
Abstract
Litchi downy blight, a destructive litchi disease caused by Peronophythora litchii, is controlled by intensive fungicide applying. Sources of resistance are used in conventional breeding approaches, but the mechanism is not well understood. Follow-up six years investigation, ‘Guiwei’ and ‘Heiye’ displayed stable susceptible and resistant against to P. litchii, respectively. After 72 hour inoculation, ‘Heiye’ showed few disease spots, while ‘Guiwei’ appeared brown and covered with white sporangia. Germination of sporangia and growth of mycelium in ‘Guiwei’ is more quickly than in ‘Heiye’. Transcript levels were measured at 6, 24, and 48 hour post-inoculation. ‘Oxidation-reduction process’ was dramatically enhanced in ‘Heiye’, which could promote its resistance to pathogen infection. A small ratio (3.78%) of common DEGs indicates that resistant and susceptible cultivars take different strategies to defense against P. litchii. At early infection stage, ‘Heiye’ induced a larger number of genes, including seven receptor-like kinases, which quickly recognized attack of pathogen and led to a rapidly resistance by regulation of degradation of proteasome, transcription factors, and cell wall remodeling. The early DGEs were exiguous in ‘Guiwei’, suggesting a weak response. Once the infection was successful, the resistance was repressed by down-regulated genes involved in phenylpropanoid metabolism, ET biosynthesis and signaling conduction in ‘Guiwei’. In conclusion, quickly recognition and early responses to pathogen, as well as minimal pathogen development and basal expression of resistance-related genes, were correlated with a high level of resistance in ‘Heiye’, while susceptible ‘Guiwei’ suffered massive infection due to lagging response and repressed signal transduction.
Similar content being viewed by others


Introduction
Litchi (Litchi chinensis Sonn.), a subtropical evergreen fruit tree of family Sapindaceae, originate from South China and has been widely cultivated in more than 20 countries due to delicious and nutritional value1. The planting area of litchi is approximately 0.59 million hectares in South China, of which yield annually 1.91 million tons fresh fruits. However, the fruits are highly susceptible to various diseases. Litchi downy blight, caused by Peronophythora litchii, is one of the major diseases in litchi2. P. litchii is an oomycete pathogen and exclusively infects litchi. The mycelium and oospore of P. litchii attacks fruits and results in watery brown spots2,3. The fungicides including carboxyl acid amide, mancozeb, cymoxanil, and metalaxyl have been extensively used to control litchi downy blight4. After a long time applying, the resistant isolates of P. litchii have been detected in some regions5. Considering both drug-resistance and environmental impact due to the fungicide applying, breeding of resistant cultivar to control the disease are required.
Detailed resistance mechanisms of plant have been described in a few model species. Plant responses to pathogens depend on recognition of microorganisms and induction of defense responses by downstream signal transduction. Pathogen invasion is firstly detected by the recognition of highly conserved pathogen-associated molecular patterns (PAMPs), leading to a basal resistance called PAMP-triggered immunity (PTI)6. The recognition of PAMPs is by cell membrane embedded pattern-recognition receptors (PRRs), which are either receptor-like kinases (RLKs) or receptor-like proteins (RLPs). PTI can be overcome by injecting type III effectors (T3Es) of pathogen into the host cells7. Recognition of T3Es is known as effector-triggered immunity (ETI)7. ETI can be accompanied by hypersensitive reaction (HR), a form of programmed cell death at the infection site that prevents pathogen spreading8. The HR thus triggers systemic acquired resistance (SAR) by salicylic acid (SA)-mediated defenses and confers broad-spectrum immunity to secondary infection9. Plant hormones are crucial systemic signals that strongly influence the level of plant resistance. SA, jasmonic acid (JA), and ethylene (ET) play vital roles in resistance to biotrophic and necrotrophic pathogens. The activation of hormone signaling will induce defense response, including stress responses, oxide-reduction processes, cell wall and wax biosynthesis processes, and pathogenesis-related proteins (PR)10. In addition, plant resistance is mediated by the proteolysis. The response of defense-related hormone, such as jasmonate, auxin, and abscisic acid signaling, are proteasome-dependent processes11. Receptor associated proteasomic degradation may be active in positive-regulation as well as negative-regulation12. Regulation of transcription factor involved in defense gene can also be regulated through the proteasome system13,14. Moreover, phytopathogenic pathogens can manipulate the proteasomal system for suppresses the PTI of host15,16.
At present, studies about litchi downy blight have mostly concentrated on biological characteristics of pathogen, chemical control and screening of resistant cultivars in litchi17,18. Little is known about the disease resistance-relevant genes involved in the interaction between host and pathogen, which requires exploration into the resistance mechanisms against P. litchii. In this paper, the disease process of litchi downy blight was firstly investigated in two litchi cultivars (‘Heiye’ and ‘Guiwei’) by Scanning Electron Microscopy (SEM). And then, transcriptome sequencing was performed via de novo RNA-seq technology at three stages (6, 24, and 48 hour post inoculation). Differential gene expression was conducted to identify the disease resistance-relevant genes and metabolic pathways involved in P. litchii infection. These genes and pathways will provide a theoretical basis for expounding the resistance mechanism against litchi downy blight in litchi.
Results
Determination of the resistance of litchi cultivars
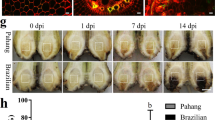
After inoculated with P. litchii, the disease severity of two cultivars was assessed according the disease index (DI) in the greenhouse (Fig. 1). A few white spots both appeared in the exocarp of two cultivars at 48 hour post inoculation (hpi) (Fig. 1a). Since then, the progress of disease accelerated. The disease severity was significantly different between two cultivars at 72 and 96 hpi (Fig. 1a). Disease resistance of two cultivars was determined by the DI at 72 hpi. The infected fruits of ‘Heiye’ showed small spots, while ‘Guiwei’ covered with masses of downy white sporangia and sporangiophores (Fig. 1b). Moreover, six years follow-up survey displayed that the DI of ‘Heiye’ were 15.8–33.0 and the DI of ‘Guiwei’ were 51.4–64.8 (Fig. 1c). The results indicate that the ‘Heiye’ is a resistant cultivar, whereas ‘Guiwei’ is a susceptible cultivar.

Diseasse symptom and disease index between two cultivars. (a) Disease index increase after inoculation with P. litchii in 2017, (b) Disease symptom of two cultivars inoculated or mocked-inoculated at 72 hpi, (c) Disease index of two cultiavrs across six years.
Microscope observation on pathogen infection
The infection process of P. litchii in two cultivars with inoculation was monitored by SEM method. The spores were both observed in the exocarp of two cultivars after inoculation with P. litchii (Fig. 2a,g). At the 6 hpi, germination of sporangia was observed in ‘Guiwei’ but not in ‘Heiye’ (Fig. 2b,h). At the 24 hpi, the sporangia extend out of the mycelium and grow along cracked lobe valley in ‘Guiwei’, while the sporangia just started to germinate in ‘Heiye’ (Fig. 2c,i). At the 48 hpi, a lot of mycelium were formed in ‘Guiwei’, while the same thing was observed in ‘Heiye’ until 72 hpi, and a few mycelia grow along the surface of exocarps in ‘Heiye’ (Fig. 2d,j,k). At the 72 hpi, the mycelium reproduced a few sporangia in the exocarp of ‘Guiwei’ (Fig. 2e). At the 96 hpi, much more sporangia were formed in ‘Guiwei’, while only a few reproduced sporangia were observed in ‘Heiye’ (Fig. 2f,l). Microscopic observations revealed that cortical infection was both present in ‘Heiye’ and ‘Guiwei’ at 6, 24 and 48 hpi (Fig. 2). For this reason, these time-points (6, 24, and 48 hpi) were selected to investigate the differential transcript changes among cultivars and inoculation.

Microscope observation of infection of P. litchii between two cultivars at six time-points. (a,g,h) The inoculated spores, (b,i) the germinated sporangia, (c,d,j,k) the mycelium, (e,f,l) the new sporangia.
