
Abstract
Reactive oxygen species (ROS) are primary effectors of cytotoxicity induced by many anti-cancer drugs. Rhythms in the pseudo-steady-state (PSS) levels of particular intracellular ROS in cancer cells and their relevance to drug effectiveness are unknown thus far. We report that the PSS levels of intracellular superoxide (SOX), an important ROS, exhibit an inherent rhythm in HCT116 colon cancer cells, which is entrained (reset) by the SOX inducer, menadione (MD). This reset was dependent on the expression of p53, and it doubled the sensitivity of the cells to MD. The period of oscillation was found to have a linear correlation with MD concentration, given by the equation, T, in h = 23.52 − 1.05 [MD concentration in µM]. Further, we developed a mathematical model to better understand the molecular mechanisms involved in rhythm reset. Biologically meaningful parameters were obtained through parameter estimation techniques; the model can predict experimental profiles of SOX, establish qualitative relations between interacting species in the system and serves as an important tool to understand the profiles of various species. The model was also able to successfully predict the rhythm reset in MD treated hepatoma cell line, HepG2.
Similar content being viewed by others


Introduction
Cancer treatment or management strategies such as chemotherapy and radiotherapy are predominantly based on the cytotoxicity caused by intracellular reactive oxygen species (ROS) such as superoxide (SOX) and hydroxyl radicals1,2,3,4. The efficacy of such strategies can be significantly improved, which would also reduce the undesirable side effects5,6. Although the effectiveness of anticancer therapeutics is known to be dependent on the circadian timing of their administration7,8,9, the chronotherapeutic methods of drug administration are not widely used probably due to a limited understanding of the underlying rhythms.
Biological rhythms have long been known to have effects on drug metabolism10,11. More than a hundred chemicals including anticancer drugs have been reported to have circadian changes in their pharmacokinetics12,13. Variation in drug pharmacokinetics, the circadian control of drug metabolism and cellular detoxification have been reported to be responsible for the temporal variation in toxicity and efficacy of drugs14.
However, the rhythmic variations of ROS, the primary effectors of cytotoxicity, and their effect on cancer drug effectiveness have not been reported thus far, probably due to the high reaction rates associated with ROS. However, it is known that the pseudo-steady-state (PSS) levels of ROS provide valuable information, which can be used to manipulate slower processes with much larger characteristic times; e.g. the resets in the rhythms of PSS levels of ROS significantly improved lipid productivity in microalgae15.
In this work, we report for the first time, a circadian rhythm in intracellular specific SOX levels in the colon cancer cell line, HCT116. No reports are available to date on rhythms of SOX in cancer cells. Further, we have studied the reset of the inherent SOX rhythm to a higher frequency by menadione (MD), which needs the expression of p53 in the cell.
MD is a well-established intracellular SOX generator16,17, which generates SOX by a redox cycling mechanism involving an enzyme catalysed single electron reduction of MD to its corresponding semiquinone (SQ) followed by the spontaneous cycling back to MD. This mechanism of redox cycling has been reported to be common to many well-known anticancer drugs18,19,20.
We have also developed a mathematical model to simulate the p53 dependent reset of SOX rhythm observed in HCT116 wt. The model generated results were found to be consistent with our experimental observations, which strengthen the possibility that the rhythms of SOX play a considerable role in the mode of action of MD in HCT116 wt.
Development of a mathematical model for the reset of SOX rhythm In HCT116 colon cancer cells
We have developed a mathematical model to better understand the mechanism of the reset in SOX rhythm by MD in HCT116 wt cells. MD is known to affect the extracellular signal -regulated kinase (ERK)21,22 and the protein p5323, which have tumour relevance. Therefore, an ERK–p53 pathway was chosen for the model development. The molecular interactions suggested by Li et al.24 through their experimental results for the effect of selenite, a redox active drug, in the apoptosis of NB4 cells, were modified by the addition of a feed-back loop for the regulation of SOX by MnSOD to derive our model for the p53–dependent, MD induced reset of SOX rhythm.
The proposed mathematical model can be said to capture the major contributing factors of the modelled phenomena if the model results are close to the experimental observations. However, the experimental system, due to its high level of complexity, may have aspects which are unaccounted for in the model. It is known that a successful mathematical model is only an abstraction of the most important features of the system, not all25. The assumptions made during the derivation of the current model are given below.
Assumptions
It was assumed that oxygen levels are saturated at a value of 20 μM26 as the system being considered was an in vitro cell culture. The translocation rate of ERK from the cytoplasm to the nucleus was considered to be much higher than the translocation rate in the reverse direction. The translocation rate of ERK into the nucleus was assumed to be second order, dependent on both ERK and SOX concentrations, instead of a first order reaction27 that does not consider SOX mediated activation of ERK. These assumptions lead to the use of a few conservation relations in the model (see Supplementary Fig. S1). The transcription of sod2 gene was assumed to be a single activator mediated activation of transcription by p53-P following Hill kinetics28. A Hill coefficient of 2 accounted for the positive cooperativity of binding of p53 to the DNA29. With these assumptions in place, the following reactions were considered for the model.
