is a central step in type 2 diabetes disease progression14. Considering that autophagy is under direct regulation by nutrient availability15 and the wide-ranging metabolic perturbations in T2DM, surprisingly few studies have investigated how type 2 diabetes affects skeletal muscle autophagy. The IGF-I/ PI3K/AKT signaling cascade directly inhibits skeletal muscle autophagy16, and as this pathway is dysregulated in skeletal muscle from T2DM individuals17, it is plausible that dysfunctional insulin signaling may lead to altered autophagy levels in skeletal muscle in type 2 diabetes. While a previous study found that both basal and insulin-stimulated autophagy levels were largely unaltered in skeletal muscle biopsies from patients with type 2 diabetes18, this has not yet been investigated in isolated muscle precursor cells during differentiation. Given the dynamic nature of autophagy it is possible that potential differences are blunted at tissue level, yet important at a cellular level. We have recently shown that differentiation is impaired in muscle precursor cells isolated from T2DM donors19. In the present study, our primary objective was therefore to investigate if muscle precursor cells isolated from type 2 diabetic donors would have altered levels of autophagy markers, suggesting altered autophagy, compared with cells from healthy control donors. Furthermore, we wanted to explore the role of autophagy in the T2DM-mediated dysregulated myogenesis.
Materials and Methods
Human muscle precursor cell donors
Muscle precursor cells were obtained from a subset of male healthy (n = 5) or type 2 diabetic (n = 5) donors (Table 1) included in a previously described study20. RNA-seq data was obtained from a previous study with a partial overlap in cell culture donors with the present study21. The WHO diagnostic criteria for type 2 diabetes were used as basis for inclusion22. All subjects gave written informed consent prior to commencement of study, which was performed according to the Declaration of Helsinki and approved by The Regional Committee on Biomedical Research Ethics in Denmark (KF 01-141/04).
Cell culture
Satellite cells were obtained from vastus lateralis muscle biopsies as previously described23. Fat and visible connective tissue was removed, and the muscle biopsy was minced into small pieces and digested in buffer containing 0.05% trypsin-EDTA, 1 mg/ml collagenase IV and 10 mg/ml BSA for 5 min at 37 °C. Digestion solution containing released muscle precursor cells was then transferred to cold FBS for trypsin inactivation. The solution was centrifuged at 800 g for 7 min. The supernatant was removed and washed with F10 nutrient mixture (HAM) medium. To minimize fibroblast contamination, the cell suspension was pre-plated in a culture plate for 3 hours in growth medium containing 20% FBS, 1% penicillin/streptomycin (PS) and 1% Fungizone antimycotic (FZ) in F10/HAM. The unattached muscle precursor cells were then seeded onto culture flasks coated with Matrigel (0.01% Matrigel in F10/HAM + supplemented with 1% PS) and cultured for 4 days in growth medium in a humidified incubator with 5% O2 and 5% CO2 at 37 °C. Cell culture medium was changed after 4 days of incubation and then after every second day. At 100% confluency, cells were transferred to intermediate medium (Dulbecco’s modified Eagle’s medium (DMEM) containing 1 g/L glucose, 10% FBS and 1% PS) to induce alignment of muscle precursors. After 2 days, medium was changed into differentiation media (DMEM containing 4.5 g/L glucose, 2% horse serum (HS) and 1% PS) to induce differentiation into myotubes (myocytes). All cells were tested negative for mycoplasma contamination. F10/ HAM, HBSS, DMEM, FBS, HS, PS and FZ were obtained from Invitrogen (Taastrup, Denmark). Bafilomycin A1 was from Invivogen (Toulouse, France). Experiments were performed on cells at passages 5 to 6.
Immunomagnetic sorting of CD56+ precursor cells
To reduce the presence of non-muscle cell types all cell cultures were sorted using CD56-conjugated magnetic microbeads (Miltenyi Biotec, Lund, Sweden). Cells were grown until ~50% confluent and incubated with CD56 primary antibody conjugated microbeads, and subsequently isolated by positive selection according to the manufacturer’s instructions.
siRNA transfection
Cells were transiently transfected with small interfering RNA (siRNA) oligo pools specifically targeting DRAM1, VAMP8 and TP53INP1 mRNA respectively, or a non-targeting scrambled siRNA pool (On-Target Plus, Dharmacon, Søborg, Denmark). Transfections were performed in >90% confluent, undifferentiated cells using a final concentration of 20 nM siRNA with Lipofectamine RNAimax (Invitrogen). A Lipofectamine-only control was included as well; all statistics for siRNA experiments were performed comparing the effects of siRNA with the effects of the scrambled control. The Lipofectamine control did not differ from the scrambled siRNA control. Transfections were performed in DMEM containing 10% FBS and no antibiotics, and subsequently incubated for 24 hours, at which point medium was changed to DMEM containing 10% FBS and 1% PS. Cells were harvested at either 48 hours (Day 0) or 7 days (Day 5) after siRNA transfection.
