
Abstract
Adaptive responses to hypoxia regulate hepatic lipid metabolism, but their consequences in nonalcoholic fatty liver disease (NAFLD) are largely unknown. Here, we show that hypoxia inducible factor-1 (HIF-1), a key determinant of hypoxic adaptations, prevents excessive hepatic lipid accumulation in the progression of NAFLD. When exposed to a choline-deficient diet (CDD) for 4 weeks, the loss of hepatic Hif-1α gene accelerated liver steatosis with enhanced triglyceride accumulation in the liver compared to wild-type (WT) livers. Expression of genes involved in peroxisomal fatty acid oxidation was suppressed significantly in CDD-treated WT livers, whereas this reduction was further enhanced in Hif-1α-deficient livers. A lack of induction and nuclear accumulation of lipin1, a key regulator of the PPARα/PGC-1α pathway, could be attributed to impaired peroxisomal β-oxidation in Hif-1α-deficient livers. The lipin1-mediated binding of PPARα to the acyl CoA oxidase promoter was markedly reduced in Hif-1α-deficient mice exposed to a CDD. Moreover, forced Lipin1 expression restored the aberrant lipid accumulation caused by Hif-1α deletion in cells incubated in a choline-deficient medium. These results strongly suggest that HIF-1 plays a crucial role in the regulation of peroxisomal lipid metabolism by activating the expression and nuclear accumulation of lipin1 in NAFLD.
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) is a common chronic liver disease that is characterized by simple steatosis, steatohepatitis, hepatic fibrosis, and cirrhosis. NAFLD is associated with systemic metabolic disorders, including obesity, type II diabetes mellitus, atherosclerosis, and dyslipidemia, and is considered to be the hepatic component of metabolic syndrome. Accumulating evidence has suggested that insulin resistance, oxidative stress, and dysregulated adipocytokine production play critical roles in the development and progression of NAFLD1. However, no effective therapies have yet been established for the disease because of an incomplete understanding of its pathogenesis.
Chronic liver hypoxia has been implicated as a cause and/or consequence of NAFLD, and has also been associated with adverse disease prognosis. Previous reports from our and other laboratories revealed disease-associated hypoxia in murine livers that had been chronically exposed to high-fat diets2,3. This was associated with mitochondrial dysfunction, including impaired fatty acid oxidation, reduced electron transport chain activity, and increased reactive oxygen species (ROS) production2. In addition, NAFLD-induced cytochrome P450 2E1 consumes a large amount of oxygen to oxidize polyunsaturated fatty acids4. This increases ROS formation, which disrupts hepatic oxygen homeostasis. These hypoxic alterations might in turn accelerate hepatic lipid accumulation and inflammatory cell infiltration, forming a vicious cycle that results in irreversible fibrotic remodeling in the liver. Intermittent hypoxia with a high-fat diet also enhances hepatic steatosis with concomitant liver inflammation and lipid peroxidation5, further supporting the aggravating effects of tissue hypoxia on NAFLD. However, the pathological significance of liver hypoxia in NAFLD has not been fully elucidated.
Mammalian cells have evolved to adapt to lowered oxygen conditions by activating a master transcriptional regulator of the hypoxic response, hypoxia inducible factor (HIF)6,7. HIF is composed of two distinct subunits: oxygen-sensitive HIFα (HIF-1α, HIF-2α, and HIF-3α) and constitutively expressed HIFβ/aryl hydrocarbon receptor nuclear translocator (ARNT). HIFα is degraded rapidly under normoxic conditions due to high prolyl hydroxylase activity, which allows the von Hippel-Lindau (VHL) tumor suppressor protein to bind to HIFα. During hypoxia, the escape of HIFα from VHL recognition results in the activation of HIF-mediated transcription. The constitutive activation of HIFα in the liver by loss of the Vhl gene evokes massive lipid accumulation in an HIF-2α-dependent manner8. In contrast, liver-specific Arnt knockout mice express increased amounts of several lipogenic genes but deposit fewer triglycerides (TG) in their livers compared with control mice9. Fatty infiltration in response to a high-fat diet occurs comparably irrespective of hepatic Hif-1α gene status, although several genes involved in hepatic fatty acid metabolisms are suppressed in the mutant mice10. These observations clearly suggest that HIF-1 plays an indispensable role in the regulation of hepatic lipid metabolism, although distinct sets of HIF-target genes might be induced or suppressed to disrupt liver lipid homeostasis in an isoform-specific and a context-dependent manner.
In the present study, we revealed that loss of Hif-1α gene suppresses peroxisomal fatty acid oxidation by inhibiting induction of the peroxisome proliferator-activated receptor (PPAR)α coactivator, lipin1, and thereby aggravating lipid accumulation in the liver after chronic exposure to a CDD. These results suggest that HIF-1 plays an endogenous protective role during the development of NAFLD.