Reactions
MD acts via the production of SOX anion radicals in the cells through a redox cycling mechanism16,17 comprising of two main steps. The first step involves the conversion of MD to its corresponding semiquinone (SQ) catalysed by the enzyme cytochrome P450 reductase (CP450R), which follows bi-substrate enzyme kinetics30 with NADPH as the second substrate. This is followed by the second step that involves the spontaneous reaction of SQ with oxygen to generate SOX31. The SOX thus generated affects the activation of ERK32 and causes its translocation into the nucleus, with the reaction kinetics dependent on both ERK and SOX concentrations. The translocated ERK acts as the catalyst for the activation of the transcription factor p53 by phosphorylation33,34 to form p53-P. Alongside, the de-phosphorylation of p53-P by nuclear p53 phosphatases also occur. Both phosphorylation and de-phosphorylation reactions follow Michaelis-Menten (MM) kinetics35,36,37. The activated p53-P binds to the promoter and acts as a transcription factor for the gene sod2 which codes for the protein Mn SOD38,39,40. Mn-SOD, formed from the msod2 mRNA, is an enzyme which follows MM Kinetics41 and catalyses the dismutation of SOX to hydrogen peroxide and oxygen and thus reduces the cellular SOX levels. The NADP+ formed during the conversion of MD to SQ is cycled back to NADPH by cellular NADP+ reductases such as Ferredoxin NADP reductase (FNR)42.
The equations were stoichiometrically balanced by considering degradations of mRNA and protein back to the cellular metabolite pool. Ordinary differential equations (ODEs) were derived for each of the reacting components, according to the general formula
The various parameters used for the model are as follows: the amplitude of the decaying sine wave (A), concentration of chromatin DNA (Dn), the constants for menadione (KM), NADPH (KN), SOX (KSOX), p53 (Kp53), p53-P (Kp53-P) and NADP+ (KNADP+), all with units of µM, the maximal possible transcription initiation rate (Imax, h−1), the rate constant for conversion of SQ to SOX (k1, µM−1 h−1), the rate constant for conversion of SOX to SQ (km1, µM−1 h−1), the rate constant for the generation of p53 (kp, h−1), the turnover number for Mn-SOD (k2, h−1), the turnover number for the phosphorylation reaction of p53 (k3, h−1), the rate constant for nuclear translocation of ERK (kt1, µM−1 h−1), the translation rate constant (ktr, h−1), the mRNA turnover rate constant (kdm, h−1), the protein turnover rate constant (kdP, h−1), the damping factor for the decaying sine wave (L, h−1), the dimensionless Hill coefficient for binding of p53-P to DNA (na), the ratio of nuclear volume to cytoplasmic volume of a cell (nv), phase of the decaying sine wave (phi, radians), the initial velocity for CP450R, with menadione and NADPH (v1, µM h−1) and the maximal rates for conversion of MD to SQ (Vm1), p53-P to p53 (Vm4) and NADP+ to NADPH (Vm5), all in µM h−1.
The reactions, rate equations and mass balances used in the model are given in Tables 1 and 2.
The formation of SOX in the cell is a complex process43. To account for the endogenous levels of SOX in the cell, a decaying sine wave term was introduced in the mass balance for SOX. Interestingly, the experimental data showed that the time period has a linear correlation with the MD concentration only when p53-P is present. Incorporating this relation, we propose the following equation for the frequency:
The parameters, a and b are estimated for the model using linear regression of initial MD concentration vs average time period (obtained from the PAST analysis), for the data corresponding to the wt case as shown in Supplementary Fig. S2. Although obtained from the dataset corresponding to the wt case, interestingly, the parameter ‘a’ is found to predict the experimental observation in the case corresponding to the absence of p53 as well.
The initial concentration used for each component is given in Supplementary Table S1.
Optimized parameter values were obtained through the parameter estimation techniques defined in detail in the supporting information (see Supplementary Table S2). It was ensured that the values are biologically meaningful – they were obtained from a search space containing the biological ranges possible for each parameter. In addition to being biologically relevant, the parameters need to account for experimental errors. So, the phase, which can account for minor experimental errors in synchronization, was adjusted for each case to ensure a better fit. The phase values for the various cases are reported in Supplementary Table S3.
Results
Endogenous SOX levels in HCT116 cells possess a circadian rhythm independent of p53 status
The SOX levels in the untreated HCT116 cells were determined as described in the materials and methods section, through dihydroethidium (DHE) fluorescence with improved strategies to reduce interferences. The software, PAST44, was used to analyse the data using a Lomb-Scargle periodogram45 (LSP) analysis to determine rhythms in the temporal variation of SOX in the wt and p53−/− variants of HCT116. Both the cell variants showed presence of an inherent circadian rhythm for SOX (Table 3, Supplementary Figs S3a, S4a). The peak frequencies obtained were 0.042 and 0.044 h−1, respectively, for wt and p53−/− cells. The period of oscillation, calculated as the inverse of the peak frequency, was about 24 h for wt cells and about 22 h for p53−/− cells.