Immunoblot analysis
Cells were washed in ice-cold PBS and lysed using lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 0.1% Triton X-100, protease inhibitor ((1 tablet/10 ml; Boehringer-Roche Diagnostics, Copenhagen, Denmark), 1% phosphatase inhibitor cocktail (Sigma-Aldrich, Brøndby, Denmark)). Cell lysates were centrifuged at 12000 g, 4 °C for 5 min and supernatants were collected. Protein concentration was determined by the Bradford reagent. 10 µg cell lysates were subjected to SDS-PAGE on 4–15% precast gels (Biorad), transferred to polyvinylidene difluoride (PVDF) membranes and immunoblotted with primary antibodies (1:1000) as indicated in the figure legends. Primary antibodies were detected using species-appropriate horseradish peroxidase-conjugated secondary IgG antibody. Protein signals were visualized using FEMTO enhanced chemiluminescence and Biorad Chemidoc XRS imager. The signal bands were quantified using Image J software (NIH, Bethesda, MD, http://rsb.info.nih.gov/ij). Anti-ULK1, ATG7, and ATG5 antibodies were from Cell Signaling (MA, US) and anti-LC3 antibody was from Nanotools (Teningen, Germany).
Immunoflourescence microscopy
In vitro differentiated human muscle stem cells were fixated with 4% Formaldehyde (Sigma) and then permeabilized with 0.5% Triton X-100. Cells were then incubated with ActinGreen 488 ReadyProbes and a nuclear counterstain was performed with Nucblue Fixed Cell stain ReadyProbes (Molecular probes). Fluorescence microscopy was performed with an EVOS FL (Thermo Fisher).
RNA isolation and quantitative real-time PCR
Total RNA was extracted from cells using TRIzol (Invitrogen, Taastrup, DK) according to manufacturer’s instructions. Total RNA was dissolved in RNase-free water and quantified using a Nanodrop ND 1000 (Saveen Biotech ApS, Arhus, Denmark). 0.2 µg of total RNA was reverse transcribed using the High Capacity Reverse Transcription kit (Applied Biosystems, Foster City, CA) according to manufacturer’s protocol. Real-time quantitative PCR was performed in triplicate using an ABI-PRISM 7900 (Applied Biosystems). Primer sequences are listed in Table 2; primer targets and genomic locations are listed in Table S1. Data analysis was performed using the comparative method (ΔΔCT).
Identification of target autophagy genes
GO-term gene members were acquired using the Bioconductor R package biomaRt as previously described21. All genes belonging to the GO-term Autophagy (GO:0006914) were cross-checked against the analysis results in21, and selected genes that were significantly differentially expressed (adjusted p-value < 0.05) in T2DM subjects compared to healthy controls were chosen for further investigation.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA). All data within figures are presented as means ± SEM. Data in tables are presented as means ± SD. For comparisons between two groups, a Student’s t-test was used. For group differences and multiple comparisons, statistical analysis was performed using two-way ANOVA with Holm-Sidak post -hoc testing.
Results
Impairment in autophagy response in differentiated T2DM muscle cells
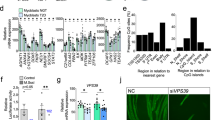
Considering the numerous metabolic defects in T2DM muscle14, we hypothesized that T2DM myocytes would show an impaired autophagy response when presented to a metabolic challenge. We therefore treated cells with HBSS medium for 4 hours to induce starvation, followed by re-addition of insulin to the starvation medium. Re-addition of insulin did not result in decrease of protein abundance of Microtubule-associated protein 1A/1B-light chain 3 II (LC3II) levels in any of the groups and there was no over-all difference between groups in response to this treatment (Fig. 1A,B). The effects of insulin on LC3II in muscle tissue have previously been shown to be dependent on glycemic status18; the lack of autophagy response to insulin in our study might therefore be due to the relatively high glucose concentration in the cell culture media. Interestingly, while both healthy and T2DM groups showed accumulation of LC3II in response to the vacuolar H+ ATPase-inhibitor Bafilomycin A (used to assess autophagy flux), only the healthy group showed a significant increase in LC3II when exposed to starvation in the presence of Bafilomycin A (Fig. 1B), suggesting that the dynamic activation of autophagy in response to a metabolic challenge is blunted in the T2DM group. Moreover, protein levels of LC3II were also differentially regulated between healthy and diabetic groups with an overall decrease in the T2DM cells (Fig. 1B) indicating a lower level of autophagy in this group.