RNA Sequencing and De novo Assembly
Approximately 11.93–17.35 million 125-bp paired-end reads were generated from the 36 samples through RNA sequencing (Table S2). The GC content of the sequence data from the 36 libraries ranged from 46.20 to 50.50%, and the Q30 values (reads with an average quality scores >30) were all ∼90%, indicating that the quality and accuracy of sequencing data was sufficient for further analysis. After sequence trimming and redundance removed, the retained high-quality reads of all samples were de novo assembled into 32614 unigenes as reference transcripts. The N50 of the assembled genes was 1466 bp, with an average length of 1110 bp and a maximum length of 11441 bp (Table 1). The percentage of sequenced reads from all libraries remapped to the assembled reference transcripts was ∼70% (Table S2). The 30006 of unigenes were functionally annotated in at least one database with an e-value cutoff of 1e−5 (Table 1, Table S3). The sequencing data has been deposited into NCBI sequence read archive (SRA) under BioProject accession PRJNA450886.
Inter-genotypes differences in basal gene expression
On the plot of Principal Component Analysis (PCA), shorter distance between two points indicates greater similarity of gene expression profiles between two samples. PCA showed that all samples of ‘Heiye’ clearly stratify away from those of ‘Guiwei’ in the first principal component, indicating large differences in the gene transcription patterns of these two cultivars. The second principal components show a further separation between 6 hpi and the others (24 and 48 hpi) in ‘Guiwei’, in which the inoculation showed no-significant changes from mock-inoculation (Fig. S1).
The uninoculated ‘Heiye’ and ‘Guiwei’ were further compared to analyze the basal gene expression pattern (Figs 3a, S2). Three pairwise comparisons of different inoculation times (HC_vs_GC_6hpi, HC_vs_GC_24hpi, and HC_vs_GC_48hpi) were generated to identify differential expression genes (DEGs) between two cultivars. Differential expression analysis revealed a total of 9081, 8811, 8784 DEGs identified respectively at 6, 24, and 48 hpi (Figs 3a, S2). The total numbers of DEGs was decreased slightly with time past. There were 12323 non-redundant DEGs and 5950 common DEGs in three time-points (Figs 3a, S2). Moreover, among all DEGs, 3997 unigenes had a much higher expression value in resistant cultivar, and a total of 1925 unigenes had a much higher expression value in susceptible cultivar (Fig. S2).

Venn diagrams showing the overlap of the differentially expressed genes (DEGs; fold change; | FC | ≥ 2). (a) The DEGs (HC; HC_vs_GC) between ‘Heiye’ and ‘Guiwei’ with mock-inoculation; (b) the DEGs (HI; HI_vs_HC) between inoculation with P. litchii and mock-inoculation in ‘Heiye’; (c) the DEGs (GI; GI_vs_GC) between inoculation with P. litchii and mock-inoculation in ‘Guiwei’; (d,f) the DEGs were up-regulated (Up) and down-regulated (Dw) at 6, 24, and 48 hpi in ‘Heiye’ (HI) and ‘Guiwei’ (GI) inoculated with P. litchii.
As inter-genotype differences may reflect the mechanisms of disease resistance and susceptibility, we also classified these DEGs according to functional categories. Following the Nr annotations, the DEGs were mapped into three Gene ontology (GO)19 categories, of which fifteen sub-categories were significantly enriched (corrected P-value ≤ 0.01) in biological process (Table S4). DEGs involved in ‘Oxidation-reduction process’ made up the largest groups, and the up-regulated DEGs were also enriched in this GO term (Table S4). The ‘Defense response’, ‘Response to stress’, and ‘Chitin metabolic process’ were both enriched in total and down-regulated DEGs analysis (Table S4).
Moreover, Kyoto Encyclopedia of Genes and Genomes (KEGG)20 analyses were performed to identify the basal level biological pathways in litchi pericarp. The 1145 out of 5922 DEGs had KEGG Orthology (KO) IDs and could be categorized into 133 pathways (data not show). A total of 7 pathways were significantly enriched (corrected P-value ≤ 0.05). Genes involved in ‘Plant-pathogen interaction’, ‘alpha-Linolenic acid metabolism’, ‘Cysteine and methionine metabolism’, ‘Brassinosteroid biosynthesis’, ‘Indole alkaloid biosynthesis’, ‘Anthocyanin biosynthesis’, and ‘Tyrosine metabolism’ were the most represented DEGs (Supplementary Table S5).
In the present study, ‘plant-pathogen interaction’ pathway was significantly enriched and exhibited the most significantly different expression levels between the resistant and susceptible cultivars. 140 DEGs were found in ‘plant-pathogen interaction’ pathway, of which 80 and 60 were respectively up- and down-regulated (Fig. 4, Tables S5–6. Among them, sixty-eight disease resistance protein RPS2 (Resistance to Pseudomonas. syringae 2) made up the largest group (Fig. 4, Table S6). Another eight disease resistance proteins RPM1 (Resistance to Pseudomonas syringae pv maculicola 1) were also differential expression between two cultivars (Fig. 4, Table S6). One RIN4 (RPM1-interacting protein 4) associating with both RPS2 and RPM1 was up-regulated. One PR1 was also up-regulated (Fig. 4, Table S6). The second large group was CNGC (cyclic nucleotide gated channel) including eighteen members, and fifteen transcription factor MYB were made up the third large groups. The number of up- and down-regulated genes were comparable in these two groups (Fig. 4, Table S6). Three sub-families of Ca2+ sensors including CALM (calmodulins), CDPK (calcium dependent protein kinases) and CML (calcium-binding protein) were identified (Fig. 4, Table S6). Two CALMs, one CDPK, and six of seven CMLs were up-regulated, while only one CDPK and one CMLs were down-regulated. One MEKK1 (mitogen-activated protein kinase kinase kinase 1), one NOS1 (nitric-oxide synthase), and one HCD1 (very-long-chain (3R)-3-hydroxyacyl-CoA dehydratase) were down-regulated. the rest six groups, which include four CTSF (cathepsin), three CERK1 (chitin elicitor receptor kinase 1), one WRKY22, one FLS2 (flagellin-sensitive 2), one EDS1 (enhanced disease susceptibility 1 protein), and one BAK1 (brassinosteroid insensitive 1-associated receptor kinase 1), were just highly expressed in ‘Heiye’ (Fig. 4, Table S6).

The diagram of the ‘Plant-pathogen interaction’ between ‘Heiye’ and ‘Guiwei’. The color rectangles represent trend of genes, in which the number indicate the amount of differentiated gene. Green represent down-regulated in ‘Heiye’, red represent up-regulated in ‘Heiye’, blue represent that both down- and up-regulated members of gene family were found in ‘Heiye’, gray represent no DEG between two cultivars.
Differentially Expressed Genes after inoculation
To find genes that were induced by P. litchii, pairwise comparisons were made between inoculation with P. litchii and mock-inoculation at the same time-point in each cultivar. A total of 1717 DEGs were identified, of which 1445 and 369 were regulated in ‘Guiwei’ and ‘Heiye’ respectively (Figs 3b,c and 5). Notably, when all DEGs of ‘Heiye’ were compared to those of ‘Guiwei’, only 97 unigenes were both regulated in two cultivars (Fig. 3d,e,f). No common DEGs was identified in two cultivars at 6 hpi (Fig. 3d). Five common DEGs were identified at 24 hpi, of which four were both up-regulated, and one was down-regulated (Fig. 3e). Sixty-four common DEGs were identified at 48 hpi, of which twenty-two were up-regulated, and thirty-eight were down-regulated, and three were up-regulated in ‘Heiye’ but down-regulated in ‘Guiwei’, and one was down-regulated in ‘Heiye’ but up-regulated in ‘Guiwei’ (Fig. 3f). At 24 hpi, there were four DEGs (c36949_c0_g1, c8306_c0_g2, c36893_c0_g1, and c13847_c0_g1) up-regulated in two cultivars. The c36949_c0_g1 and c8306_c0_g2 were encoded EXL2 (EXORDIUM-like 2 protein). The c36893_c0_g1 was encoded an AOC (allene oxide cyclase). The function of c13847_c0_g1 was unknown. One gene (c5397_c0_g3) was down-regulated in two cultivars at 24 hpi, which function was unknown (Table S7).