MD induces a p53-dependent reset in SOX rhythm in HCT116 cells
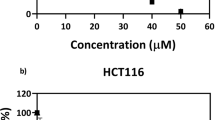
Both the wt and p53−/− cell variants were treated with various concentrations of MD. The IC50 values were determined 48 h post drug treatment (Fig. 1) using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay, and the IC50 values were in the earlier reported range46. The IC50 value was around 15 µM for HCT116 wt cells whereas the efficacy of the drug on the p53−/− variant was 50% less with an IC50 value of around 30 µM.

Variation of cell viability of HCT116 wt and p53−/− cells with increasing concentrations of menadione (MD). The cells were seeded at 1 × 104 cells per well. The cell viability was measured using MTT, 48 h post drug treatment. The values are expressed as mean ± SD, n = 3. Paired two tailed Student’s t test showed the data to be significant with p = 0.049.
The temporal variations of SOX levels were markedly different for wt and p53−/− cells (Fig. 2). The LSP analysis using PAST for wt cells showed that the frequency of SOX rhythm shifted from 0.042 h−1 for untreated cells to 0.112 h−1 for 15 µM MD treated cells. However, no significant change in rhythm was observed for the p53−/− cells (see Table 3, Supplementary Figs S3 and S4).

Temporal variations in SOX levels in (a) HCT 116 wt cells and (b) HCT 116 p53−/− cells for untreated control, 3 µM, 9 µM and 15 µM menadione (MD) treatment. The cells were seeded in 6 well plates and synchronized for 24 h in serum free medium. 0 h corresponds to the time of medium change to DMEM with 10% FBS with (treated) or without (untreated) MD. The time where SOX levels peak are different for different concentrations of MD in the wt cells whereas the trend is unchanged in the p53−/− cells. The values are expressed as mean ± SD, n = 3. The data was shown to be significant using two way ANOVA with p < 0.001.
The mathematical model predicts SOX rhythm reset
The model generated profiles were found to fit the experimental data well for all the initial MD concentrations, for both wt and p53−/− cases (Fig. 3). The peak frequencies for the experimental and model generated time series were obtained using spectral analysis with LSP and are reported in Table 3.

Comparison of experimental and model derived profiles of superoxide (SOX) in HCT116 wt cells and p53−/− cells. The experimental values are expressed as mean ± SD, n = 3. The experimental data was shown to be significant using two way ANOVA with p < 0.001.
The model was also used to make new predictions for a different initial MD concentration with and without p53 (Supplementary Fig. S5). The frequencies of these cases are also reported in Table 3. We find that they fit into the trends followed by the frequency as a function of MD induced cytotoxicity.
Robustness and sensitivity analysis
The robustness limits for all the parameters, along with the species affected the most by each parameter are reported in Supplementary Table S4. The initial MD concentration of 15 μM in the presence of p53 was used for the analysis. The robustness bound used was 20% as it represented a good span of the experimental data, as indicated in Supplementary Fig. S6.
Experimental investigations into aspects of the mathematical model
When the HC116 wt cells were pre-treated for 2 h with 10 µM of U0126 (inhibitor of mitogen-activated protein kinase kinase (MEK), the kinase which activates ERK) in a serum starved medium, there was a drastic effect on the rhythm reset observed in MD treated cells with the period of oscillation shifting from 8.9 h to 21.8 h for 15 µM MD treated cells. (Fig. 4a).

(a) Near circadian rhythmicity of SOX levels in HCT116 wt cells pre-treated with U0126 to inhibit ERK activation. The control rhythm is not affected, whereas the menadione induced rhythm reset does not occur when ERK activation is inhibited (b) In gel activity assay for manganese superoxide dismutase (MnSOD) in HCT116 wt and p53−/−. The graph shows the fold change in the MnSOD levels in menadione treated cells, pre-treated or untreated with U0126 for two hours to inhibit ERK activation, as compared to its respective untreated control for wt and p53−/− cells. The analysis was done using ImageJ software. The fold changes shown are the band intensities which have been normalized to their respective control values. The full length images are given in Supplementary Fig. S12. (c) Fold changes in p53 luciferase activity in HCT116 wt cells at the indicated concentrations of menadione, with (black) or without (grey) U0126 pre-treatment to inhibit ERK activation. The values have been normalized to their respective control values to obtain the fold changes. Two-way ANOVA was used to analyse the significance (p < 0.001) in the differences. The experimental values are expressed as mean ± SD, n = 3.
To determine whether Mn-SOD levels showed any up-regulation on MD treatment, in gel activity analysis was performed with the cell lysate (Fig. 4b). The results were similar to model predicted results, with Mn SOD levels being up-regulated by about 1.6 fold in the HCT116 wt cells treated with MD, 48 h after MD treatment and the p53−/− cells showing no significant change upon MD treatment. Pre-treatment of the cells with U0126, resulted in decrease in MnSOD levels in 15 µM MD treated cells to a level similar to that of control in wt cells, whereas they were unaffected in p53−/− cells.
MD treated cells showed a 4 fold increase in the activated p53 levels as indicated by pG13 luciferase reporter assay, which significantly decreased to 1.1 fold upon pre-treatment with U0126 (Fig. 4c). Further, the western blots of p53, ERK and pERK were analysed (Supplementary Fig. S7). The results show that the p53 levels between control and MD treated HCT116 wt cells were not significantly different at least up to 24 h, whereas MD significantly up-regulated ERK and pERK levels by 1.4-fold and 1.3-fold, respectively. But, p53 levels were down-regulated upon treatment with U0126 (Supplementary Fig. S7).