Results
Loss of the hepatic Hif-1α gene aggravates CDD-induced liver steatosis in mice
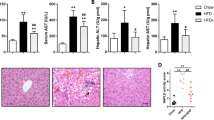
We first investigated if exposure to a CDD for 4 weeks can activate HIF-1 transcriptional activity in mouse liver. Wild-type (WT) mice modestly increased HIF1α protein levels in liver by a CDD (Supplementary Fig. S1a). Liver expression of Dmt1 (divalent metal transporter 1) and Phd3 (prolyl hydroxylase domain-containing protein 3), well-known target genes of HIF-1, was elevated in WT mice exposed to a CDD, but these responses were almost abolished by inactivation of Hif-1α gene (Supplementary Fig. S1b). On the other hand, levels of Vegf (vascular endothelial growth factor), another target gene of HIF-1, were modestly, but significantly reduced in CDD-treated WT liver, while this was further enhanced in hepatocyte-specific Hif-1α-deficient (HIFKO) one (Supplementary Fig. S1b). We next examined effects of loss of hepatic Hif-1α gene on CDD-induced tissue damage, steatosis, and fibrosis in mouse liver. Serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), indicative of tissue damage, were markedly elevated by a CDD, but were not different between WT and HIFKO mice (Supplementary Fig. S2a). Massive lipid accumulation occurred in the periportal hepatocytes of WT mice fed a CDD; these alterations were aggravated and expanded toward the midzonal region of lobules in HIFKO (Fig. 1a). Consistent with these findings, TG levels were elevated significantly in HIFKO livers compared with WT ones (Fig. 1b). The levels of liver non-esterified fatty acids (NEFA) increased substantially by CDD treatment in WT and HIFKO mice, but there was no significant difference between groups (Fig. 1b). Conversely, serum TG and NEFA levels did not show any remarkable change in response to a CDD, regardless of Hif-1α gene status (data not shown). Disruption of Hif-1α gene has been reported to reduce liver fibrosis when exposed to a high fat diets for 6 months10. However, in our model, sirius red staining of livers and gene expression analysis showed that liver fibrosis occurred comparably in both groups fed a CDD (Supplementary Fig. S2b,c). Collectively, these results suggest that deletion of hepatic Hif-1α gene selectively enhances fatty infiltration with increased liver TG content after exposure to a CDD.

Hepatic Hif-1α deficiency enhances lipid accumulation in CDD-treated livers. (a) Representative H&E staining image of livers from WT and HIFKO mice fed a CDD. CV, central venule. Scale bar = 20 μm. (b) Liver lipid content in mice fed normal chow (NC) or a CDD. n = 5–10 mice per group. *P < 0.05. TG, triglycerides; NEFA, non-esterified fatty acid.
Decreased peroxisomal β-oxidation causes severe liver steatosis in HIFKO mice
We next examined the changes in lipid metabolism-related gene expression in CDD-induced fatty livers. Consistent with a previous report11, the expression of genes responsible for very low-density lipoprotein (VLDL) production, including Mtp (microsomal triglyceride transfer protein) and ApoB (apolipoprotein B), was reduced markedly by CDD treatment in WT and HIFKO mice (Fig. 2a). However, no differences were observed between groups. The suppression of de novo fatty acid synthesis-related genes such as Fasn (fatty acid synthase), Dgat2 (diacylglycerol acyltransferase2), and Scd1 (stearoyl CoA desaturase1) also occurred in the livers of WT mice fed a CDD (Fig. 2a), presumably due to an increased flux of NEFA into the liver (Fig. 1b). Surprisingly, HIFKO mice had further decreased expression of these lipogenic genes in response to a CDD. Consistent with these findings, the expression of a master regulator of lipogenic genes, Srebp1c (sterol regulatory element-binding protein1c), was reduced dramatically in steatotic livers compared with normal livers, and Hif-1α deficiency showed a trend toward further decreased expression of Srebp1c (Fig. 3). CD36, a member of the class B scavenger receptor responsible for transport of long-chain fatty acids, was significantly reduced in Hif-1α-deficient liver exposed to a CDD (Fig. 2a).

CDD-induced suppression of peroxisomal β-oxidation is aggravated in Hif-1α-deficient livers. (a) Analyses of the expression of hepatic lipid oxidation-related genes. NC, mice fed normal chow. n = 5–10 mice per group. *P < 0.05. (b) β-oxidation enzymatic activity in peroxisomes (AOX) and mitochondria (CPT2) in the livers of CDD-fed mice. NC, mice fed normal chow. n = 5–10 mice per group. *P < 0.05.

Loss of Hif-1α reduces the expression of PPARα/RXRα co-activators in CDD-treated livers. Quantification of mRNA of transcriptional factors and their associated transcriptional co-regulators for hepatic lipid metabolism in the livers of mice fed a CDD. NC, mice fed normal chow. n = 5–10 mice per group. *P < 0.05.