DEGs response to P. litchii in susceptible cultivar
Thirteen (0.90%), 753 (52.11%), and 1045 (72.32%) DEGs were identified in ‘Guiwei’ at 6, 24 and 48 hpi, respectively. Only 8 (61.54%), 411 (54.58%), and 345 (33.01%) were up-regulated (Figs 3c and 5). There were six DEGs induced across three time-points, of which one (c20888_c0_g1) encoded (+)-neomenthol dehydrogenase (SDR1), one (c3313_c0_g1) encoded LRR receptor-like serine/threonine-protein kinase (LRR-RLK, GSO2), two (c22648_c0_g1 and c22646_c0_g1) encoded galactinol synthase 2 (GolS2), one (c44404_c0_g1) encoded galactinol sucrose galactosyltransferase 5, and one (c3790_c0_g1) encoded stachyose synthase (Figs 3c, S3). The two of former were up-regulated at 6 hpi, and down-regulated at 24 hpi, and up-regulated at 48 hpi (Fig. S3). The four of latter were down-regulated at 6 and 48 hpi, and up-regulated at 24 hpi (Fig. S3).

The number of DEGs comparison inoculation with mock-inoculation in two cultivars at three time-points. Green represent down-regulated after inoculation, red represent up-regulated after inoculation. The upper number indicate the amount of down-regulated DEGs, the lower number indicate the amount of up-regulated DEGs.
KEEG enrichment show that twenty-one pathways were significant difference after inoculation with P. litchii in ‘Guiwei’ (Fig. 6, Table S8). The ‘Galactose metabolism’ (ko00052) was down-regulated enriched at 6 hpi but up-regulated at 24 hpi (Fig. 6, Table S8). The major DEGs involved in ‘MAPK signaling pathway - plant’ (ko04014) and ‘Plant hormone signal transduction’ (ko04075) were down-regulated consistently at 24 and 48 hpi (Fig. 6, Tables S8, S9, S10). The ‘Monoterpenoid biosynthesis’ (ko00902) was up-regulated enriched at 6 and 24 hpi but down-regulated at 48 hpi (Fig. 6, Table S8). The major DEGs involved in ‘Photosynthesis’ (ko00195), ‘Carbon fixation in photosynthetic organisms’ (ko00710), and ‘Diterpenoid biosynthesis’ (ko00904) were up-regulated at 24 hpi but down-regulated 48 hpi (Fig. 6, Table S8). While ‘Phenylalanine metabolism’ (ko00360), ‘Selenocompound metabolism’ (ko00450), ‘Zeatin biosynthesis’ (ko00908), and ‘Phenylpropanoid biosynthesis’ were down-regulated at 24 hpi but up-regulated at 48 hpi (Fig. 6, Table S8). The major DGEs involved in ‘Cysteine and methionine metabolism’ (ko00270) and ‘alpha-Linolenic acid metabolism’ (ko00592) were down-regulated at 24 hpi (Fig. 6, Table S8). Additionally, nineteen of thirty-four DEGs involved ‘Plant-pathogen interaction’ (ko04626) were also down-regulated at 48 hpi (Fig. 6, Table S8).

The significantly enriched KEGG pathways of DEGs respond to P. litchii infection in two cultivars. Area of circle represent the number of DEGs in related pathways. Green represent down-regulated, red represent up-regulated, blue represent that both down- and up-regulated members of pathway were found after inoculation.
The ‘MAPK signaling pathway - plant’ was the most enrichment pathway response to P. litchii in ‘Guiwei’ at 24 hpi (Figs 6, S4, Table S8). Twenty-one DEGs were identified in this pathway at 24 hpi, of which 16 and 5 were down- and up-regulated respectively (Fig. 6, Table S8), whereas twenty-one DEGs in 48 hpi, while 11 and 10 were respectively down- and up-regulated (Fig. 6, Table S8). Among them, one NDK (nucleoside-diphosphate kinase) and five PP2Cs (protein phosphatase 2C) were up-regulated, while the rest of fifteen DEGs, three FLS2s, two RBOHs (respiratory burst oxidase), one PR1, one PYL (abscisic acid receptor), three ETRs (ethylene receptor), three EBF1_2s (EIN3-binding factor), and two ERF1s (ethylene-responsive transcription factor 1), were down-regulated (Figs S4–6, Table S9). The ‘Plant hormone signal transduction’ and ‘Phenylpropanoid biosynthesis’ both contained nineteen DEGs and enriched significantly (Figs S6–8, Tables S8, 10–11). In ‘Plant hormone signal transduction’, except the DEGs mentioned in ‘MAPK signaling pathway-plant’, one ARF (auxin response factor), one TCH4 (xyloglucan:xyloglucosyl transferase), and one NPR1 (non-expressor of pathogenesis-related genes 1) were up-regulated and one BSK (BR-signaling kinase) was down-regulated (Fig. S6, Table S10). In ‘Phenylproanoid biosynthesis’ pathway, except one COMT (caffeic acid 3-O-methyltransferase) and one POD (peroxidase) were up-regulated, the rest of seventeen DEGs, such as three PALs (phenylalanine ammonia-lyase), one HST (shikimate O-hydroxycinnamoyltransferase), one CCoAOMT (caffeoyl-CoA O-methyltransferase), one CSE (caffeoylshikimate esterase), and one 4CL (4-coumarate–CoA ligase), were down-regulated at 24 hpi (Figs S7–8, Table S11). Interestingly, the profile of phenylpropanoid metabolism, ET related genes, and PP2Cs at 24 hpi was reversed at 48 hpi (Tables S6–7).
DEGs response to P. litchii in resistant cultivar
‘Heiye’ inoculated with P. litchii showed 57 (15.45%), 139 (37.67%), and 183 (49.59%) DEGs at 6, 24 and 48 hpi, respectively; among them 44 (77.19%), 46 (33.09%), and 80 (43.72%) were up-regulated (Figs 3b and 5). The results showed that the remarkable changes in the transcriptome profile occurred at 6 hpi in ‘Heiye’. The number of DEGs at 6 hpi in ‘Heiye’ was markedly more than those in other two time-points. One DEGs (c13031_c0_g1) encoding a receptor-like kinase EP1 with mannose-binding domain was regulated at three time-points, which was up-regulated (FC = 2.28) at 6 hpi, then down-regulated (FC = −1.58) at 24 hpi, and up-regulated (FC = 1.60) at 48 hpi (Fig. S3).
As a KEEG enrichment of DEGs response to P. litchii in ‘Heiye’, only six pathways showed significant difference (corrected P-value ≤ 0.01) (Fig. 6, Table S8). The up-regulated DEG was enriched in ‘Amino sugar and nucleotide sugar metabolism’ pathway at 6 hpi, and the down-regulated DEGs in this pathway enriched at 24 hpi (Table S8). In fact, the chitinases (all five at 6 hpi and seven out of eight at 24 hpi) made up the most of DEGs in ‘Amino sugar and nucleotide sugar metabolism’ pathway. At the 48 hpi, ‘Photosynthesis’ and ‘Diterpenoid biosynthesis’ were down-regulated, and ‘Selenocompound metabolism’ was up-regulated (Fig. 6, Table S8).
To obtain an overview of the process respond to infection of P. litchii during early stages, the DEGs were further analysis in ‘Heiye’ at 6 hpi. According profile of expression and function, the 44 of 57 DEGs with annotation were classified into five groups (Fig. 7). Except the group 1 was down-regulated, the other groups were all up-regulated. The group 1 were made up with five DEGs including a GolS (c32732_c0_g1), SAMT (salicylate carboxymethyltransferase, c626_c0_g1), and 3-ketoacyl-CoA synthase 1 (KCS1, c32732_c0_g1). The group 2 were consisted with seven RLKs (c1814_c0_g3, c22078_c0_g2, c21337_c0_g1, c17471_c0_g2, c13031_c0_g1, c13040_c0_g1, and c41194_c0_g1). The group 3 were consisted with seven protease proteins, including one E3 ubiquitin-protein ligase (c15582_c0_g2), two U-box domain-containing protein (c28190_c0_g2, c27506_c0_g2, and c14430_c0_g1), two Subtilisin-like protease (SBTs, c16100_c0_g1 and c11051_c1_g1), and one NEDD8-specific protease (c30629_c0_g1). The group 4 were transcription factor including three cyclic dof factor (c17755_c0_g1, c41186_c0_g1, and c672_c0_g1), one bHLH100 (c43842_c0_g1), and one transcription repressor OFP4 (c2512_c0_g1), and one zinc finger protein CONSTANS-LIKE 2 (c21414_c0_g1). The group 5 were the rest such as five chitinases (c38647_c0_g1, c16554_c0_g1, c37038_c0_g1, c30435_c0_g2, and c38648_c0_g1), and one cysteine-rich repeat secretory protein 38 (c15826_c0_g1), expansin-A4 (c44531_c0_g1), Glucomannan 4-beta-mannosyltransferase 2 (CSLA2, c33025_c1_g1), Glucan 1,3-beta-glucosidase (c31763_c0_g1), Feruloyl CoA ortho-hydroxylase 2 (F6’H2, c43692_c0_g1), L-ascorbate oxidase (c26250_c0_g1), and galactosidase (c33001_c0_g1) etc.