Since the model predicted variations were close to the actual observed changes in case of experimentally determined concentrations, the variations in the other species involved could also be reliable. Therefore, the model serves as an important tool for elucidating the reaction mechanism of MD by predicting the time profiles of the various species involved in this action, which would be difficult to determine experimentally. These profiles are given in Supplementary Fig. S8.
We have observed that the redox cycling compound, doxorubicin, also induces SOX rhythm reset in HCT116 wt cells, which is reversed upon inhibition of ERK activation (Supplementary Fig. S9). SOX rhythms in the hepatoma cell line HepG2 which expresses wt p53 were also analysed. HepG2 shows an inherent SOX rhythm of 25.6 h which was reset to 11 h upon treatment with MD (Supplementary Fig. S10).
Discussion
In this work, we report for the first time, a circadian rhythm in intracellular specific superoxide levels in cancer cells. The model cell line used for most of the studies was a human colon cancer cell line, HCT116. The temporal variations of SOX levels in the colon cancer cell line HCT116 which expressed p53 (wt) as well as its p53−/− variant, showed near-circadian rhythms of 24 h and 22 h, respectively. The ROS levels were higher in cancer cells compared to normal cells. However, survival of cancer cells requires regulation of these ROS levels below a certain threshold47,48. Since SOX have been reported to be important signalling molecules in the cell, the presence of these endogenous circadian rhythms suggests a possibility for tight regulation in the redox homeostasis to enable the cancer cell survival. Since ROS are crucial regulators of cell death1,2,3,4, the above leads to a speculation that alterations in SOX rhythms could be a mechanism for inducing cancer cell death. Also, ROS have been implicated in the cell death induced by multiple anti-cancer agents1,2,3,4,5,6. Thus, a better understanding could lead to the use of ROS rhythms as a generic target for better effective cancer chronotherapy.
MD is a well-established SOX generator16,17 that generates SOX through a redox cycling mechanism. The generation of additional SOX in the system by MD perturbs the SOX homeostasis in the cells and could be used to study the variations of SOX rhythm. We found that the exposure of HCT116 cells to a single pulse of MD reset the SOX rhythm to a higher frequency; the frequency increased with increase in MD concentration. Also, we found that the reset was dependent on the presence of the tumour suppressor protein, p53, since the p53−/− cell variant did not show any reset in the SOX rhythms. It is interesting to note that the cytotoxicity induced by MD in HCT116 cells was also p53 dependent with the p53−/− variant requiring double the concentration (IC50 = 30 µM) of MD to induce 50% cell death as compared to HCT116 wt (IC50 = 15 µM).
Experimental evidence is available for the involvement of an ERK–p53- MnSOD pathway in the action of selenite24 treatment on cancer cells. Also, ERK has been reported to be activated by MD21,22 and multiple other redox cycling anti-cancer drugs49,50. Since we have found the presence of p53 to be required for the rhythm reset, the possible involvement of an ERK –p53-MnSOD pathway in the SOX rhythm reset was hypothesized. MnSOD also was an ideal component to be involved in a feedback regulation of SOX, implied by the oscillatory nature of SOX concentrations. Since MD has been well studied, the use of MD for the model provided good initial values for optimization of parameters.
We found that the system was extremely sensitive to changes in na (the Hill coefficient), as it altered the functional form of the system. The system was highly sensitive to parameters Im, k3, Kp53, Vm4, Kt1, nv, phi, k1 and km1, moderately sensitive to parameters A, L, k2, ktr, Vm1, kna and Vm5, and less sensitive to the parameters Ksox, Kp53p, km and KNADP+. It can also be seen that the highly sensitive parameters are mostly involved in the nuclear events such as translocation of ERK or in the formation of p53-P and mSOD2.
The parameter that showed a very high sensitivity was na, the Hill coefficient for the binding of p53 to the DNA. The reported literature values are between 1.7 and 2, signifying a positive cooperativity for binding of p53 to the DNA. The changes in na values, even within these limits, significantly affected the concentration profiles of multiple species, especially the transcriptional formation of sod2 mRNA, in the model.
The model generated as described earlier, was able to predict the reset of SOX rhythm in HCT116 wt cells. The near circadian rhythm observed in the p53−/− case for all concentrations of MD is predicted satisfactorily as well. When the HCT116 wt cells were pre-treated with U0126 to inhibit ERK activation, the observed rhythm was near circadian in both control and 15 µM MD treated cells, implying the involvement of ERK activation in the MD induced rhythm reset.
The MnSOD levels were analysed using an in gel activity assay. HCT116 wt cells showed a 1.6 fold change in the MnSOD levels upon treatment with MD, which was similar to the model predicted values. This increase in MnSOD levels were not observed in MD treated cells when the HCT116 wt cells were pre-treated with U0126. The p53−/− cell variant did not show any changes in the MnSOD levels upon treatment with MD, in the presence or absence of U0126 pre-treatment. Therefore the presence of activated ERK seems to be required for MnSOD up-regulation in HCT116 wt cells. However, the basal levels of MnSOD in p53−/− cells seem to be higher. This could possibly be due to the complex cellular regulation of SODs involving multiple signalling networks51. The reason for the higher basal expression of MnSOD in p53−/− variant requires further in depth investigations, which are beyond the scope of the current study.