Autophagy protein expression in myoblasts from healthy and T2DM donors. (A,B) Fully differentiated myotubes from healthy control or T2DM groups were starved in HBSS medium (4 hours), and treated with 100 nM insulin (30 minutes) and/ or Bafilomycin A1 (10 nM, 4 hours). Protein levels of LC3I/II were measured by immunoblotting; representative blots are shown. (C) Myoblasts from healthy controls (n = 5) or T2DM donors (n = 5) were harvested under proliferation, at confluence (day -2), as aligned, undifferentiated myoblasts (day 0), at early differentiation, after 2 days in differentiation medium (day 2) and as fully differentiated myotubes, after 5 days in differentiation medium (day 5). Protein levels of ULK-1, ATG7, ATG5 and LC3I/II were measured by immunoblotting. Representative blots are shown. Two samples harvested from proliferating cells were not included in analysis, as protein levels were too low. A Reactive Brown stain was used to visualize total protein. (D) Quantifications of immunoblots shown in (c). Protein levels were normalized to proliferating healthy controls. Data are means ± SEM. * indicates P < 0.05; ***P < 0.001.
Given that muscle precursor cells derived from people with T2DM have a dysregulated p53-dependent differentiation19, we next aimed to determine whether the disturbed regulation of autophagy was reflected by dysregulation of autophagy-associated proteins during human myoblast differentiation. To address this question, cells were grown in culture and harvested at the time points indicated in Fig. 1. Immunoblot analysis of Autophagy related 7 and 5 (ATG7 and ATG5), unc-51 like autophagy activating kinase 1 (ULK1) and LC3II revealed a marked upregulation of all four proteins as cells progressed through differentiation (Fig. 1C,D). These markers of autophagy were chosen for analysis to ensure that proteins involved in both early and later steps of the autophagic process would be assessed. At day 5, when myotubes were fully differentiated, ATG7 was markedly lower expressed in the cells derived from people with T2DM, whereas the rest of the assessed proteins did not differ between groups (Fig. 1D).
Thus, we show that basal autophagy increases during myoblast differentiation also in humans. We also found that muscle cells from T2DM donors have an blunted autophagy response and reduced levels of ATG7, a rate limiting protein of the conjugation system and therefore, of LC3 lipidation and autophagosome formation. Beside autophagy, ATG7 has been previously shown to be important for handling metabolic stress by targeting p53 activity24.
DRAM1, VAMP8 and TP53INP1 are differentially expressed in T2DM muscle precursor cells
To further investigate the molecular dysregulations behind the reduced autophagy response to metabolic stress, we took on a global approach. In a previous study, we performed RNA sequencing to describe the transcriptional profiles of muscle cells derived from healthy or T2DM donors21. Based on these previous results, we identified several autophagy-related genes as differentially expressed between groups (Fig. 2A). The genes Damage-Regulated Autophagy Modulator protein 1 (DRAM1) and Vesicle Associated Membrane Protein 8 (VAMP8) were two of the most up- or down-regulated genes respectively (Fig. 2A) and were therefore chosen for further analysis. The autophagy markers analyzed by immunoblotting in Fig. 1 were not detected as differentially expressed in the RNA-seq dataset when correcting for multiple testing, nor were markers of myogenesis or apoptosis. Another differentially expressed gene, Tumor Protein 53-inducible nuclear protein 2 (TP53INP2) was previously shown to regulate muscle mass and autophagy and is downregulated in muscle tissue in type 2 diabetes25. We chose to also include the TP53INP2 homologue Tumor Protein 53-inducible nuclear protein 1 (TP53INP1) for analysis, as it was previously identified as a diabetes susceptibility locus26, yet had no described role in skeletal muscle to date.