The DEGs respond to infection of P. litchii in ‘Heiye’ at early stage. The forty-four DEGs with annotation divide into five groups. I, down-regulated DEGs, II, receptor-like kinases, III, proteases, IV, transcription factors, V, other up-regulated DEGs.
Validation of transcripts by real-time PCR
To validate the expression profiling by RNA-seq, the expression levels of six genes, including two peroxidase genes, one phenylalanine ammonia-lyase gene, one beta-glucosidase gene, one caffeoyl-CoA O-methyltransferase gene, and one feruloyl-CoA ortho-hydroxylase gene, were further analysed by qRT-PCR (Fig. S9, Table S12). All the genes showed differential expression levels between ‘Guiwei’ and ‘Heiye’. Correlation analysis was performed between FPKM by RNA-seq and relative expression by qRT-PCR for each gene. The Pearson’s correlation coefficient between the data generated from the two platforms was high (R2 value in range of 0.75 to 0.97), indicating that the RNA-seq approach provided reliable differential gene expression information (Fig. S9).
Discussion
In the present study, the distinct colonization of P. litchii on litchi pericarps was first revealed by SEM over a time series. In susceptible cultivar, P. litchii was directly penetrated the exocarps and established a primary restricted infection before 6 hpi (contact phase and penetration phase). Subsequently, the pathogen initiated a massive outgrowth before 72 hpi (incubation phase) and formed a lot of sporangium until 96 hpi (symptom appearance phase). Conversely, penetration of P. litchii on resistant cultivar was not detected at 6 hpi (contact phase) and showed a substantial delay until 24 hpi (penetration phase). This lagging of infection resulted in markedly lower germination and infection rates in resistant cultivar than susceptible cultivar at the following stages. Additionally, the sporangium was also formed but much smaller in resistant than susceptible cultivar at 96 hpi (symptom appearance phase). The resistant cultivar is characterized as a long contact phase and a short incubation phase (24–72 hpi) with a less DI. It indicates that the germination of sporangia and the growth of mycelium in susceptible cultivar is more quickly than in resistant cultivar. It should be noted that the inoculation pore suspension (1 × 104 spores·mL−1) of P. litchii were higher content than the spores in the fields. The high spore content can accelerate the progress of disease under lab condition. In fact, the downy blight was less happened in resistant cultivar ‘Heiye’ than in susceptible cultivar ‘Guiwei’ in fields investigation (data not showed). Therefore, it seems that the disease is obstructed through blocked colonization of P. litchii on resistant cultivar during early infection stages.
There was a small ratio (3.78%, 65/1717) of common DEGs respond to infection of P. litchii between two cultivars. Especially, there is no common DEGs at early infection stage (6 hpi). It indicates that resistant and susceptible cultivar take different strategies to defense against P. litchii at contact phase. At 24 hpi, the infection was both complete in ‘Guiwei’ and ‘Heiye’. In this study, two EXL2 and one AOC were both up-regulated response to P. litchii attack in two cultivars at 24 hpi. EXL2 play a role in a brassinosteroid (BR)-dependent regulation. BR can stimulate photosynthesis and cell expansion in plant. One EXL2 is highly expression and involved in cell wall remodeling in litchi21. In Arabidopsis, EXL1, homolog of EXL2, is involved in a primary response to energy deprivation and reduced BR-dependent growth22. AOC is involved in the production of 12-oxo-phytodienoic acid, a precursor of jasmonic acid. It suggests that the BR and JA take part in response to infection of P. litchii in litchi. There were sixty common DEGs between two cultivars at post-incubation phase (48 hpi). Four methyltransferases and two dehydratases were up-regulated, while seventeen genes related to chloroplast were down-regulated. It suggests that the cell wall of litchi was degrade by activated methyltransferase activity and photo reaction were repress at post-incubation phase.
After establishing that ‘Heiye’ could effectively block P. litchii and that ‘Guiwei’ was a favorable host, the underlying possible mechanisms of resistance in ‘Heiye’ and susceptibility in ‘Guiwei’ were investigated. PCA analysis indicated that the major difference between two cultivars attributed to genotype variation. Further analysis showed that the ‘plant-pathogen interaction’ pathway was significantly enriched as an inter-genotype difference. Plant defense responses are divided into two categories: PTI and ETI7. PTI is triggered by the recognition of generally conserved elicitors such as chitin and flagellins23. FLS2 is an RLK recognized flagellin of bacterial and acts as a key regulator of PTI triggered by diverse PAMPs. BAK1, a ligand-independent coreceptor of RLK, and CERK1 are also a central regulator of PTI. In agreement with this condition, One FLS2, one BAK1, and three CERK1 genes were higher expression in resistant cultivar than in susceptible cultivar. The downstream signaling activates one WRKY22, which driving highly expression of PR1. Furthermore, eight genes involved in Ca2+ signaling pathway, which of two CALMs and six of seven CMLs, were up-regulated in ‘Heiye’. It suggests that high FLS2 activity together with BAK1, CERK1, WRKY22 and Ca2+ signaling may contribute to PTI in basal defense in resistant cultivar.
Host resistance can regulate through the reactive oxygen species (ROS). CERK1, as well as GNGCs, could activated ROS signaling pathway. In our study, eighteen of CNGCs were differentially expressed between two cultivars. The ‘Oxidation-reduction process’ was dramatically enhanced in ‘Heiye’, which was consistent with the responses of wheat resistance against Heterodera avenae24. It seems that the reliably high level of ROS produced by basal gene expression in ‘Heiye’ could, at least in part, promote its resistance to pathogen infection, while the weak ROS in ‘Guiwei’ may contribute to its susceptibility. On other hand, CNGCs and ETI induced the hypersensitive response to block the pathogen attack. A large of ETI related genes, such as RIN4, MYB, EDS1, RPM1, and RPS2, were differential expressed between two cultivars. It’s important to note that one EDS1 was highly expressing in ‘Heiye’. EDS1 acts redundantly with salicylic acid and positive regulate both basal resistance and effector-triggered immunity25,26. The differential expression of ETI-related genes may also contribute to basal defense in resistant cultivar.
The pathogen infected successfully exocarp of ‘Guiwei’ at 6 hpi. At this time, only a few DEGs responding to infection of P. litchii were detected, indicating that response to infection of P. litchii is feeble in susceptible cultivar. Six DEGs expressed differential at three point-times. Among them, one SDR1 encoding (+)-neomenthol dehydrogenase and one GSO2 encoding LRR-RLK were up-regulated. SDR1 not only catalyze a menthone reduction to produce neomenthol but also possess activity for neomenthol oxidation. AtSDR1 positively regulate defenses against a broad spectrum of pathogens in Arabidopsis27. GSO2 can positively regulate cell proliferation by intercellular signaling28. Moreover, as an LRR-RLKs, GSO2 may have a function to recognize attack of pathogen. These two genes reflect feedback to infection of P. litchii in susceptible cultivar. Whereas another four genes, which were involved in synthesis of raffinose family of oligosaccharides (RFOs), were all down-regulated. RFOs are currently emerging as crucial molecules during stress response in plants. Biosynthesis of RFOs begin with the activity of galactinol synthase (GolS). The GolS1-overexpressing transgenic tobacco plants enhanced resistance against the pathogens Botrytis cinerea and Erwinia carotovora29. Further study demonstrated that galactinol and RFOs act as signaling component for induced systemic resistance against B. cinerea infection in Arabidopsis30. It suggests that the successful infection of P. litchii disturb the resistant action by down-regulated RFOs related genes at penetration phase in susceptible cultivar.