The expression of genes involved in mitochondrial fatty acid oxidation such as Cpt1 (carnitine palmitoyltransferase1), Mcad (medium-chain acyl CoA dehydrogenase) and Lcad (long-chain acyl CoA dehydrogenase) was not changed significantly after exposure to a CDD (Fig. 2a). The loss of Hif-1α showed marginal effects on the expression of these genes. In contrast, the expression of peroxisomal fatty acid oxidation-related genes such as Aox (acyl CoA oxidase), Fatp2 (fatty acid transport protein 2), and Abcd1 (ATP-binding cassette, sub-family D1) was reduced significantly in CDD-treated WT livers, whereas these alterations were enhanced markedly by the deletion of hepatic Hif-1α (Fig. 2a). Collectively, these findings strongly suggest that the aggravated lipid accumulation in HIFKO mice could be partly attributed to severely impaired fatty acid oxidation in peroxisomes.
To further examine the importance of peroxisomal fatty acid oxidation in the aberrant lipid accumulation in HIFKO mice, in vitro enzymatic activity assays were performed. The hepatic activity of CPT2, which plays a role in the commitment step of mitochondrial β-oxidation, and de novo lipogenic activity, as assessed by FASN activity, were comparable between WT and HIFKO mice, even after exposure to a CDD (Fig. 2b). In contrast, deletion of Hif-1α caused a modest but significant reduction in AOX activity in CDD-treated livers (Fig. 2b), supporting the hypothesis that the enhanced suppression of peroxisomal β-oxidation by Hif-1α deficiency aggravates fatty infiltration in mice fed a CDD. However, expression of genes responsible for peroxisomal biogenesis (Pex3, Pex11α and Pex14) and functions (Uox: urate oxidase, and catalase) did not differ between WT and HIFKO mice even when exposed to a CDD (Supplementary Fig. S3a). Consistent with these results, protein levels of PEX14 and Catalase were comparable between the two groups (Supplementary Fig. S3b).
The induction of Lipin1, a PPARα/PGC1α coactivator, is abolished in the aggravated fatty livers of HIFKO mice
PPARα is a transcription factor that regulates the expression of genes involved in peroxisomal fatty acid oxidation; its dysfunction causes severe hepatic steatosis12. We next assessed if the expression of Pparα and/or its transcriptional partner and coactivators is disturbed during the enhanced lipid accumulation in HIFKO mice fed a CDD. Choline deprivation significantly suppressed the levels of Pparα and Rxrα (retinoid X receptor α) in both WT and HIFKO mice (Fig. 3). The loss of Hif-1α diminished expression of a highly inducible transcriptional coactivator of PPARα, PPARγ coactivator-1α (Pgc-1α), markedly after exposure to a CDD (Fig. 3). However, the protein levels of PGC-1α were comparable between WT and HIFKO mice fed a CDD (data not shown). The expression of Lipin1, a key regulator of the PPARα/PGC-1α pathway, was induced significantly by choline deprivation in WT mice, whereas this induction was totally abolished in HIFKO mice (Fig. 3). There are two splice variants of Lipin1: Lipin1α and Lipin1β. The expression of hepatic Lipin1β, but not Lipin1α, increased in mice fed a CDD, which was dependent on HIF-1 expression (Supplementary Fig. S4). In addition, Pparγ, a key transcription factor for the pathogenesis of NAFLD, substantially increased by CDD treatment in both mice, but the levels were comparable between the groups (Fig. 3). Taken together, these results suggest that a lack of HIF-1 mediated Lipin1 induction might stimulate aberrant lipid accumulation in the livers of mice fed a CDD by inhibiting PPARα transcriptional activity.
Reduced lipin1 nuclear accumulation in the livers of HIFKO mice fed a CDD
Lipin1 exerts different functions in response to nutrients depending on its localization; endoplasmic reticulum (ER)-associated lipin1 acts as a lipid phosphatase that is essential for the synthesis of TG, whereas nuclear lipin1 functions as a coactivator for PPARα/PGC1α13. The total and cytosolic lipin1 protein levels increased significantly by a CDD in WT livers, whereas this induction was completely abolished in Hif-1α-deficient livers (Fig. 4a,b). ER-localized lipin1 levels were comparable between WT and HIFKO mice that had been fed a CDD (Fig. 4a,b), although microsomal lipin1 increased in response to a CDD in both groups. In contrast, nuclear lipin1 levels were elevated substantially in the livers of mice fed a CDD compared with normal livers, but the loss of Hif-1α abolished these alterations completely (Fig. 4a,b). These results suggest that HIF-1 induces lipin1 expression in response to a CDD, accumulating nuclear lipin1 and thereby sustaining peroxisomal fatty acid oxidation.