DRAM1, VAMP8 and TP53INP1 expression in myoblast derived from humans with T2DM and healthy control subjects. (A) Gene counts from 21for identified differentially expressed autophagy-related genes (adjusted p-value < 0.5) in T2DM subjects (n = 6) versus healthy controls (n = 6). (B) Myoblasts from healthy controls or T2DM donors were harvested under proliferation, at confluence (day -2), as aligned, undifferentiated myoblasts (day 0), at early differentiation, after 2 days in differentiation medium (day 2) and as fully differentiated moytubes, after 5 days in differentiation medium (day 5). DRAM1, VAMP8 and TP53INP1 mRNA was measured by RT-qPCR in healthy or T2DM muscle precursor cells. mRNA expression was normalized to the geometric mean of 18 s, B2M and PPIA mRNA. Data are means ± SEM. * indicates P < 0.05; **P < 0.01.
Using qPCR analysis, we verified that DRAM1, VAMP8 and TP53INP1 were indeed differentially regulated in T2DM cells (Fig. 2B), whereas TP53INP2 showed differential expression in proliferating cells only (Fig. S1). Only VAMP8 was consistently downregulated over time in the T2DM group, whereas DRAM1 was upregulated in fully differentiated T2DM muscle cells and TP53INP1 was downregulated in T2DM muscle precursors at day 0 of differentiation (Fig. 2B). Interestingly, both DRAM1 and TP53INP1 have previously been reported to positively regulate the capacity of p53 to control cell cycle exit and induce apoptosis27,28. p53 also plays a role in the context of myogenesis, since activation of the p53 target gene p21is a mandatory step for muscle precursor cells to exit the cell cycle and proceed through the myogenic programme29,30. Since we have previously shown that muscle precursor cells from humans with type 2 diabetes have impairments in p53/p21-dependent myogenesis19, we therefore decided to further investigate the role of DRAM1, VAMP8 and TP53INP1 in myogenesis.
Loss of TP53INP1 results in enlarged myotubes
In accordance with previous studies4,9 we found that autophagy appeared to increase during muscle cell differentiation, indicating that autophagy plays a role in this process. We therefore hypothesized if knock down of the autophagy-associated genes DRAM1, VAMP8 or TP53INP1 would have an impact on myogenesis. To assess this, cells were grown until confluent and siRNAs against DRAM1 in T2DM cells and against VAMP8 and TP53INP1 in cells from healthy donors were used to knock down their respective target mRNAs. Cells were harvested 48 hours and 7 days after knockdown respectively (indicated as day 0 and day 5). mRNA levels of DRAM1 (Fig. S3A) and VAMP8 and TP53INP1 (Fig. 3A,B) were significantly reduced after siRNA treatment and remained low throughout differentiation suggesting that siRNA treatment was effective. Knockdown of either DRAM1 or VAMP8 or TP53INP1 did not appear to affect cell viability as assessed by light microscopy at day 0 or day 5 of differentiation, since siRNA-treated cells all proceeded to form large, multinucleated myotubes during differentiation (Figs S2, S3B). However, whereas DRAM1 and VAMP8 siRNA treatment did not affect myotube morphology, knock down of TP53INP1 lead to formation of visibly larger myotubes when compared to control cells (Figs 3D; S2). We next measured the mRNA expression of myogenic markers PAX7, MyoD, myogenin and myosin heavy chain 2 a (MYH2), to see if loss of DRAM1, VAMP8 or TP53INP1 would affect their expression. However, while all myogenic markers showed significant changes over time, we did not detect any effect of DRAM1, VAMP8 or TP53INP1 knockdown on their expression (Figs 3C; S2C).

Effect of VAMP8 and TP53INP1 knock down on myogenic marker expression. Myoblasts from healthy controls (n = 5) were treated with 20 nM siRNA oligonucleotides targeting VAMP8 or TP53INP1, or with a non-targeting scrambled control. A lipofectamine-only control was included for each experiment (data not shown) to confirm no off-target effects of the scrambled siRNA control. Cells at >90% confluence were treated with siRNA and harvested 48 hours or 7 days after siRNA treatment; indicated as day 0 or day 5 respectively. (A,B) VAMP8 and TP53INP1 mRNA levels were measured by RT-qPCR and normalized to TBP mRNA. The same control was used for both knock-downs. (C) Light microscopy images of siRNA-treated cells at 4X magnification; scale bar indicates 1000 µM. (D) Expression of myogenic markers PAX7, MYOD, MYOG and MYH2 in healthy control myoblasts treated with VAMP8, TP53INP1 or control siRNA. Data are means ± SEM. * indicates P < 0.05; **P < 0.01, ***P < 0.001.