At 24 hpi, most genes related to ET biosynthesis and signaling conduction were suppressed in ‘Guiwei’. ET plays diverse roles in plant defense response9. One SAM and two ACO genes, which involved in the ethylene biosynthesis, were strongly down-regulated in ‘Guiwei’ after infection of P. litchii. Not only the synthesis of ET was inhibited but also the ethylene signaling was repressed. Three ETRs were down-regulated. ETRs percept the ET and act as regulator of ethylene signaling. ERFs are the major downstream regulatory factors of ET signaling pathway in stress-responses. The expression of ERF1 was induced by the transcription factor EIN3 (ETHYLENE INSENSITIVE3)31. EIN3-binding F-box protein (EBF1_2) may mediate E3 ubiquitin ligase complexes and subsequent degradation of target proteins. Three EBF1_2s and two ERF1 genes were down-regulated upon P. litchii attack at 24 hpi. Moreover, one PR-1 gene mediated by ERF1 was negatively-regulated. On the other hand, phenylpropanoid metabolism, which is central to secondary metabolite production of defense-related compounds, was inhibited. The phenylpropanoid genes in key lignin formation, such as PAL, HST, CCoAOMT, CSE, 4CL, POD, were mostly repressed in susceptible cultivar at 24 hpi. This change not only reduced the defense-related compounds but also effected the content of related hormones. In plants such as poplar, SA synthesis mainly takes place through phenylalanine ammonia-lyase (PAL)-dependent phenylpropanoid pathway32. In our study, three PAL-encoding genes were down-regulated in ‘Guiwei’ at 24 hpi. It suggests that infection of P. litchii repress the resistant action by down-regulated phenylpropanoid metabolism related genes, ET-related biosynthesis and signaling conduction at early stage of incubation phase in susceptible cultivar.
ABA is known to stimulate responses to several abiotic stresses (drought, salt and cold) as well as seed germination and plant growth33. The application of exogeneous ABA increases the susceptibility to pathogens. Additionally, ABA-deficient mutants showed a reduction in susceptibility to Hyaloperonospora parasitica in Arabidopsis34. Collectively, ABA behaves as a negative regulator of defence responses. ABA binding to PYR/PYL/RCAR intracellular receptors leads to inhibition of PP2Cs, causing the activation of the ABA signaling pathway. There were one down-regulated PYL and five up-regulated PP2Cs in ‘Guiwei’ after inoculation, indicating that the ABA mediated susceptibility was repressed. ABA deficiency activates NPR1-dependent resistance to Psm ES4326 in Arabidopsis35. The transcription coactivator NPR1 is a master regulator of basal and systemic acquired resistance in plants. Overexpression of NPR1 in Brassica juncea enhanced resistance to Alternaria brassicae and Erysiphe cruciferarum36. One NPR1 were up-regulated in ‘Guiwei’ after infection of P. litchii. NPR1 regulates positively the majority of salicylic acid (SA)-dependent signaling pathway and negatively regulates JA-dependent signaling pathway. It suggests that infection of P. litchii stimulate the resistant action by up-regulated NPR1 and PP2Cs at early stage of incubation phase in susceptible cultivar.
Interestingly, the profile was reversed, which the four RFOs related genes were up-regulated whereas the GSO2 and SDR1 were down-regulated in ‘Guiwei’ at 24 hpi. Similarly, the profile of phenylpropanoid metabolism, ET related genes, and PP2Cs at 24 hpi was reversed at 48 hpi. The promotion and repression of resistance both exist at the same time and fluctuated at the next time in susceptible cultivar. It speculates that the successful infection of P. litchii in susceptible cultivar at penetration phase (6 hpi) restrain the resistance by down-regulated RFOs related genes, which in turn restrain the GSO2, SDR1, ET-related genes, and phenylpropanoid metabolism related genes at 24 hpi (Fig. S10). Meanwhile, GSO2 sense the infection of P. litchii and enhance resistance by up-regulated SDR1, which in turn induce resistance by up-regulated RFOs related genes, NPR1, and PP2Cs at 24 hpi. At 48 hpi, RFOs related genes and PP2Cs were down-regulated due to GSO2 and SDR1 down-regulated at 24 hpi. The GSO2, SDR1, ET-related genes, and phenylpropanoid metabolism related genes were up-regulated due to RFOs related genes up-regulated at 48 hpi. This trend suggests that susceptible cultivar experiencing P. litchii infection fundamentally repression of RFOs at penetration phase and disturbance signal transduction by altered endogenous hormone balance at incubation phase.
The pathogen infected unsuccessfully exocarp of ‘Heiye’ until 6 hpi. At this time, a relative abundance of DEGs interacted with P. litchii were detected, indicating that response to infection of P. litchii is strong in resistant cultivar. Chitin is a typical pathogen-derived molecule from fungal cell walls which elicits plant immune responses. The former study indicated that the cell of oomycete species was composed of lignin as the major component and with few chitins or without chitin. As markers of response to fungal infection, five chitinase genes were up-regulated in ‘Heiye’ after inoculation with P. litchii. Higher up-regulation of chitinase genes might be a consequence of recognition pathogen and subsequent activation of pathogen reaction. In actual, the expression of five chitinases was lower in resistance cultivar than in susceptible cultivar. Therefore, the increasing of chitinases in ‘Heiye’ is not an effective reaction to block infection of P. litchii but a biomarker effectively linked to defense response of litchi. SAMT converts SA to SA methyl ester (MSA), which act as an airborne signal that triggers defense responses in uninfected plants. SAMT was induced specifically around the lesions. In this study, one SAMT was down-regulated in ‘Heiye’ at 6 hpi. It suggests that the SA-mediated resistance is not the major mechanism against P. litchii in resistant cultivar.
Plant employ PRRs (RLKs and RLPs) for sensitive and rapid detection of the potential danger caused by pathogens. RLKs is surface-localized and contain various ligand-binding ectodomains that perceive PAMPs. RLKs contain three functional domains: an extracellular domain, a transmembrane domain, and an intracellular serine/threonine kinase domain. External signal ligands are recognized by the extracellular domain which triggers phosphorylation activity of the intracellular cytoplasmic kinase domain. The intracellular cytoplasmic kinase domain then activates the downstream signaling pathways. One RLP and six RLKs were up-regulated in resistance cultivar at early infection phase (6 hpi). One RLK with a mannose-binding domain was regulated at three time-points, which was up-regulated (FC = 2.28) at 6 hpi. LRR receptor-like serine/threonine-protein kinase (LRR-RLKs) regulates cell wall composition and structure and required for callose deposition upon infection in Arabidopsis37. LRR-RLKs confers resistance to the pathogenic bacteria Ralstonia solanacearum and to the necrotrophic fungi Plectosphaerella cucumerina38,39,40. The rice receptor-like kinase protein Xa21 confers resistance to bacterial blight disease, caused by infection of Xanthomonas oryzae pv. oryzae (Xoo)41. Xa21 perceives the presence of Xoo and relays the signal to the nucleus through multi-step signal cascades involving some key proteins such as XA21 Binding Protein 3 (XB3), mitogen-activated protein kinase 5 (MAPK5), and transcription factors (TFs) including OsWRKY62. XB3 is an E3 ubiquitin ligase protein that binds to the kinase domain of XA2113.
Interaction of E3 ligase proteins with the kinase domain of RLKs appears to be a conserved mechanism for the regulation of various plant processes42. Protein degradation mediated plays critical roles in plant immunity. The ubiquitin-proteasome system (UPS) is used for selectively degrading proteins, in which E3 ligases determine the substrate specificity43. E3 ubiquitin ligases can be classified into different groups based on the presence of specific HECT, RING, or U-box domains (PUBs)43. There were seven up-regulated protease genes in ‘Heiye’, which including one E3 ubiquitin ligase, two PUBs, two SBTs, and one NEDD8-specific protease. E3 ubiquitin ligases have been involved in the pathogen perception, as they appear to modulate the PPRs. In addition, E3 ubiquitin ligases are also involved in the signaling conduction. OsBBI1 is an E3 ubiquitin ligase protein that mediate broad-spectrum disease resistance against the blast fungus in rice. OsBBI1-overexpressing plants accumulate hydrogen peroxide and phenolic compounds and display enhanced cross-linking of proteins in cell walls at infection sites by Magnaporthe oryzae44. CaRING is another active E3 ligase in pepper and is induced by an avirulent strain of Xanthomonas campestris pv vesicatoria45. CaRING overexpression in Arabidopsis induces enhanced resistance to Pseudomonas and Hyaloperonospora arabidopsidis46. PUB17 is positive regulators of ETI responses in Arabidopsis. Plant over-expressing OsPUB15 at early stage in rice display a constitutive activation of plant basal defense responses, including excessive accumulation of hydrogen peroxide, up-regulated expression of PR47. SBTs are serine proteases that fulfill highly specific functions in plant development and signaling cascades48. It has been shown that several SBTs are specifically induced following pathogen infection48. SBT3.3 was hypothesized to function as a receptor located in the plasma membrane activating downstream immune signaling processes in Arabidopsis48. NEDD8-specific protease 1 processes the preform of the ubiquitin-like protein, while their function related to pathogen was not reported49.