Deletion of Hif-1α attenuates the nuclear accumulation of Lipin1 in CDD-treated livers. (a) Representative immunoblots of lipin1 levels in different subcellular fractions of the livers of mice fed a CDD. NC, mice fed normal chow. (b) Protein levels of lipin1 in different subcellular fractions of the livers of mice fed a CDD quantified using densitometry; n = 5–7 mice per group. *P < 0.05. (c) ChIP-qPCR analysis of lipin1 levels at the promoter of PPARα target genes in the livers of mice fed a CDD; n = 7–8 mice per group. *P < 0.05.
Loss of Hif-1α abolishes the lipin1-mediated binding of PPARα to the Aox promoter region
We next investigated if the inhibition of increase in nuclear lipin1 accumulation resulted in decreased PPARα transcriptional activity in the livers of CDD-fed HIFKO mice using ChIP assays with anti-lipin1 antibodies. Two putative PPARα binding sites in Aox gene promoter, Aox-1 and Aox-2, were found to be enriched constitutively in both WT and HIFKO mice fed normal chow (Fig. 4c). Feeding with a CDD showed a trend toward decreased enrichment of Aox-2 in the liver irrespective of Hif-1α gene status. On the other hand, CDD treatment greatly reduced the binding of lipin1 to Aox-1 in HIFKO, but not WT mice (Fig. 4c). In addition, CDD feeding and Hif-1α gene status did not affect the lipin1-mediated binding to the PPAR response element of Pparα and Mcad gene (Fig. 4c). Taken together, these results suggest that sustained PPARα-mediated transcription of peroxisomal fatty acid oxidation-related genes inhibits aberrant lipid accumulation in response to a CDD by activating lipin1 in a HIF-1-dependent manner.
Forced Lipin1β expression inhibits Hif-1α deletion-induced aberrant lipid accumulation
Choline deficiency enhanced lipid deposition in cultured AML12 hepatocytes in vitro (Fig. 5b,c), confirming the cell-autonomous effects of choline deprivation on lipid accumulation. Lipin1 protein expression increased modestly, and was accumulated predominantly in the nuclei after exposure to choline-deficient medium (Supplementary Fig. S5a,c). In addition, we observed HIF-1α protein also increased modestly in cells by choline deprivation (Supplementary Fig. S5b). To confirm effects of loss of Hif-1α on choline deficiency-stimulated lipid accumulation, cells were infected with shRNA adenovirus against Hif-1α. Expression of Hif-1α significantly decreased by treating with an adenovirus expressing shHif-1α (Fig. 5a). Consistent with the in vivo results, inactivation of Hif-1α aggravated the lipid accumulation and decreased lipin1 expression in cells treated with choline-deficient medium compared with shβ-galactosidase-infected control cells (Fig. 5b,c, Supplementary Fig. S5a). This aberrant lipid deposition was abolished completely by forced expression of Lipin1β (Fig. 5b,c). In addition, suppression of Lipin1 by introducing siRNA against Lipin1 gene alone was observed to successfully enhance lipid accumulation in response to choline deficiency (Supplementary Fig. S5d,e). Collectively, these results support the importance of lipin1 expression in the HIF-1-mediated protection against lipid deposition.

Forced Lipin1 expression abolishes choline deprivation-induced lipid accumulation in cells lacking Hif-1α. (a) Quantification of Hif-1α mRNA by qPCR in AML12 cells infected with adenovirus expressing shHif-1α; n = 3 per group. *P < 0.05 (b) Nile Red staining of AML12 cells exposed to choline-deficient medium. Lipid accumulation (red) is enhanced in cells lacking Hif-1α (middle), but reduced by Lipin1β overexpression (right). DAPI staining is shown in blue. Scale bar = 25 μm. (c) Densitometric quantification of lipid accumulation in A; n = 5–7 plates per group. *P < 0.05.
Discussion
Excessive lipid accumulation in hepatocytes is a hallmark of NAFLD. The metabolic alterations in NAFLD not only interfere with intracellular oxygen diffusion but also prevent the delivery of sufficient oxygen by narrowing the sinusoidal blood vessels in liver acinus2,3. The induction of Cyp2E1 expression in diet-induced steatosis has been well-documented in both rodents and humans4. This increased expression stimulates hepatic oxygen consumption and increases ROS production, which further impairs oxygen homeostasis in NAFLD livers. These metabolic and structural changes result in chronic liver hypoxia, and thereby induce varied adaptive responses to hypoxic stress by activating HIF-1 expression in the liver. However, the effect of this pathophysiological activation in NAFLD remains largely unclear. In the current study, we have demonstrated that the loss of Hif-1α in hepatocytes aggravates lipid infiltration in the liver in response to a CDD by reducing VLDL secretion and impairing peroxisomal β oxidation, the latter of which could explain the further increased lipid accumulation in the livers of HIFKO mice fed a CDD. These metabolic disturbances occur exclusively via the lack of HIF-1-mediated lipin1 expression and the resulting decrease in PPARα/PGC-1α-dependent expression of the genes involved in peroxisomal lipid metabolism.