The finding that the p53 levels were not significantly different between control and MD treated cells is consistent with the outcome of another study done by Ham et al.52 where the authors have demonstrated that p53 level remains unchanged upon MD treatment in SK-hep-1 cells. However, increased level of ERK1/2 and pERK in MD treated HCT116 cells in this study suggests the activation of ERK1/2. As ERK1/2 are well known for stabilizing and enhancing p53 transcriptional activity through phosphorylation at different sites, the increased level of luciferase activity (Fig. 4c) may suggest the increase in ERK1/2 upon MD treatment. Another study by Ham et al.53 suggests a mechanistic basis of ERK activation upon MD treatment as MD inactivates MKP-1, a phosphatase, which mediates the de-phosphorylation event of MAP and hence stabilizing the phosphorylated form. However, confirming p53 activation by MD in HCT116 cells requires detailed investigations and would be beyond the scope of the present work. Nonetheless, the present data confirm the activation of ERK by MD, thereby providing a mechanistic basis for the increased MnSOD by MD.
The model described a possible mechanism for the p53-dependent SOX rhythm reset induced by MD in HCT116 colon cancer cells. Since the model was able to explain the experimentally observed rhythm aspects, the modelled mechanism seems to be mostly responsible for the observed rhythms, although it may not be solely responsible. To check whether the model’s predictive capability could be expanded to other cancer cell lines, we studied the SOX rhythm in untreated and MD treated hepatoma cells, HepG2, which has been reported to express wt p5354. Interestingly, HepG2 cells also showed an inherent, near-circadian SOX rhythm of 25.6 h which was reset to about 11 h upon treatment with MD at its IC50 value, which were similar to the model predicted results for SOX rhythm reset in HepG2 (Fig. S10). The proposed model mechanism could also be applicable to other redox cycling chemicals such as doxorubicin which induces a SOX rhythm reset in HCT 116 wt cells, reversible upon inhibition of ERK activation (Fig. S9).
In summary, the temporal variations of PSS SOX values seem to be rhythmic in HCT116 and the rhythm was reset by MD in a concentration dependent fashion in the presence of p53. Experimental SOX data from HCT116, HCT116 p53−/−, and HepG2 cell lines have been shown to be explained well by the model developed in this work. HCT116 wild type and HCT116 p53−/− cells are well known syngenic model systems, which have the same genetic background, whereas the HepG2 system is genetically different from the HCT116 colon cancer system. The model, summarized in Fig. 5, was able to predict well the results obtained from the studied syngenic systems as well as a genetically different system, which shows its general applicability. Further, in depth studies with multiple cell lines and multiple drugs need to be performed before the model could be used as a reliable tool to predict SOX rhythm reset induced by redox cycling chemicals, in general. This could also be useful in the manipulation of ROS rhythms as a target for cancer chronotherapy.

Proposed mechanism for menadione (MD) induced reset of circadian superoxide rhythms. SOX indicates superoxide, ERK – Extracellular signal-regulated kinase, p53 - Tumour suppressor protein, MnSOD - Manganese Superoxide dismutase.
Materials and Methods
Growth conditions and quantification
The cell line used in this study was a human colon cancer cell line, HCT116 and the hepatoma cell line HepG2. Both the wild type (wt) and p53 null (p53−/−) variants of HCT116, were gifted by Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, USA). The p53 status of the cells were validated (Supplementary Fig. S9) by western blotting55 using antibodies against p53 and vinculin (Cell Signaling Technology, USA). HepG2 cells were obtained from NCCS Pune, India. The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Himedia Labs, India) containing 10% heat-inactivated Fetal Bovine Serum (FBS, Invitrogen USA) and 1X penicillin/streptomycin solution (Himedia Labs, India). Cells were maintained at 37 °C with 5% CO2 in saturated humidity, and passaged upon reaching 80–90% confluency.
Determination of MD cytotoxicity
HCT116 wt and p53−/− cells were seeded at 1 × 104 cells/well in a 96-well microplate. The cells were treated with varying concentrations of MD (Sigma Aldrich, USA), 24 h after seeding. 48 h post drug treatment, the cytotoxic effect was measured using MTT (Sigma Aldrich, USA) assay56 by dual wavelength measurement in a microplate reader (Model 680, Bio-Rad Laboratories).
Intracellular reactive species induction
The cells were seeded at a concentration of 4 × 105 cells into 6 well dishes (Thermo Scientific, USA) in serum starved media to synchronize the cells57. To induce intracellular reactive species, 24 h after seeding, the spent medium was replaced with fresh DMEM containing 10% serum and varying concentrations of MD, in the test plates. Considering the drug addition time to be zero time, samples were collected every hour, up to 48 h. The control cultures, without MD were maintained at otherwise same conditions as the treated cultures.