Loss of TP53INP1 increases pro-and antiapoptotic markers
Growth factor withdrawal signals to myoblasts to exit the cell cycle and undergo differentiation; however, loss of growth factor signaling can also induce myoblast apoptosis31,32. TP53INP1 is a p53 target gene involved in the regulation of p53-mediated apoptosis27,33,34. We therefore hypothesized that loss of TP53INP1 combined with reduced presence of serum in the cell culture medium would alter the cells’ propensities to undergo apoptosis versus differentiate. We consequently measured the mRNA expression of pro-apoptotic markers BCL2-associated agonist of cell death (BAD) and BCL2-associated X (BAX), as well as the anti-apoptotic marker BCL2-like 1 (BCL-X) in TP53INP1 siRNA-treated muscle cells. Interestingly, mRNA expression of BAD was unaltered whereas BAX expression was markedly upregulated after knock-down of TP53INP1 with a simultaneous increase in BCL-X expression (Fig. 4A), thus indicating increases in both pro- and anti-apoptotic gene expression. Beyond regulating apoptosis, TP53INP1 has previously been reported to regulate the transcriptional activation of p53 target genes involved in myogenesis, including the cyclin-dependent kinase inhibitor 1 (p21)33. Interestingly, although P53 expression was not affected by TP53INP1 knock-down, p21 mRNA expression was robustly upregulated in TP53INP1 siRNA-treated cells, while the expression of RB1, another p53-induced gene required for myogenesis35,36, was unaltered (Fig. 4B). Hence, despite lack of change in myogenic transcription factors, our data suggest that TP53INP1 may influence apoptosis and cell cycle exit during early differentiation, with a visible effect on myotube size.

Effect of TP53INP1 knock down on P53 and apoptosis-related gene expression. Myoblasts from healthy controls (n = 5) were treated with 20 nM siRNA oligonucleotides targeting TP53INP1, or with a non-targeting scrambled control. A lipofectamine-only control was included (data not shown) to confirm no off-target effects of the scrambled siRNA control. Cells at >90% confluence were treated with siRNA, and harvested 48 hours or 7 days after siRNA treatment. (A) mRNA expression of pro-apoptotic markers BAD and BAX or anti-apoptotic marker BCL-X. (B) mRNA expression of P53, MDM2, RB1 and p21 in healthy control myoblasts treated with TP53INP1 or control siRNA. (B) Data are means ± SEM. * indicates P < 0.05; **P < 0.01, ***P < 0.001.
Discussion
In the present study we demonstrated reduced autophagy response to metabolic stress in human muscle precursor cells from humans with T2DM. Upon in vitro differentiation, these cells had lower protein levels of ATG7, up-regulated mRNA levels of DRAM1 and down-regulated mRNA levels of VAMP8. In addition, TP53INP1 was downregulated in the early differentiation state. Through a loss-of-function approach we demonstrated that this downregulation results in an increase in the apoptosis genes BAX and BCL-X and the cell cycle regulator p21 at early differentiation, and enlarged myotubes once the cells are fully differentiated. Our data thus provide novel insight into the mechanisms manifested in muscle precursor cells in T2DM and highlight that a dysregulation of autophagy genes may contribute to the muscular phenotype of T2DM in humans.
The study was limited to in vitro experiments and thus only reflects the dysregulations in autophagy that are manifested in the muscle precursor cells even after in vitro expansion. However, an advantage is that we have utilized isolated cells from multiple human donors, thus including a biological variation from individuals that are suffering from T2DM. Moreover, using a cell-based approach allows us to study autophagy flux, which importantly cannot be done in muscle biopsies. In line with this, attempts to assess whether baseline autophagy is dysregulated in skeletal muscle biopsies in type 2 diabetes patients have resulted in conflicting findings, reporting either down-regulation37 or no difference from healthy controls18. These results might reflect the difficulties of measuring a dynamic mechanism such as autophagy in a biopsy including several cell types as well as muscle cells in different stages from precursor cell to differentiated myotube.
An increase in basal autophagy during C2C12 myogenesis has been previously demonstrated in several studies4,9,38; the current study provides the first evidence that this also occurs during human myogenesis. We previously showed that myogenesis is impaired in T2DM muscle cells, and that this dysregulation was associated with impaired signaling of p53 and the p53 target gene p2119. The current study support this concept of altered p53 signaling in T2DM muscle cells by demonstrating that two direct transcriptional targets of p53 (DRAM1 and TP53INP1)27,39 are dysregulated in T2DM cells. While we expected to detect a difference in basal autophagy between cells from healthy and T2DM donors, we only found a difference in expression of ATG7, although this was not reproducible in our global gene expression analysis. ATG7 is a critical regulator of autophagy40; however, further studies are required to explore the potential dysregulation of autophagy in T2DM muscle cells. Nevertheless, as ATG7 has previously been shown to regulate the response to metabolic stress through interaction with p53 and subsequent induction of p21 transcription24, our finding that ATG7 is downregulated in T2DM muscle cells further supports the notion of altered p53 signaling in T2DM muscle cells.