The results of the current study clearly demonstrate that loss of Hif-1α in the liver augments hepatic steatosis in mice fed a CDD. This is consistent with our previous report showing that the alcoholic fatty liver is worsened in Hif-1α-deficient mice14. Although hepatic HIF-1 exerts anti-steatotic effects in these different disease conditions, the underlying mechanisms are distinct. In response to excessive alcohol exposure, HIF-1 suppresses de novo fatty acid synthesis and subsequent TG accumulation by inhibiting the aberrant activation of SREBP1c, a master lipogenic transcription factor. In contrast, HIF-1 prevents the massive reduction in fatty acid oxidation caused by choline deprivation by alleviating the diet-induced suppression of PPARα activity. This suggests that HIF-1 regulates hepatic lipid metabolism in a context-dependent manner. Given that lipid accumulation occurs in the pericentral and periportal regions of the liver acinus after exposure to alcohol-containing and choline-deficient diets, respectively, the different roles of HIF-1 in the regulation of hepatic lipid metabolism could be attributable to differences in its intrahepatic localization. Hepatocytes show distinct metabolic activity profiles depending on their localization in the liver acinus15: pericentral hepatocytes exhibit increased fatty acid synthesis, whereas periportal hepatocytes predominantly oxidize fatty acids. Therefore, it is possible that HIF-1 regulates lipid metabolism in an intrahepatic zone-specific manner by sensing and responding to distinct cellular microenvironments in different disease conditions.
Choline deficiency suppresses VLDL secretion and fatty acid oxidation, resulting in severe liver steatosis. A drastic reduction in the expression of Mtp and ApoB, which are essential for VLDL formation, occurs irrespective of Hif-1α gene status, suggesting that VLDL-related genes also play essential roles in the enhanced lipid accumulation in HIFKO livers. In contrast, the present findings revealed a marked reduction in Aox, Fatp2, and Abcd1 expression as well as decreased AOX activity in HIFKO mice fed a CDD, providing evidence to suggest that aggravated fatty acid oxidation in peroxisomes leads to aberrant lipid accumulation in HIFKO mice. Long-chain and very long-chain fatty acids, which are preferred substrates for peroxisomal β-oxidation, have been implicated in the induction of steatohepatitis16. Aox-deficient mice develop severe steatosis spontaneously, in spite of the increased expression of mitochondrial β-oxidation-related genes17. This supports the current findings, whereby the aggravated CDD-induced inhibition of peroxisomal fatty acid oxidation caused aberrant lipid accumulation in Hif-1α-deficient livers. In contrast to a previous report showing impaired mitochondrial fatty acid oxidation in Vhl-deficient livers8, the ability of mitochondria to oxidize fatty acids was not affected severely in WT and HIFKO mice fed a CDD in the current study. This further highlights the importance of peroxisomal β-oxidation in the progression of CDD-induced fatty livers in HIFKO mice. The selective protection of HIF-1α against the CDD-induced suppression of peroxisomal fatty acid oxidation might help reduce the metabolic burden of excessive fatty acid oxidation in the mitochondria, resulting in decreased ROS production and the subsequent mitigation of ROS-induced disease progression toward steatohepatitis, as catalase enriched in peroxisome can efficiently reduce hydrogen peroxide produced in peroxisomal fatty acid oxidation.
Lipin1 is a multifunctional protein that plays roles in lipid metabolism18. Its phosphatidate phosphatase activity, which converts phosphatidate to diacylglycerol on the ER membrane, is a key process for de novo TG synthesis. In contrast, nuclear lipin1 serves as a transcriptional coactivator for PPARα and PGC-1α to induce expression of genes related to fatty acid oxidation. In the current study, exposing mice to a CDD resulted in the elevated microsomal lipin1 accumulation. This suggests a possible role for lipin1-mediated phosphatidate phosphatase activity in CDD-stimulated lipid accumulation in the liver. Consistent with a previous report showing hypoxia-induced HIF-1-dependent lipin1 expression19, CDD-induced lipin1 expression was abolished completely in HIFKO mice in the current study. However, HIFKO deteriorated excessive fatty infiltration without affecting microsomal lipin1 accumulation compared with CDD-treated WT mice, suggesting that lipin1-mediated TG synthesis is not critical for the aggravated phenotype in HIFKO mice. Instead, the loss of Hif-1α reduced nuclear lipin1 expression in the livers of CDD-fed mice compared with the WT liver, suggesting that the reduced nuclear accumulation of lipin1 could diminish its lipolytic transcriptional coactivator activity and thus augment lipid accumulation in HIFKO livers. A major isoform of Lipin1 in the liver, Lipin1β, was reported to increase the expression of the PPARα target genes that are involved in fatty acid oxidation by associating directly with PGC-1α in the liver13. In addition, liver-specific Lipin1-deficient mice showed aggravated lipid accumulation by inhibiting VLDL excretion and fatty acid oxidation during the development of alcoholic liver steatosis20. Consistent with these reports, silencing of Lipin1 gene alone enhanced choline deficiency-evoked lipid accumulation in vitro, further supporting the pathological importance of lipin1 induction against development of NAFLD.