Intracellular SOX quantification
The intracellular SOX concentrations were quantified by a fluorescent assay using the cell permeable dye, dihydroethidium ((DHE) (Invitrogen, USA)), with an excitation/emission pair of 405/570 nm to ensure specificity58. The calibration curve was obtained with superoxide radicals generated by the reaction of xanthine and xanthine oxidase59. Both control and treated cells were trypsinized, centrifuged at 2,000 rpm for 5 min, and re-suspended in PBS, containing 10 µM of DHE, to a final concentration of 2.5 × 105 cells mL−1. The cells were incubated at 37 °C for 30 minutes, centrifuged and re-suspended in the same volume of PBS. Fluorescence measurements were taken with 200 µL of cell suspension in each well in 96 well plates, with PBS as blank, in a multimode reader (Enspire, PerkinElmer, UK).
Determination of superoxide dismutase levels
The two types of superoxide dismutases (SOD), Cu/Zn SOD and Mn SOD, levels were determined using an in gel activity assay60. Both control and treated cells were trypsinized and collected at the respective time points, centrifuged at 3,000 rpm for 5 min, washed with PBS, and re-suspended in PBS containing 0.1% Nonidet P-40 (NP-40, Himedia Labs, India) for sonication. Sonication was carried out with a sonicator (Q700, a Q-Sonica, USA) at an amplitude of 70% for a total on time of 90 seconds with pulses of 10 seconds on and 10 seconds off. 200 µg protein was loaded to each lane. The gel was stained using a mixture of Nitro Blue Tetrazolium (NBT), N, N, N′, N′-tetramethylethane-1,2-diamine (TEMED) and Riboflavin 5′ phosphate (Himedia Labs, India), the bands were developed under white light using GelDoc™ and the gel picture was analysed using ImageJ software.
Activation of p53 through a luciferase reporter assay
A pG13 luciferase assay vector with p21 promoter construct, gifted by Dr. Ghanshyam Swarup (CCMB, Hyderabad, India), was used to determine the activation of p53 in MD treated HCT116 cells. Cells were transfected with pG13 using polyethylenimine (PEI, Polysciences Inc., USA). After transfection, the cells were pre-treated with 10 µM U0126 (Promega Life Sciences, USA) or serum free medium in case of control for 2 h and then were incubated with varying concentrations of MD. 48 h after MD treatment, the cells were collected, lysed and analysed for luciferase activity using a luminometer (Bio-Rad Laboratories, USA).
Analysis of rhythms of SOX
The temporal data obtained for SOX were analysed using the free software PAST, which uses the Lomb Scargle Periodogram (LSP) analysis to obtain the statistically significant (p < 0.05) rhythm frequency for evenly or unevenly spaced data. The software fits the data to sinusoidal curves of increasing incremental frequencies and gives the power for each, the statistically significant frequency with the highest power being the most probable frequency for the data set.
Mathematical modelling of superoxide rhythms
The mathematical modelling of SOX rhythms was done using MATLAB 2016b.
We developed a mathematical model comprising of 11 ordinary differential equations (ODEs) (Tables 1 and 2) and 5 species mass conservation relations (Fig. S1). The development and validation of the mathematical model involves finding a parameter set that yields SOX profiles that fit the observed experimental data. The set of ODEs comprises 23 parameters which we estimate using temporal experimental data for SOX as input. We use the case with p53 with initial MD concentration of 6 µM as the training dataset for model parameter estimation and all other datasets as test data for model validation. The model is simulated by solving the initial value problem using initial concentrations, obtained from literature (Supplementary Table S1), of the species in the system as initial conditions. Other initial conditions are used to obtain the behavior of the system under different conditions such as MD drug dosage and absence of p53.
The multi scale nature of the given system of equations makes it highly stiff, i.e. there exist nearby solutions that vary rapidly when compared to the required solution. Thus, for effective performance, we require a stiff ode solver which chooses an appropriate step size for the numerical integration of such a problem. Ode15s was used as the solver as the algorithm provides the inbuilt option of forcing only non-negative solutions, which is required in our case as we are dealing with concentrations. The genetic algorithm was found to give satisfactory parameters while maintaining good convergence and was therefore used to obtain the final parameter set. The detailed parameter estimation and robustness analysis is given in supplementary information.
Statistical analysis
The error bars in the graphs depict standard deviation from mean for three independent experiments. The rhythm frequency was taken at p < 0.05 using the software PAST. Graph Pad PRISM 7 was used to analyse all other data by two tailed Students t test or two-way ANOVA, as applicable, to test for statistical significance of the data with alpha being set at 0.05 and p < 0.05 being considered as significant. The statistical analysis for each figure is explained in the legend.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Conklin, K. A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr Cancer Ther. 3, 294–300 (2004).
Liou, G. Y. & Storz, P. Reactive oxygen species in cancer. Free Radic Res. 44, 479–496 (2010).
Wang, H. et al. Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance. Nat. Commun. 9, 562, https://doi.org/10.1038/s41467-018-02915-8 (2018).
Kasiappan, R. & Safe, S. ROS-inducing agents for cancer chemotherapy. Reactive Oxygen Species. 1, 22–37 (2016).
Moding, E. J., Kastan, M. B. & Kirsch, D. G. Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov 12, 526–542 (2013).
Saeidnia, S. & Abdollahi, M. Antioxidants: friends or foe in prevention or treatment of cancer. The debate of the century. Toxicol. Appl. Pharmacol. 271, 49–63 (2013).
