
Abstract
Silencing of anticoagulant protein C using RNA interference (siProc) evokes low incident but spontaneous atherothrombosis in the aortic root of apolipoprotein E–deficient (Apoe−/−) mice. The aims of the current study were (1) to analyze if plaque characteristics or circulating factors could be linked to atherothrombosis susceptibility, (2) to increase the incidence of atherothrombosis by transiently increasing blood pressure, and (3) to direct atherothrombosis to an additional predefined vascular site by applying a semi-constrictive collar around the carotid artery. siProc-driven spontaneous atherothrombosis in the aortic root of Apoe−/− mice was reproduced and occurred at an incidence of 23% (9 out of 39 mice), while the incidence of collar-induced atherothrombosis in the carotid artery was 2.6% (1 out of 39 mice). Treatment with phenylephrine, to transiently increase blood pressure, did not increase atherothrombosis in the aortic root of the Apoe−/− mice nor in the carotid arteries with collars. Plaques in the aortic root with an associated thrombus were lower in collagen and macrophage content, and mice with atherothrombosis had significantly more circulating platelets. Plasma protein C, white blood cell counts, total cholesterol, fibrinogen, serum amyloid A, and IL-6 were not different amongst siProc treated mice with or without thrombosis. Remarkably, our data revealed that thrombus formation preferably occurred on plaques in the right coronary sinus of the aortic root. In conclusion, there is a predilection of low protein C-induced spontaneous atherothrombosis in Apoe−/− mice for the right coronary sinus, a process that is associated with an increase in platelets and plaques lower in collagen and macrophage content.
Similar content being viewed by others
Introduction
Atherothrombosis, characterized by superimposed thrombus formation overlying a ruptured or eroded atherosclerotic lesion, is the cause of death for more than 14 million individuals per year worldwide (in 2015, World Health organization, www.who.int). Atherogenesis, which can eventually lead to atherothrombosis, has been studied extensively in genetically modified mouse models: When mice are deficient for genes involved in cholesterol metabolism, such as in apolipoprotein E knockout (Apoe−/−) mice or low-density lipoprotein receptor knockout (Ldlr−/−) mice, are fed a cholesterol-rich diet, they rapidly develop atherosclerotic plaques1,2,3. These mouse models have proven to be valuable for unraveling the pathophysiology of atherosclerosis4,5. However, unlike in humans, atherothrombosis does not occur spontaneously in mice, although some signs of rupture or erosion and intraplaque hemorrhage have been recorded6,7,8.
Recently, we showed that transient (7 days) siRNA mediated lowering of the natural anticoagulant protein C (siProc) in atherosclerotic Apoe−/− mice induced superimposed thrombus formation on atherosclerotic plaques in the aortic root9. Although the incidence of this unique phenotype was low (1 out of 4 and 3 out of 25 in two independent experiments, cumulative incidence of 14%), our novel mouse model might be of use to better understand the pathophysiology of atherothrombosis. Moreover, a mouse model of atherothrombosis will be instrumental for the development of novel strategies for the treatment and prevention of atherothrombosis in humans in the future.
Currently, the factors which determine when and where a thrombus develops on top of an atherosclerotic plaque are still largely unknown. We hypothesized that the incidence of atherothrombosis may increase when an additional risk factor for atherothrombosis is introduced on top of the impaired anticoagulant activity by knockdown of protein C. An important risk factor for the development of atherothrombosis and consequent cardiovascular hospitalization is pulse pressure10. A transient increase in blood pressure can be achieved in animal models by the administration of phenylephrine (PE), a selective α1-adrenergic receptor agonist. This will increase the strain on atherosclerotic plaques and thereby stimulate the risk of plaque rupture/erosion and subsequent atherothrombosis. In line, PE administration was associated with increased rupture of p53 treated plaques in Apoe−/− mice11. In the current study we therefore also applied PE treatment to increase the strain on the atherosclerotic plaques and stimulate atherothrombosis incidence in the siProc treated Apoe−/− mice. Moreover, we attempted to direct atherothrombosis to the carotid artery by placing perivascular collars around the carotid artery, a procedure which induces rapid atherogenesis proximal of the collars12. This allowed us to investigate the impact of low protein C on plaques at an additional predefined vascular site and of a different origin than the plaques in the aortic root.
Results
At the time of sacrifice, all Apoe−/− mice treated with PBS or phenylephrine (PE) and siRNA (both siNEG and siProc) appeared healthy and did not show any abnormalities. Histological analysis of the aortic root of 10 weeks WTD-fed siProc treated mice demonstrated the presence of atherosclerotic plaque-associated thrombi, with a similar size and composition as previously reported9. In total, 9 out of 39 siProc treated Apoe−/− mice (23.0%, CI (95%): 16.7–47.9) were categorized as atherothrombosis-positive in the aortic root (siProc + THR, Figs 1A–D and 2). In addition to the 9 siProc + THR mice with typical plaque-associated thrombi, 2 mice developed atypical clots in the aortic root. One atypical clot, although fibrin-positive and clearly associated with an atherosclerotic plaque, did not have the layered structure and did not contain leukocytes as the typical thrombi. These are histological arguments for being a fresh thrombus (Supplemental Fig. 1A). The second atypical thrombus was associated with a valve, although present in the aortic sinus. Despite its unusual location, the thrombus was similar to the typical thrombi found on top of the atherosclerotic plaques. The valve with which it was associated contained a high number of leukocytes, but this was not unique for this specific heart valve (Supplemental Fig. 1B). Mice with atypical thrombi were included in the siProc mice without a thrombus (siProc − THR) during further analyses.