Consistent with previous reports and the current in vivo results, knocking down Hif-1α in hepatocytes enhanced lipid accumulation and reduced lipin1 expression in choline-deficient medium. Moreover, the increased lipid deposition was abolished by the forced expression and enhanced nuclear accumulation of lipin1β, further highlighting the importance of nuclear lipin1 for the prevention of excessive and aberrant lipid accumulation in the liver. In fact, the increased nuclear lipin1 successfully sustained the binding of PPARα to a distal PPARα-responsive site in the Aox promoter of a CDD-treated WT liver, even though hepatic expression of Pparα and Rxrα substantially decreased. In addition, this hypothesis is supported by the current results showing the reduced association of lipin1 with the same PPARα-responsive site in the Aox promoter in the livers of HIFKO mice fed a CDD. However, this lipin1-mediated transcriptional regulation appeared to be selective for genes related to peroxisomal, but not mitochondrial, fatty acid oxidation because a putative PPARα-binding site in the Mcad promoter was not enriched by the CDD. This contradicts previous reports, where lipin1 activated several PPARα-target genes related to both peroxisomal and mitochondrial fatty acid oxidation in the liver13. Although the molecular mechanisms behind the differential binding of lipin1 to promoter regions remains unclear, the present findings suggest that the selective association between lipin1 and PPARα with the PPARα target genes that are involved in hepatic lipid metabolism after exposure to a CDD might play a role.
In conclusion, the current study revealed that HIF-1 protects against choline deprivation-induced NAFLD. Specifically, HIF-1-induced lipin1 expression inhibits aberrant lipid accumulation by preventing the suppression of peroxisomal fatty acid oxidation. Although these data suggest that HIF-1 might have therapeutic potential in liver steatosis during NAFLD, future studies are needed to clarify the pathological significance of HIF-1 in the development of non-alcoholic steatohepatitis and fibrosis because HIF-1 has diverse and complicated roles in the regulation of inflammation and tissue remodeling in different organs.
Methods
Animals and experimental procedures
Hepatocyte-specific Hif-1α-deficient (HIFKO) mice were generated by crossing HIF-1αloxP/loxP (WT) mice with Alb-Cre transgenic mice, as described previously21. Male mice (5–6 weeks old) were exposed to a CDD (Oriental Yeast; Tokyo, Japan) for 4 weeks. All experiments were conducted in accordance with the Waseda University Animal Welfare Guidelines and approved by the Animal Experimentation Committee of the Waseda University (Protocol number: 2015-A032).
Liver histology
Livers were fixed overnight in 10% phosphate-buffered formalin, and were embedded in paraffin. Five-micrometer-thick sections were then stained with hematoxylin and eosin (H&E) to assess lipid infiltration. Picro-sirius red staining was performed for collagen staining.
Biochemical analyses
Hepatic lipid fractions were extracted using Folch solution (a mixture of 1 vol. of methanol and 2 vol. of chloroform), as described previously14. The amounts of TG, total cholesterol, and NEFA in liver and serum were then measured using colorimetric enzyme assays (Wako Pure Chemical Industries; Tokyo, Japan). Liver injury was assessed by measuring serum ALT and AST levels using a diagnostic kit (Wako Pure Chemical Industries).
Quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from the livers using an RNeasy Micro Kit (Qiagen; Tokyo, Japan). Complementary DNA was then synthesized using GoScriptTM Reverse transcription system (Promega; Tokyo, Japan) using 1 μg total RNA as the template. Quantitative PCR was performed using either GoTaq qPCR Master Mix (Promega) or FastStart TaqMan® Probe Master (Roche Diagnostics Japan; Tokyo, Japan) in a StepOnePlusTM system (Applied Biosystems Japan; Tokyo, Japan). TaqMan gene expression assays (Applied Biosystems) and the sequences of the primers used in the current study are listed in Supplementary Table S1 in the supplemental material. Gene expression was normalized to either β-actin (for Hif-1α) or 18 S ribosomal RNA (for other genes) as an internal control.