The proteasome pathway plays a key role in turning off transcription mediate. Transcription factor families commonly reported to be involved in plant defense. Four TFs were up-regulated during pathogen infection in ‘Heiye’. Two TF (CYCD3-1 and OFP4) involved in regulation of cell wall remodeling were up-regulated in ‘Heiye’ at 6 hpi. CYCD3-1 involved in the control of the cell cycle at the G1/S transition and the induction of mitotic cell division. Plants overexpressing CYCD3-1 show extensive leaf curling and disorganized meristems50. CYCD3-1 is mediated by a proteasome-dependent pathway. OFP4 forms a transcription repression complex with KNAT7 to regulate secondary cell wall formation. Plants over-expressing OFP4 show curled leaves in Arabidopsis51. CYCD3-1 and OFP4 may play an important role in regulation of cell wall remodeling to block the infection of pathogen in resistant cultivar at early stage. Moreover, several functional DEGs related to cell wall remodeling respond to infection of P. litchii in ‘Heiye’ at 6 hpi. Expansin-A4 can loosen and extension of plant cell walls by disrupting non-covalent bonding between cellulose microfibrils and matrix glucans52. CSLA2 possesses biosynthesis galactomannan, which is a non-cellulosic polysaccharides of plant cell wall53. Glucan 1,3-beta-glucosidase was involved in plasmodesmal callose degradation and implicated in the defense of plants against pathogens54. F6’H2 was involved in scopoletin biosynthesis (included in Phenylproanoid biosynthesis)55. Another gene, KCS1, contributes to cuticular wax and suberin biosynthesis56. It suggests that the inoculation with P. litchii lead to cell wall remodeling in resistant cultivar at early stage.
Another up-regulated transcription factor bHLH100 belongs to a basic helix-loop-helix DNA-binding superfamily. Arabidopsis transformants overexpressing bHLH100 showed increased tolerance to high Zn and nickel compared to wild-type plants, confirming their role in metal homeostasis57. The high expression of bHLH explain the mechanism of resistance to Dryocosmus kuriphilus in Castanea mollissima Shuhe-WYL strain58. NaMYC2, a member of bHLH family, enhance resistance by regulating the biosynthesis of nicotine and phenolamides in Nicotiana attenuate59. On the other hand, the mutant of Arabidopsis, bhlh99, exhibited enhanced disease susceptibility to both Botrytis cinerea and Plectosphaerella cucumerina60. It speculates that the up-regulated bHLH100 will enhance disease resistance against P. litchii in resistance cultivar.
In generally, ‘Guiwei’ has a feeble response to infection of P. litchii due to lagging transcriptional modulation at early incubation stage. Once the infection was successful, P. litchii repressed resistant action in ‘Guiwei’ by down-regulated genes involved in phenylpropanoid metabolism, ET biosynthesis and signaling conduction. Whereas, ‘Heiye’ resist infection through sensing pathogens by receptor-like kinases at early infection stage, upon which they activate defense responses, such as transcription factors, degradation of proteasome, that lead to cell wall remodeling and activating expression of resistance genes to prevent disease.
Methods
Pathogen
The pathogen of P. litchii isolate (hk-1) was preserved in our lab. The isolate was cultured on 10% V8 agar media for 7 days at 25 ± 2 °C. The plates were flooded with 10 mL of sterile water and kept for 2 hour at 4 °C to promote zoospores release. The spore suspension was filtrated through double sterile layer and adjusted to 1 × 104 spores·mL−1 for inoculation.
Plant materials and treatment
In present study, freshly harvested mature fruits of Litchi chinensis cv. ‘Guiwei’ and ‘Heiye’ were obtained from orchards in Zhanjiang, China from 2012 to 2017. Fruits were selected for uniformity of shape, color, size and free of blemish or disease. Destalking of fruits was done by sharp scissor leaving about 4 mm pedicel. Prior to inoculation, fruits were precleaned with sterile water for twice. The fruits of each cultivar were divided into two groups, of which infiltrated respectively into a solution contained sterile water (mock-inoculation) or 1 × 104 spores·mL−1 spore suspension of P. litchii (inoculation) for 10 min. After air-drying, the fruits of each group were divided into three biological replications, which were placed in polyethylene packets (280 × 200 × 150 mm, 20 fruits per packet), and stored with 85–90% relative humidity at 25 ± 2 °C. At 2016, the pericarps of thirty fruits inoculated with P. litchii from each replication were collected respectively at 0, 6, 24, 48, 72, and 96 hpi, as well as mock-treated samples. Samples for SEM study was immediately treated as descripted by Hong et al.61. Samples for RNA isolated were stored immediately in liquid nitrogen and then stored at −80 °C until use.
Disease index
The DI was calculated according to the standard described by Cao et al.62 and with little modification. The disease grade was measured by monitored 60 fruits at 72 hpi. The disease grade was recorded the percentage of pathogen infection and accessed by the following scale: 0 = no disease; 1 = less than 1/10 disease; 3 = 1/10 to 1/4 disease; 5 = 1/4 to 1/2 disease; 7 = 1/2 to 3/4 disease; and 9 = more than 3/4 disease. The disease index was calculated as:
Disease resistance levels of the different genotypes were classified as: Highly Resistant (HR: scores of 0–25.00); Resistant (R: scores of 25.01–50.00); Susceptible (S: scores of 50.01–75.00); or Highly Susceptible (HS: scores of 75.01–100.00)62.
Scanning electron microscopy
We observe the disease process on the exocarp of two cultivars by SEM. According to methods outlined by Hong61, pericarp samples (5 mm × 5 mm) were fixed in 4% glutaraldehyde at 4 °C for over 6 hour, and then washed three times with 0.1 M phosphate buffer solution (PBS) for 10 min. The samples were then transferred into 1% osmic acid at 4 °C for 2 hour and washed three times with 0.1 M PBS for 10 min. The samples were then dehydrated with a series of ethanol mixtures (30%, 50%, 70%, 85%, and 100%) for 20 min respectively, followed by a series of tert‐butyl alcohol mixtures (50%, 75%, 100%) twice to remove the ethanol. After being dried, samples were sprayed with a 12.5–15 nm gold layer. Samples were examined and photographed using a HITACHI S-4700 Scanning Electron Microscope at an accelerating voltage of 2.0 kV and working distance of 12.0 mm.
RNA Extraction and Sequencing
Total RNA was extracted from 6, 24, and 48 hpi sample of three biological replicates using the Quick RNA Isolation Kit (Huayueyang, China) according to the manufacturer’s instructions. DNAase I (Takara, Otsu, Japan) was given to remove genomic DNA contamination. The integrity of total RNA was verified through RNase-free agarose gel electrophoresis, and the concentration was measured using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). A cDNA library was constructed for high-quality RNA (2 μg) from each sample and was sequenced with Illumina HiSeq™ 2500 (San Diego, CA, USA) using a 125 paired-end module at Berry Genomics Corporation (Beijing, China, http://www.berrygenomics.com).
De novo assembly
Before assembly, adapter sequences were removed from the raw reads. Low-quality reads (>50% bases with quality scores ≤5) and unknown bases (>10% N bases) were removed from each dataset. The high-quality clean reads from all 36 samples is too huge to assemble. To reduce memory requirements and improve upon runtimes, three runs of in silico normalization were carried out prior to assemble. The sequences from each cultivar were respectively normalized using insilico_read_normalization.pl in Trinity with default parameters, and then the former results were normalized finally63. The reduced reads were de novo assembled using Trinity pipeline (Trinity-v2.4.0)63. The contigs were further processed with cd-hit for removes redundancy64. The normalization reads were mapping to the de-redundancy contigs with botiwe2 and calculated the expression with RSEM65,66. The isoforms with highest expression were selected as candidate genes and merged via corset to construct unique consensus sequences (unigenes) as ref.67.