Atherothrombosis in the aortic root of siProc treated Apoe−/− mice. (A) Representative section of the aortic root of a mouse treated with siNEG. (B) Representative section of the aortic root of a mouse treated with siProc, without a thrombus (siProc − THR). (C and D) Representative section of the aortic root of a mouse treated with siProc, with a thrombus associated with an atherosclerotic plaque (siProc + THR, panel D is a magnification of the black square in panel C). (E) Representative section of the common carotid artery containing an atherosclerotic lesion upon collar placement, derived from an siProc treated mouse. (F) A thrombus-like structure associated with an atherosclerotic lesion in the common carotid artery. All sections are hematoxylin and eosin (HE) stained. The scattered black lines in panels A, B, and C mark borders of the plaque. LCS: Left coronary sinus, NCS: Non-coronary sinus, RCS: Right coronary sinus. *Thrombus. Black bars represent 200 μm.

Overview of all siProc associated atherothrombotic events in the aortic root of Apoe−/− mice. (A) In 5 mice in the siProc group without phenylephrine (siProc − PE) treatment atherothrombosis in the aortic root was observed. (B) In 4 mice in the siProc group with phenylephrine (siProc + PE) treatment atherothrombosis in the aortic root was observed. All sections were HE stained. Black bars represent 500 μm. LCS: Left coronary sinus, NCS: Non-coronary sinus, RCS: Right coronary sinus. *Thrombus.
siProc mice treated with PE had a similar incidence of atherothrombosis compared to PBS treated siProc mice (4 out of 19 vs. 5 out of 20 mice, respectively, Fig. 2).
To investigate if an additional predefined site allowing atherothrombosis development could be created, perivascular collars were placed around both the carotid arteries of the Apoe−/− mice, 4 weeks prior to siRNA treatment. In line with previous studies12,13, histological analysis of the carotid arteries showed that collar placement resulted in atherosclerotic plaque formation proximal to the collar. Although gross visual inspection revealed a large variation in plaque size and composition within groups, these were not different at the site of maximal stenosis between mice in the different groups (Supplemental Fig. 2). In total, 1 out of 39 siProc mice presented a structure identified as a thrombus in one of the carotid arteries (Fig. 1E,F). The thrombus was associated with the plaque, was rich in erythrocytes, and had a layered structure, and it was comparable to the thrombi found in the aortic root. siNEG treated mice did not show events or structures identified as a thrombus, neither in the aortic root and nor in the carotid arteries. It should however be noted that in our long term experience with collar placement around the carotid arteries of Apoe−/− mice, the occurrence of thrombosis as a complication of the procedure is not a rare event. Hence, atherothrombosis induced by silencing of Proc seems to be restricted to the aortic root.