Western blotting
Western blotting was performed using 50–100 μg of nuclear fraction, microsomal fraction, cytosolic fraction or whole extract of the liver, as described previously14. In some experiments, AML12 cells exposed to choline-deficient medium were lysed in 50 mM HEPES, pH 7.5, 150 mM NaCl, 1% CHAPS, 5 mM EDTA, and protease inhibitors (Nacalai Tesque, Kyoto, Japan), and resultant cell lysates (15 μg) were used. The expression of PGC-1α, lipin1, HIF-1α, lamin, and tubulin was detected using specific antibodies against the respective antigens as follows: PGC-1α (2178 S, Novus Biologicals; Littleton, CO, USA), lipin1 (AF3885, R&D Systems; Minneapolis, MN, USA), HIF-1α (36169 and 14179, Cell Signaling Technology, Danvers, MA, USA), lamin A/C (sc-20681, Santa Cruz Biotechnology; Santa Cruz, CA, USA), and tubulin (T9026, Sigma-Aldrich, St. Louis, MO, USA). Images of Western blots are shown in Supplementary Fig. S6.
Enzyme assays
Livers (30 mg) were homogenized in 10 vol. of buffer (0.25 M sucrose, 1 mM EDTA, 3 mM Tris-HCl, pH7.2) supplemented with protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 1 μg/mL aprotinin, 1 μg/mL pepstatin A, and 2 μg/mL leupeptin). The lysates were then centrifuged at 500 × g for 10 min to obtain the supernatant as the peroxisome fraction. The peroxisome fraction was further centrifuged at 9000 × g for 10 min to collect the supernatant and pellet as the lipid synthesis fraction and the mitochondrial fraction, respectively. Hepatic AOX activity was determined based on the change in absorbance at 500 nm by incubating the peroxisome fraction (0.2 mg protein) in assay buffer (58 mM phosphate potassium buffer [pH 7.4], 10.6 mM phenol, 0.82 mM 4-aminoantipyrine, 10 μM FAD+, 4 units of peroxidase, 0.1 mM palmitoyl-CoA, and 0.2 mg albumin) for 5 min. To measure CPT2 and FASN activity in the liver, the mitochondrial (0.1 mg protein) and lipid synthesis fractions (0.15 mg protein) were mixed with the appropriate buffer (58 mM Tris-HCl [pH 8.0], 1.25 mM EDTA, 0.25 mM 5, 5′-dithiobis(2-nitrobenxoic acid), 0.04 mM palmitoyl-CoA, 0.1% TritonX-100, 1.25 mM l-carnitine for CPT2; and 0.1 M phosphate potassium buffer [pH 7.0], 0.2 mM EDTA, 0.3 mM NADPH, 0.05 mM acetyl-CoA, and 0.2 mM malonyl-CoA for FAS) for 3 min, and the light absorbance at 412 nm and 340 nm, respectively, was measured.
Chromatin immunoprecipitation (ChIP)-qPCR
Livers (300 mg) were lysed in a buffer containing 10 mM Tris-HCl pH 7.5, 10 mM NaCl, 0.5% NP-40, and protease inhibitors. The samples were crosslinked using 1% formaldehyde for 10 min at room temperature, and were then prepared for ChIP as described previously22. The crosslinked samples were incubated with anti-lipin1 antibodies (R&D Systems) and protein G-Sepharose beads (Life Technologies Japan; Tokyo, Japan) overnight at 4 °C. The beads were then washed several times, and bound proteins were eluted in elution buffer (1% sodium dodecyl sulfate in 0.1 M NaHCO3). The elutes were treated with pronase (1 mg/mL; Roche) for 2 h at 42 °C, and were then were incubated at 65 °C overnight to remove the crosslinks. The samples were further purified using nucleospin extract II (TaKaRa; Tokyo, Japan). qPCR was performed using the primers listed in Table S1 in the supplemental material.
Cell culture, adenovirus infection, and siRNA treatment
Normal mouse liver AML12 cells were purchased from ATCC (Manassas, VA, USA). The cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Life Technologies) with or without choline chloride (Kohjin Bio, Saitama, Japan) containing penicillin (100 U/mL), streptomycin sulfate (100 μg/mL), and 10% fetal bovine serum for 72 h at 37 °C in 5% CO2. Cells were infected with adenovirus expressing either shRNA containing repeat mouse Hif-1α sequences (GCAACTGTCATATATAATACG), which were separated by a four-nucleotide spacer from the reverse complement of the same 21 nucleotide sequences (Life Technologies), or His-tagged-mouse Lipin1 (Life Technologies) 24 h before experiments. Viruses encoding shRNA against β-galactosidase were used as a control. A multiplicity of infection of 100 for each virus was used for the virus infection. In some cases, cells were transfected with siRNA targeting for mouse Lpin1 (50 nM, 5′-AGGUUGACGCCAAAGAAUAACCUGG-3′; Integrated DNA Technologies KK, Tokyo, Japan) using Lipofectamine 2000 (Life Technologies) 24 h before experiments.