Function annotation and DEG analysis
The assembled unigenes were annotated by BLASTx (E-value < 10−5) against the NCBI non-redundant (Nr) database68, the Swiss-Prot protein database (Swiss-Prot)69, GO19, and KEGG20. The sequencing reads for each sample were remapped to the reference sequences using bowtie265. After the number of reads mapped to each unigene was counted, differential expression analysis was conducted using edgeR70. The library was recalibrated, and the lowly expressed genes (<5 counts) were filtered in each sample. The threshold of DEGs was set as |log2FC| ≥ 1, P-value < 0.001, and FDR < 0.01 in each comparison.
Plants of two litchi cultivars ‘Guiwei’ (susceptible; referred as ‘G’) and ‘Heiye’ (resistant; referred as ‘H’) were either inoculated (‘I’) with the pathogen (GI and HI), or mock-inoculated (‘C’) with sterile deionized water (GC and HC) at three time-points (6, 24, and 48 hpi). For example, the ‘GC_6 hpi’ represent the sample of ‘Guiwei’ with mock-inoculated at 6 hpi. To infer the transcriptional changes in the two genotypes, DEGs were respectively identified by comparing the expression levels bewteen ‘Heiye’ and ‘Guiwei’ with mock-treated (HC_vs_GC) at 6, 24, and 48 hpi. For convenience, DEGs showing higher expression levels in ‘Heiye’ were designated ‘up-regulated’, whereas the rest were designated ‘down-regulated’. To infer the transcriptional changes responding to infection of P. litchii, DEGs were respectively identified by comparing the expression levels between inoculation and mock-inoculation in cultivar (HI_vs_HC or GI_vs_GC) at 6, 24, and 48 hpi. DEGs showing higher expression levels in inoculation with P. litchii were designated ‘up-regulated’, whereas the rest were designated ‘down-regulated’. The DEGs of each comparison was further subjected to GO and KEGG enrichment analysis to verify biological significance with the threshold (Q-value ≤ 0.05). PCA was performed on the normalized gene expression values using R statistical software (version 3.1.0)71. The heat map was constructed using the R package ‘pheatmap’72.
Real time PCR validation
Total RNA was extracted as above description. Two μg of RNA was reverse-transcribed using M-MLV reverse transcriptase (Invitrogen, USA) and an oligo (dT18) primer according to the manufacturer’s protocol. Six DEGs involved in ‘Phenylpropanoid biosynthesis’ were analysed using qRT-PCR. Primers for selected transcripts were designed by Primer3 (http://frodo.wi.mit.edu/primer3)(Table S12). The qRT-PCR reactions were performed with 10 μL of SYBR Green master mix, 50 ng of cDNA, and 500 nM each of the sense and antisense primers, in a total volume of 20 μL (Takara). Actin was used as an internal control for calculating relative abundances using the 2−△△CT method. The expression ratio was the average of three technical replicates and three biological replicates.
Data Availability
Additional data sets generated and/or analysed during the current are available from the corresponding author on reasonable request.
References
Wei, Y. Z. et al. Phenological growth stages of lychee (Litchi chinensis Sonn.) using the extended BBCH-scale. Scientia Horticulturae 161, 273–277 (2013).
Sun, J. et al. Transcriptome analysis of Phytophthora litchii reveals pathogenicity arsenals and confirms taxonomic status. PLoS One 12, e0178245 (2017).
Yi, C. et al. ATP-regulation of antioxidant properties and phenolics in litchi fruit during browning and pathogen infection process. Food Chemistry 118, 42–47 (2010).
Wang, H. C., Sun, H. Y., Stammler, G., Ma, J. X. & Zhou, M. G. Baseline and differential sensitivity of Peronophythora litchii (lychee downy blight) to three carboxylic acid amide fungicides. Plant Pathology 58, 571–576 (2009).
Wang, H., Sun, H., Stammler, G., Ma, J. & Zhou, M. Generation and characterization of isolates of Peronophythora litchii resistant to carboxylic acid amide fungicides. Phytopathology 100, 522–527 (2010).
Mackey, D. & McFall, A. J. MAMPs and MIMPs: proposed classifications for inducers of innate immunity. Molecular Microbiology 61, 1365–1371 (2006).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–239 (2006).
Coll, N. S., Epple, P. & Dangl, J. L. Programmed cell death in the plant immune system. Cell Death and Differentiation 18, 1247–1256 (2011).
Bari, R. & Jones, J. D. Role of plant hormones in plant defence responses. Plant Mol Biol 69, 473–488 (2009).
Grant, M. & Lamb, C. Systemic immunity. Curr Opin Plant Biol 9, 414–420 (2006).
Santner, A. & Estelle, M. The ubiquitin‐proteasome system regulates plant hormone signaling. The Plant Journal 61, 1029–1040 (2010).
Furlan, G., Klinkenberg, J. & Trujillo, M. Regulation of plant immune receptors by ubiquitination. Front Plant Sci 3, 1–6 (2012).
Wang, Y. S. et al. Rice XA21 binding protein 3 is a ubiquitin ligase required for full Xa21-mediated disease resistance. Plant Cell 18, 3635–3646 (2006).
Huang, W. et al. SlNAC1, a stress‐related transcription factor, is fine‐tuned on both the transcriptional and the post‐translational level. New Phytologist 197, 1214–1224 (2013).
Yaeno, T. et al. Phosphatidylinositol monophosphate-binding interface in the oomycete RXLR effector AVR3a is required for its stability in host cells to modulate plant immunity. Proc. Natl. Acad. Sci. USA 108, 14682–14687 (2011).
Üstün, S. & Börnke, F. Interactions of Xanthomonas type-III effector proteins with the plant ubiquitin and ubiquitin-like pathways. Front Plant Sci 5, 1–6 (2014).
Jiang, L. et al. A Puf RNA-binding protein encoding gene PlM90 regulates the sexual and asexual life stages of the litchi downy blight pathogen Peronophythora litchii. Fungal Genet Biol 98, 39–45 (2017).
Xu, D. et al. Biological activity of pterostilbene against Peronophythora litchii, the litchi downy blight pathogen. Postharvest Biol. Tec. 144, 29–35 (2018).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 45, D353–D361 (2017).
Ying, P. et al. Identification and molecular characterization of an IDA-like gene from litchi, LcIDL1, whose ectopic expression promotes floral organ abscission in Arabidopsis. Scientific Reports 6, 37135 (2016).
Schröder, F., Lisso, J. & Müssig, C. EXORDIUM-LIKE1 promotes growth during low carbon availability in Arabidopsis. Plant Physiology 156, 1620–1630 (2011).
Liu, W., Liu, J., Triplett, L., Leach, J. E. & Wang, G.-L. Novel insights into rice innate immunity against bacterial and fungal pathogens. Annu Rev Phytopathol 52, 213–241 (2014).
Kong, L.-A. et al. Large-scale identification of wheat genes resistant to cereal cyst nematode Heterodera avenae using comparative transcriptomic analysis. BMC Genomics 16, 801 (2015).
Venugopal, S. C. et al. Enhanced disease susceptibility 1 and salicylic acid act redundantly to regulate resistance gene-mediated signaling. PLOS Genetics 5, e1000545 (2009).
Bhattacharjee, S., Halane, M. K., Kim, S. H. & Gassmann, W. Pathogen effectors target Arabidopsis EDS1 and alter its interactions with immune regulators. Science 334, 1405–1408 (2011).
Choi, H. W. et al. A Role for a menthone reductase in resistance against microbial pathogens in plants. Plant Physiology 148, 383–401 (2008).
Racolta, A., Bryan, A. C. & Tax, F. E. The receptor‐like kinases GSO1 and GSO2 together regulate root growth in Arabidopsis through control of cell division and cell fate specification. Developmental Dynamics 243, 257–278 (2014).
Kim, M. S. et al. Galactinol is a signaling component of the induced systemic resistance caused by Pseudomonas chlororaphis O6 root colonization. Mol Plant Microbe In 21, 1643–1653 (2008).