Genome-wide association studies have identified TP53INP1 as a diabetes susceptibility locus26,41 and interestingly, tp53inp1-deficient mice have chronic oxidative stress accompanied by insulin resistance and an increased number of dysfunctional mitochondria due to PINK1/ PARKIN-mediated impaired mitophagy42. We found that TP53INP1 was downregulated in early differentiation, when cells were confluent but not yet fused. Muscle precursor cells committed to differentiation are prone to undergo apoptosis43 and TP53INP1 can promote apoptosis in an autophagy-dependent manner34. Paradoxically, we found that TP53INP1 knockdown increased mRNA expression of both pro-and-antiapoptotic genes, while also increasing p21 expression. This may be explained by previous studies showing that although TP53INP1 promotes apoptosis, TP53INP1 deficiency leads to ROS accumulation and consequentially upregulation of p21 and BAX expression44. In turn, p21 conveys anti-apoptotic effects to differentiating muscle cells31 and promotes BCL-X expression45,46; thus the observed increased expression of BCL-X after TP53INP1 knock-down might reflect a compensatory p21-mediated anti-apoptotic response. Given that p21 is involved in cell cycle exit during early differentiation, this up-regulation might also explain our observation of enlarged myotubes following TP53INP1 knockdown. Thus, despite the lack of alterations in the measured myogenic markers, we propose that the downregulation of TP53INP1 in muscle precursor cells from people with T2DM might affect the early state of myogenesis leading to an imbalance between apoptosis and cell cycle exit, which may disturb proper differentiation. Nevertheless, our studies were limited to mRNA expression analysis; the use of more functional apoptosis assays would be necessary to explore the role of TP53INP1 in myogenesis-associated apoptosis. In line with this, further mechanistic studies examining exactly how TP53INP1 interacts with the autophagic machinery, such as co-localization experiments would be required to elucidate the specific roles of TP53INP1 in autophagy regulation.
Although we identified both VAMP8 and DRAM1 to be dysregulated in muscle cells derived from humans with T2DM, we were unable to further pinpoint their function in the current cell model. VAMP8 is a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) involved in autophagosome-lysosome fusion47, which has also been described in adipocyte GLUT4 trafficking48,49,50. Interestingly, Vamp8 knock-out mice have markedly increased insulin sensitivity and muscle glucose uptake51, suggesting that the downregulation in human muscle cells from people with T2DM, might be a compensatory action to ameliorate impaired insulin action. This idea however remains to be explored. DRAM1 is required for p53-regulated autophagy and is upregulated in response to genotoxic stress28. The fact that we found DRAM1 to be upregulated in fully differentiated T2DM myotubes may thus be a consequence of cellular stress in the T2DM cells, and it may explain why loss of DRAM1 in undifferentiated muscle cells had no effect on myogenic progression.
Conclusion
Our data suggest that muscle precursor cells from humans with T2DM have an altered expression of autophagy markers during metabolic stress and we identify a gene expression profile of autophagy-associated genes which are dysregulated in these cells. One of these genes, TP53INP1, seems to act in early myogenesis to regulate the balance between apoptosis and cell cycle exit and might thus be important in vivo to mediate proper muscle differentiation.
Data Availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Le Grand, F. & Rudnicki, M. A. Skeletal muscle satellite cells and adult myogenesis. Current Opinion in Cell Biology 19, 628–633 (2007).
Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 9, 493–5 (1961).
Wagers, A. J. & Conboy, I. M. Cellular and Molecular Signatures of Muscle Regeneration: Current Concepts and Controversies in Adult Myogenesis. Cell 122, 659–667 (2005).
Sin, J. et al. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 12, 369–380 (2016).
Mizushima, N. & Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 147, 728–741 (2011).
Yang, L., Li, P., Fu, S., Calay, E. S. & Hotamisligil, G. S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–78 (2010).
Levine, B. & Kroemer, G. Autophagy in the Pathogenesis of Disease., https://doi.org/10.1016/j.cell.2007.12.018.