Nile Red staining and immunocytochemistry
After treatment with choline-deficient DMEM for 72 h, cells were fixed with 4% paraformaldehyde for 20 min at 4 °C. Lipid droplets were then visualized by staining cells with Nile Red (250 ng/mL) for 10 min followed by DAPI. Lipin1 expression was detected by incubating cells with anti-lipin1 antibodies followed by Alexa-Fluor® 488 goat anti-rabbit IgG (A-11034, Life Technologies). Cells were then observed using confocal microscopy (FV-1000, Olympus; Tokyo, Japan).
Statistical analyses
Results are expressed as means ± SE. Statistical analyses were performed using Student’s t-test for all experiments. P values < 0.05 were considered to be significant.
References
Bechmann, L. P. et al. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol 56, 952–964 (2012).
Mantena, S. K. et al. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J 417, 183–193 (2009).
Kondo, K. et al. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, improves hepatic microcirculatory patency and oxygen availability in a high-fat-diet-induced fatty liver in mice. Adv Exp Med Biol 662, 77–82 (2010).
Leung, T. M. & Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J Hepatol 58, 395–398 (2013).
Drager, L. F. et al. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obesity (Silver Spring) 19, 2167–2174 (2011).
Aragones, J., Fraisl, P., Baes, M. & Carmeliet, P. Oxygen sensors at the crossroad of metabolism. Cell Metab 9, 11–22 (2009).
Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 (2012).
Qu, A. et al. Hypoxia-inducible transcription factor 2alpha promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 54, 472–483 (2011).
Wang, X. L. et al. Ablation of ARNT/HIF1beta in liver alters gluconeogenesis, lipogenic gene expression, and serum ketones. Cell Metab 9, 428–439 (2009).
Mesarwi, O. A. et al. Hepatocyte Hypoxia Inducible Factor-1 Mediates the Development of Liver Fibrosis in a Mouse Model of Nonalcoholic Fatty Liver Disease. PLoS One 11, e0168572 (2016).
Yao, Z. M. & Vance, D. E. Reduction in VLDL, but not HDL, in plasma of rats deficient in choline. Biochem Cell Biol 68, 552–558 (1990).
Chakravarthy, M. V. et al. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell 138, 476–488 (2009).
Ryu, D. et al. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metab 9, 240–251 (2009).
Nishiyama, Y. et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J Hepatol 56, 441–447 (2012).
Hijmans, B. S., Grefhorst, A., Oosterveer, M. H. & Groen, A. K. Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences. Biochimie 96, 121–129 (2014).
Rao, M. S. & Reddy, J. K. Peroxisomal beta-oxidation and steatohepatitis. Semin Liver Dis 21, 43–55 (2001).
Fan, C. Y. et al. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem 273, 15639–15645 (1998).
Bou Khalil, M., Blais, A., Figeys, D. & Yao, Z. Lipin - The bridge between hepatic glycerolipid biosynthesis and lipoprotein metabolism. Biochim Biophys Acta 1801, 1249–1259 (2010).
Mylonis, I. et al. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J Cell Sci 125, 3485–3493 (2012).
Hu, M. et al. Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology 58, 1953–1963 (2013).
Tajima, T. et al. HIF-1alpha is necessary to support gluconeogenesis during liver regeneration. Biochem Biophys Res Commun 387, 789–794 (2009).
Okamura, M. et al. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc Natl Acad Sci USA 106, 5819–5824 (2009).
Acknowledgements
We are grateful to members of the Goda lab, especially Y. Kato, K. Sayama, Y. Ono, M. Kanai and S. Shinjo for technical support and discussion. This work was supported in part by the MEXT-Supported Programme for the Strategic Research Foundation at Private Universities (No. S1201006 to N.G.), a Grant-in-Aid for Scientific Research (C) (No. 17570119 to N.G.), and a Grant-in-Aid for Scientific Research on Innovative Areas “Oxygen Biology: a new criterion for integrated understanding of life” (No. 2611103 to N.G.).
Author information
Authors and Affiliations
Contributions
T.A. and N.G. performed the experiments, analysed the data, and wrote the paper. M.T. performed western blot analysis for HIF-1α.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arai, T., Tanaka, M. & Goda, N. HIF-1-dependent lipin1 induction prevents excessive lipid accumulation in choline-deficient diet-induced fatty liver. Sci Rep 8, 14230 (2018). https://doi.org/10.1038/s41598-018-32586-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32586-w
Keywords
This article is cited by
-
Role of lipins in cardiovascular diseases
Lipids in Health and Disease (2023)
-
The role of hypoxia-inducible factor 1α in hepatic lipid metabolism
Journal of Molecular Medicine (2023)
-
IκBζ regulates the development of nonalcoholic fatty liver disease through the attenuation of hepatic steatosis in mice
Scientific Reports (2022)
-
Danlou Tablet Improves Chronic Intermittent Hypoxia-Induced Dyslipidemia and Arteriosclerosis by HIF-1α-Angptl4 mRNA Signaling Pathway
Chinese Journal of Integrative Medicine (2022)
-
How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease?
Hepatology International (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