Cho, S. M. et al. Jasmonate-dependent expression of a galactinol synthase gene is involved in priming of systemic fungal resistance in Arabidopsis thaliana. Botany 88, 452–461 (2010).
Solano, R., Stepanova, A., Chao, Q. & Ecker, J. R. Nuclear events in ethylene signaling: a transcriptional cascade mediated by Ethylene-insensitive 3 and ethylene-response-factor 1. Genes & Development 12, 3703–3714 (1998).
Yuan, Y. et al. Alternative splicing and gene duplication differentially shaped the regulation of isochorismate synthase in Populus and Arabidopsis. Proc. Natl. Acad. Sci. USA 106, 22020–22025 (2009).
Chen, L. & Yu, D. ABA regulation of plant response to biotic stresses. in Abscisic Acid: Metabolism, Transport and Signaling (ed. Zhang, D.-P.) 409–429 (Springer Netherlands, Dordrecht, 2014).
Mohr, P. G. & Cahill, D. M. Abscisic acid influences the susceptibility of Arabidopsis thaliana to Pseudomonas syringae pv. tomato and Peronospora parasitica. Funct Plant Biol 30, 461–469 (2003).
Ding, Y., Dommel, M. & Mou, Z. Abscisic acid promotes proteasome‐mediated degradation of the transcription coactivator NPR1 in Arabidopsis thaliana. The Plant Journal 86, 20–34 (2016).
Ali, S. et al. Overexpression of NPR1 In Brassica juncea confers broad spectrum resistance to fungal pathogens. Front Plant Sci 8, 1–16 (2017).
Sanchez-Rodriguez, C. et al. The ERECTA receptor-like kinase regulates cell wall-mediated resistance to pathogens in Arabidopsis thaliana. Mol Plant Microbe In 22, 953–963 (2009).
Llorente, F., Alonso‐Blanco, C., Sánchez‐Rodriguez, C., Jorda, L. & Molina, A. ERECTA receptor‐like kinase and heterotrimeric G protein from Arabidopsis are required for resistance to the necrotrophic fungus Plectosphaerella cucumerina. The Plant Journal 43, 165–180 (2005).
Godiard, L. et al. ERECTA, an LRR receptor‐like kinase protein controlling development pleiotropically affects resistance to bacterial wilt. The Plant Journal 36, 353–365 (2003).
Hanemian, M. et al. Arabidopsis CLAVATA1 and CLAVATA2 receptors contribute to Ralstonia solanacearum pathogenicity through a miR169‐dependent pathway. New Phytologist 211, 502–515 (2016).
Song, W. Y. et al. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science 270, 1804–1806 (1995).
Marino, D., Peeters, N. & Rivas, S. Ubiquitination during plant immune signaling. Plant Physiol 160, 15–27 (2012).
Vierstra, R. D. The ubiquitin-26S proteasome system at the nexus of plant biology. Nat Rev Mol Cell Biol 10, 385–397 (2009).
Li, W. et al. Rice RING protein OsBBI1 with E3 ligase activity confers broad-spectrum resistance against Magnaporthe oryzae by modifying the cell wall defence. Cell Res 21, 835–848 (2011).
Hong, J. K., Choi, H. W., Hwang, I. S. & Hwang, B. K. Role of a novel pathogen-induced pepper C3-H-C4 type RING-finger protein gene, CaRFP1, in disease susceptibility and osmotic stress tolerance. Plant Mol Biol 63, 571–588 (2007).
Lee, D. H., Choi, H. W. & Hwang, B. K. The pepper E3 ubiquitin ligase RING1 gene, CaRING1, is required for cell death and the salicylic acid-dependent defense response. Plant Physiol 156, 2011–2025 (2011).
Wang, J. et al. The E3 ligase OsPUB15 interacts with the receptor-like kinase PID2 and regulates plant cell death and innate immunity. BMC Plant Biol 15, 49–63 (2015).
Figueiredo, A., Monteiro, F. & Sebastiana, M. Subtilisin-like proteases in plant-pathogen recognition and immune priming: a perspective. Front Plant Sci 5, 1–4 (2014).
Schwechheimer, C. & Mergner, J. The NEDD8 modification pathway in plants. Front Plant Sci 5, 1–15 (2014).
Schnittger, A. et al. Ectopic D-type cyclin expression induces not only DNA replication but also cell division in Arabidopsis trichomes. Proc. Natl. Acad. Sci. USA 99, 6410–6415 (2002).
Liu, Y. & Douglas, C. J. A role for OVATE FAMILY PROTEIN1 (OFP1) and OFP4 in a BLH6-KNAT7 multi-protein complex regulating secondary cell wall formation in Arabidopsis thaliana. Plant Signaling & Behavior 10, e1033126 (2015).
Cosgrove, D. J. Plant expansins: diversity and interactions with plant cell walls. Curr Opin Plant Biol 25, 162–172 (2015).
Liepman, A. H., Wilkerson, C. G. & Keegstra, K. Expression of cellulose synthase-like (Csl) genes in insect cells reveals that CslA family members encode mannan synthases. Proc. Natl. Acad. Sci. USA 102, 2221–2226 (2005).
Zavaliev, R., Ueki, S., Epel, B. L. & Citovsky, V. Biology of callose (β-1,3-glucan) turnover at plasmodesmata. Protoplasma 248, 117–130 (2011).
Kosuke, K. et al. Scopoletin is biosynthesized via ortho‐hydroxylation of feruloyl CoA by a 2‐oxoglutarate‐dependent dioxygenase in Arabidopsis thaliana. The Plant Journal 55, 989–999 (2008).
James, T., Dusty, P. B. & JanG., J. KCS1 encodes a fatty acid elongase 3‐ketoacyl‐CoA synthase affecting wax biosynthesis in Arabidopsis thaliana. The Plant Journal 17, 119–130 (1999).
E., V. D. M. J. et al. Expression differences for genes involved in lignin, glutathione and sulphate metabolism in response to cadmium in Arabidopsis thaliana and the related Zn/Cd‐hyperaccumulator Thlaspi caerulescens. Plant, Cell & Environment 31, 301–324 (2008).
Geng, G. M., Zhu, C. C. & Zhou, J. Y. Resistance of Castanea mollissima Shuhe-WYL strain to Dryocosmus kuriphilus and its molecular mechanism. Genet Mol Res 14, 11456–11461 (2015).
Woldemariam, M. G. et al. NaMYC2 transcription factor regulates a subset of plant defense responses in Nicotiana attenuata. BMC Plant Biology 13, 73–86 (2013).
Dobon, A. et al. Novel disease susceptibility factors for fungal necrotrophic pathogens in Arabidopsis. PLoS Pathog 11, e1004800 (2015).
Hong, L. et al. The morphology and anatomy of the haustoria of the holoparasitic angiosperm cuscuta campetris. Pakistan Journal of Botany 43, 1853–1859 (2011).
Cao, L., Sun, J. & Wang, J. Establishment and preliminary application of the resistance evaluation system of litchi fruit to downy blight. Chinese Journal of Tropical Crops 38, 126–130 (2017).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature biotechnology 29, (644–652 (2011).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323–338 (2011).
Davidson, N. M. & Oshlack, A. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biology 15, 410–423 (2014).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35, D61–D65 (2007).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 45–48 (2000).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Team, R.C. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (2017).
Kolde, R. pheatmap: Pretty Heatmaps. R package version 1.0.8 (2015).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation (31301766) and the Special Funds of the Modern Agricultural Industry Technology System of China (CARS-33-03).
Author information
Authors and Affiliations
Contributions
Jinhua Sun and Jiabao Wang conceived and designed the experiments; Jinhua Sun and Lulu Cao performed the experiments of ESM; Jinhua Sun and Xiaoxiao Zou participated in bioinformatics analysis; Huanling Li contributed the P. litchii strain; Guo Wang contributed the treatment of fruits; Shujun Wang contributed the extraction of RNA; Fang Li contributed the survey of fruit disease; and Jiabao Wang provided guidance in the experiment design and writing the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, J., Cao, L., Li, H. et al. Early responses given distinct tactics to infection of Peronophythora litchii in susceptible and resistant litchi cultivar. Sci Rep 9, 2810 (2019). https://doi.org/10.1038/s41598-019-39100-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39100-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