Salemi, S., Yousefi, S., Constantinescu, M. A., Fey, M. F. & Simon, H.-U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 22, 432–5 (2012).
Fortini, P. et al. The fine tuning of metabolism, autophagy and differentiation during in vitro myogenesis. Cell Death Dis. 7, e2168 (2016).
Masiero, E. et al. Autophagy is required to maintain muscle mass. Cell Metab. 10, 507–15 (2009).
De Palma, C. et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 3, e418 (2012).
He, C. et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–5 (2012).
Kim, K. H. et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 19, 83–92 (2012).
Phielix, E. & Mensink, M. Type 2 diabetes mellitus and skeletal muscle metabolic function. Physiol. Behav. 94, 252–8 (2008).
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T. & Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–11 (2004).
Zhao, J. et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–83 (2007).
Krook, A., Roth, R. A., Jiang, X. J., Zierath, J. R. & Wallberg-Henriksson, H. Insulin-Stimulated Akt Kinase Activity Is Reduced in Skeletal Muscle From NIDDM Subjects. Diabetes 47, 1281–1286 (1998).
Kruse, R., Vind, B. F., Petersson, S. J., Kristensen, J. M. & Højlund, K. Markers of autophagy are adapted to hyperglycaemia in skeletal muscle in type 2 diabetes. Diabetologia 58, 2087–2095 (2015).
Henriksen, T. I. et al. Dysregulation of a novel miR-23b/27b-p53 axis impairs muscle stem cell differentiation of humans with type 2 diabetes. Mol. Metab., https://doi.org/10.1016/j.molmet.2017.04.006 (2017).
Pedersen, M. et al. Cognitive functions in middle aged individuals are related to metabolic disturbances and aerobic capacity: a cross-sectional study. PLoS One 7, e51132 (2012).
Väremo, L. et al. Type 2 diabetes and obesity induce similar transcriptional reprogramming in human myocytes. Genome Med. 9, 47 (2017).
WHO. Definition and diagnosis of diabetes mellitus and intermediate hyperglycaemia. World Heal. Organ (2006).
Green, C. J., Pedersen, M., Pedersen, B. K. & Scheele, C. Elevated NF-κB Activation Is Conserved in Human Myocytes Cultured From Obese Type 2 Diabetic Patients and Attenuated by AMP-Activated Protein Kinase. Diabetes 60, 2810–2819 (2011).
Lee, I. H. et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 336, 225–8 (2012).
Sala, D. et al. Autophagy-regulating TP53INP2 mediates muscle wasting and is repressed in diabetes. J. Clin. Invest. 124, 1914–1927 (2014).
Voight, B. F. et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat. Genet. 42, 579–589 (2010).
Okamura, S. et al. p53DINP1, a p53-Inducible Gene, Regulates p53-Dependent Apoptosis. Mol. Cell 8, 85–94 (2001).
Crighton, D. et al. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 126, 121–134 (2006).
Hawke, T. J. et al. p21 is essential for normal myogenic progenitor cell function in regenerating skeletal muscle. Am. J. Physiol. Cell Physiol. 285, C1019–27 (2003).
Guo, K., Wang, J., Andrés, V., Smith, R. C. & Walsh, K. MyoD-induced expression of p21 inhibits cyclin-dependent kinase activity upon myocyte terminal differentiation. Mol. Cell. Biol. 15, 3823–9 (1995).
Wang, J. & Walsh, K. Resistance to apoptosis conferred by Cdk inhibitors during myocyte differentiation. Science 273, 359–61 (1996).
McClearn, D., Medville, R. & Noden, D. Muscle cell death during the development of head and neck muscles in the chick embryo. Dev. Dyn. 202, 365–377 (1995).
Tomasini, R. et al. TP53INP1s and Homeodomain-interacting Protein Kinase-2 (HIPK2) Are Partners in Regulating p53 Activity. J. Biol. Chem. 278, 37722–37729 (2003).
Seillier, M. et al. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 19, 1525–35 (2012).
Porrello, A. et al. p53 regulates myogenesis by triggering the differentiation activity of pRb. J. Cell Biol. 151, 1295–304 (2000).
Wang, J., Guo, K., Wills, K. N. & Walsh, K. Rb functions to inhibit apoptosis during myocyte differentiation. Cancer Res. 57, 351–4 (1997).
Møller, A. B. et al. Altered gene expression and repressed markers of autophagy in skeletal muscle of insulin resistant patients with type 2 diabetes. Sci. Rep. 7, 43775 (2017).