Eriguchi, M. et al. Chronotherapy for cancer. Biomed. Pharmacother. 57, 92s–95s (2003).
Lévi, F. et al. Implications of circadian clocks for the rhythmic delivery of cancer therapeutics. Adv. Drug Deliv. Rev. 59, 1015–1035 (2007).
Mormont, M. C. & Levi, F. Cancer chronotherapy: principles, applications, and perspectives. Cancer. 97, 155–169 (2003).
Holcslaw, T. L. et al. Circadian rhythms in drug action and drug metabolism in the mouse. J Pharmacol Exp Ther. 195, 320–32 (1975).
Griffett, K. & Burris, T. P. The mammalian clock and chronopharmacology. Bioorg. Med. Chem. Lett. 23, 1929–1934 (2013).
Baraldo, M. The influence of circadian rhythms on the kinetics of drugs in humans. Expert Opin Drug Metab Toxicol. 4, 175–192 (2008).
Sallam, H. et al. The effect of circadian rhythm on pharmacokinetics and metabolism of the Cdk inhibitor, roscovitine, in tumor mice model. Chronobiol Int. 32, 608–14 (2015).
Lévi, F. & Okyar, A. Circadian clocks and drug delivery systems: impact and opportunities in chronotherapeutics. Expert Opin Drug Deliv. 8, 1535–41 (2011).
Balan, R. & Suraishkumar, G. K. UVA-Induced reset of hydroxyl radical ultradian rhythm improves temporal lipid production in Chlorella vulgaris. Biotechnol Prog. 30, 673–680 (2014).
Criddle, D. N. et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 281, 40485–40492 (2006).
Fukui, M., Choi, H. J. & Zhu, B. T. Rapid generation of mitochondrial superoxide induces mitochondrion-dependent but caspase-independent cell death in hippocampal neuronal cells that morphologically resembles necroptosis. Toxicol. Appl. Pharmacol. 262, 156–166 (2012).
Zhu, H. et al. Doxorubicin redox biology: redox cycling, topoisomerase inhibition, and oxidative stress. React Oxyg Species(Apex). 1, 189–198 (2016).
Wang, Y. et al. Distinct roles of cytochrome P450 reductase in mitomycin c redox cycling and cytotoxicity. Mol Cancer Ther. 9, 1852–1863 (2010).
Shimada, H. et al. Possible mechanism of superoxide formation through redox cycling of plumbagin in pig heart. Toxicol In Vitro. 26, 252–257 (2012).
Loor, G. et al. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic Biol Med. 49, 1925–1936 (2010).
Abdelmohsen, K., Gerber, P. A., Von Montfort, C., Sies, H. & Klotz, L. O. Epidermal growth factor receptor is a common mediator of quinone-induced signaling leading to phosphorylation of connexin-43. Role of glutathione and tyrosine phosphatases. J Biol Chem. 278, 38360–38367 (2003).
Chowdhury, R., Chowdhury, S., Roychoudhury, P., Mandal, C. & Chaudhari, K. Arsenic induced apoptosis in malignant melanoma cells is enhanced by menadione through ROS generation, p38 signaling and p53 activation. Apoptosis. 14, 108–123 (2009).
Li, Z. et al. ROS leads to MnSOD upregulation through ERK2 translocation and p53 activation in selenite-induced apoptosis of NB4 cells. FEBS Lett. 584, 2291–2297 (2010).
Aris, R. Mathematical modelling techniques. (Dover Inc., New York, 1994).
Wagner, B. A., Venkataraman, S. & Buettner, G. R. The Rate of Oxygen Utilization by Cells. Free Radic Biol Med. 51 (2011).
Fujioka, A. et al. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J. Biol. Chem. 281, 8917–8926 (2006).
Santillan, M. On the use of the Hill functions in mathematical models of gene regulatory networks. Math. Model. Nat. Phenom. 3, 85–97 (2008).
Follis, A. V. et al. The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat Struct Mol Biol. 21, 535–543 (2014).
Sem, D. S. & Kasper, C. B. Kinetic mechanism for the model reaction of NADPH-cytochrome P450 oxidoreductase with cytochrome C. Biochemistry. 25, 4438–43 (1986).
Song, Y. & Buettner, G. R. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic. Biol. Med. 49, 919–962 (2010).
Son, Y. et al. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J. Signal Transduct. 2011, 1–6 (2011).
She, Q. B., Chen, N. & Dong, Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 275, 20444–20449 (2000).
Wang, S. & Shi, X. Mechanisms of Cr (VI) -induced p53 activation: the role of phosphorylation, mdm2 and ERK. Carcinogenesis 22, 757–762 (2001).
Barcia, R., López-Borges, S., Vega, F. M. & Lazo, P. A. Kinetic properties of p53 phosphorylation by the human vaccinia-related kinase 1. Arch. Biochem. Biophys. 399, 1–5 (2002).
Shaltiel, I. A. et al. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc. Natl. Acad. Sci. 111, 7313–7318 (2014).
Takekawa, M. et al. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19, 6517–26 (2000).
Gibellini, L. et al. A. Interfering with ROS metabolism in cancer cells: the potential role of quercetin. Cancers (Basel). 2, 1288–1311 (2010).