McMillan, E. M. & Quadrilatero, J. Autophagy is required and protects against apoptosis during myoblast differentiation. Biochem. J. 462, 267–277 (2014).
Xie, X., Le, L., Fan, Y., Lv, L. & Zhang, J. Autophagy is induced through the ROS-TP53-DRAM1 pathway in response to mitochondrial protein synthesis inhibition. Autophagy 8, 1071–1084 (2012).
Komatsu, M. et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169 (2005).
Cauchi, S. et al. European genetic variants associated with type 2 diabetes in North African Arabs. Diabetes Metab. 38, 316–323 (2012).
Seillier, M. et al. Defects in mitophagy promote redox-driven metabolic syndrome in the absence of TP53INP1. EMBO Mol. Med. 7, 802–18 (2015).
Sandri, M. & Carraro, U. Apoptosis of skeletal muscles during development and disease. Int. J. Biochem. Cell Biol. 31, 1373–1390 (1999).
Cano, C. E. et al. Tumor Protein 53–Induced Nuclear Protein 1 Is a Major Mediator of p53 Antioxidant Function. Cancer Res. 69 (2008).
Wu, Y.-C. M. & O’Reilly, M. A. Bcl-XL is the primary mediator of p21 protection against hyperoxia-induced cell death. Exp. Lung Res. 37, 82–91 (2011).
Staversky, R. J. et al. p21Cip1/Waf1/Sdi1 protects against hyperoxia by maintaining expression of Bcl-XL. Free Radic. Biol. Med. 41, 601–609 (2006).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 151, 1256–1269 (2012).
Zhao, P. et al. Variations in the requirement for v-SNAREs in GLUT4 trafficking in adipocytes. J. Cell Sci. 122, 3472–3480 (2009).
Williams, D. & Pessin, J. E. Mapping of R-SNARE function at distinct intracellular GLUT4 trafficking steps in adipocytes. J. Cell Biol. 180, 375–387 (2008).
Sadler, J. B. A., Bryant, N. J. & Gould, G. W. Characterization of VAMP isoforms in 3T3-L1 adipocytes: implications for GLUT4 trafficking. Mol. Biol. Cell 26, 530–536 (2015).
Zong, H. et al. Enhanced energy expenditure, glucose utilization, and insulin sensitivity in VAMP8 null mice. Diabetes 60, 30–8 (2011).
Acknowledgements
Sarah Heywood and Ricardo Soares from The Centre for Physical Activity Research (CFAS) are acknowledged for their technical assistance. CFAS is supported by a grant from TrygFonden. This study was supported by a grant from the Novo Nordisk Foundation (C.S.), by a PhD scholarship from Faculty of Health Sciences, University of Copenhagen (T.I.H.) and by funding from the Knut and Alice Wallenberg Foundation (J.N. and L.V.W). The Centre for Physical Activity Research (CFAS) is supported by a grant from Trygfonden. The Novo Nordisk Foundation Center for Basic Metabolic Research (http://www.metabol.ku.dk) is supported by an unconditional grant from the Novo Nordisk Foundation to University of Copenhagen.
Author information
Authors and Affiliations
Contributions
T.I.H. and C.S. designed the study; C.S. directed the study; T.I.H. and L.V.W. collected and analyzed data; T.I.H. drafted the manuscript; T.I.H., L.V.W., J.N., B.K.P., S.M. and C.S. interpreted data and edited and approved the final version of the article.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Henriksen, T.I., Wigge, L.V., Nielsen, J. et al. Dysregulated autophagy in muscle precursor cells from humans with type 2 diabetes. Sci Rep 9, 8169 (2019). https://doi.org/10.1038/s41598-019-44535-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44535-2
This article is cited by
-
Nutri-stress, mitochondrial dysfunction, and insulin resistance—role of heat shock proteins
Cell Stress and Chaperones (2023)
-
The CEBPA-FGF21 regulatory network may participate in the T2DM-induced skeletal muscle atrophy by regulating the autophagy-lysosomal pathway
Acta Diabetologica (2023)
-
VPS39-deficiency observed in type 2 diabetes impairs muscle stem cell differentiation via altered autophagy and epigenetics
Nature Communications (2021)
-
The effects of different doses of exercise on pancreatic β-cell function in patients with newly diagnosed type 2 diabetes: study protocol for and rationale behind the “DOSE-EX” multi-arm parallel-group randomised clinical trial
Trials (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