Hussain, S. P. et al. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 64, 2350–2356 (2004).
Olovnikov, I. A., Kravchenko, J. E. & Chumakov, P. M. Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Semin Cancer Biol. 19, 32–41 (2009).
Hsu, J. L. et al. Catalytic properties of human manganese superoxide dismutase. J Biol Chem. 271, 17687–17696 (1996).
Wang, S. et al. NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J Bacteriol. 192, 5115–5123 (2010).
Halliwell, B. & Gutteridge, J. M. C. Free radicals in biology and medicine. 896p (Oxford University Press, 2015).
Hammer, Ø., Harper, D. A. T. & Ryan, P. D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron. 4, 1–9 (2001).
Refinetti, R., Lissen, G. C. & Halberg, F. Procedures for numerical analysis of circadian rhythms. Biol Rhythm Res. 38, 275–325 (2007).
Basri, D. F. et al. Mechanism of cytotoxic activity of Canarium odontophyllum Miq. (dabai) against human colon cancer cell line HCT 116. J. Chem. Pharm. Sci. 10, 109–115 (2017).
Acharya, A., Das, I., Chandhok, D. & Saha, T. Redox regulation in cancer: A double-edged sword with therapeutic potential. Oxid Med Cell Longev. 3, 23–34 (2010).
Hawk, M. A., Schafer, Z. T. & Dame, N. Redox metabolism during ECM-detachment. J Biol Chem, https://doi.org/10.1074/jbc.TM117.000260 (2018).
Navarro, R. & Busnadiego, I. Superoxide Anions Are Involved in Doxorubicin-Induced ERK Activation in Hepatocyte Cultures. Ann. N.Y. Acad. Sci. 1090, 419–428 (2006).
Chou, S., Chang, S. & Chuang, J. Expression via Mitogen-Activated Protein Kinases in Corneal Fibroblasts. Invest Ophthalmol Vis Sci. 48, 2009–2016 (2007).
Xu, Y., Porntadavity, S. & St Clair, D. Transcriptional regulation of the human manganese superoxide dismutase gene: the role of specificity protein 1 (Sp1) and activating protein-2 (AP-2). Biochem. J. 362, 401–412 (2002).
Ham, S. W., Jin, S. & Song, J. H. Mechanism of cell growth inhibition by menadione. Bull. Korean Chem. Soc. 23, 1371–1372 (2002).
Ham, S. W., Song, J. H., Kim, H. I. & Jin, S. Mechanism of cell cycle arrest by menadione. Bull. Korean Chem. Soc. 21, 1173–1174 (2000).
Lee, Y. R. & Park, S. Y. P53 expression in hepatocellular carcinoma: influence on the radiotherapeutic response of the hepatocellular carcinoma. Clin Mol Hepatol. 21, 230–231 (2015).
Thacker, P. & Karunagaran, D. Emodin suppresses Wnt signaling in human colorectal cancer cells SW480 and SW620. Eur. J. Pharmacol. 742, 55–64 (2014).
Suresh, S., Raghu, D. & Karunagaran, D. Menadione (Vitamin K3) induces apoptosis of human oral cancer cells and reduces their metastatic potential by modulating the expression of epithelial to mesenchymal transition markers and inhibiting migration. Asian Pac J Cancer Prev. 14, 5461–5465 (2013).
Balsalobre, A., Damiola, F. & Schibler, U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 93, 929–37 (1998).
Nazarewicz, R. R., Bikineyeva, A. & Dikalov, S. I. Rapid and specific measurements of superoxide using fluorescence spectroscopy. J Biomol Screen. 18, 498–503 (2013).
Galbusera, C., Orth, P., Fedida, D. & Spector, T. Superoxide radical production by allopurinol and xanthine oxidase. Biochem. Pharmacol. 71, 1747–1752 (2006).
Weydert, C. J. & Cullen, J. J. Measurement of superoxide dismutase, catalase and glutathione peroxidase in cultured cells and tissue. Nat. Protoc. 5, 51–66 (2010).
Acknowledgements
The authors would like to thank Department of Science and Technology, Government of India (Grant Number: EMR/2016/002069), for financial assistance; Dr. Bert Vogelstein, Johns Hopkins University, Baltimore, USA for both the variants of the cell line; and Dr. Ghanshyam Swarup, Centre for Cellular & Molecular Biology, Hyderabad, India, for the pG13 luciferase vector with p21 promoter construct.
Author information
Authors and Affiliations
Contributions
U.K., D.K. and G.K.S. conceived and designed the study. U.K. and P.S. conducted the experiments and processed the data. U.K. and M.V.P. coded and optimized the model and prepared the manuscript. R.R. refined the mathematical model. G.K.S. refined the interpretation and provided overall direction for the study. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kizhuveetil, U., Palukuri, M.V., Sharma, P. et al. Entrainment of superoxide rhythm by menadione in HCT116 colon cancer cells. Sci Rep 9, 3347 (2019). https://doi.org/10.1038/s41598-019-40017-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40017-7
This article is cited by
-
Live-cell imaging of circadian clock protein dynamics in CRISPR-generated knock-in cells
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