Livers and blood/plasma were analyzed to determine whether specific markers could be correlated to atherothrombosis susceptibility. As expected, siProc treatment strongly decreased hepatic Proc transcript levels, compared to siNEG treated mice (remaining Proc transcript: siProc − THR: 32.0% (18.0–63.7), siProc + THR: 25.8% (18.8–40.0), P < 0.001, Fig. 3A as compared to siNEG treated mice). Interestingly, the Proc transcript in siProc treated mice was significantly lower in siProc + THR, compared to the siProc − THR mice (P = 0.013, Fig. 3A). Protein C plasma levels were also reduced in siProc treated mice, as compared to the siNEG treated mice (remaining plasma protein C: siProc − THR: 49.4% (18.4–118.7), siProc + THR: 40.5% (33.2–50.3), P < 0.001, Fig. 3B as compared to siNEG treated mice). However, in contrast to the Proc transcript data, protein C plasma levels did not reach statistical significance between siProc − THR and siProc + THR mice (P = 0.11, Fig. 3B). No differences were observed in mice treated with PE in both the siNEG or siProc group (Supplemental Fig. 3).

Proc liver transcript and plasma protein C levels of siNEG and siProc treated Apoe−/− mice. (A) Proc transcript in the liver upon sacrifice (7 days after siProc treatment) compared to the mean value of siNEG treated mice (100%). (B) Plasma protein C levels, measured by ELISA, compared to the mean value of siNEG treated mice (100%). Black bars indicate the median. **P < 0.01 for siNEG vs. siProc. $P < 0.05 for siProc − THR vs. siProc + THR.
Blood cell counts (total white blood cells, neutrophils, lymphocytes, monocytes, eosinophils, and red blood cells) were comparable between siNEG, siProc − THR, and siProc + THR mice (Fig. 4). Interestingly, platelet levels were significantly higher in siProc treated mice compared to siNEG treated mice (siNEG: 800 × 109/L (508–1112), siProc − THR: 1280 × 109/L (728–1632), siProc + THR: 1512 × 109/L (1004–1696), P < 0.001, Fig. 5A). Within the siProc treated groups, siProc + THR mice had even more platelets (P = 0.043 as compared to siProc − THR, Fig. 5A). To follow-up on this observation, we determined mRNA levels of megakaryocytes in the spleen of genes associated with platelet production. In agreement with the increased platelet counts, upon si Proc treatment,expression of some of these genes (Gata1, Itga2b, Nfe2, and Fog1) were increased as compared to siNEG treated mice (data not shown). siProc + THR treated mice showed an increased expression for Fog1, while for the other spleen-expressed genes expression levels were not different between siProc − THR and siProc + THR mice.

Blood cell populations of siNEG and siProc treated Apoe−/− mice. Blood cell numbers from the siProc treated group is divided in plaques without a thrombus (siProc − THR) and plaques containing a thrombus (siProc + THR). (A) Total white blood cells, (B) neutrophils, (C) lymphocytes, (D) monocytes, (E) eosinophils, and (F) red blood cells are displayed. Black bars indicate the median. No significant differences between the groups were scored.

Blood platelet levels and plasma analysis of siNEG and siProc treated Apoe−/− mice. (A) Total blood platelet levels, (B) plasma total cholesterol levels, (C) plasma fibrinogen levels, measured by ELISA, and expressed as % of normal pool plasma. We did not have any reason to exclude outliers with a low value (e.g. due to coagulation upon blood withdrawal). (D) Serum Amyloid A levels, and (E) plasma IL-6 levels (negative measurements were set to 0 pg/mL). Black bars indicate the median. **P < 0.01 for siNEG vs. siProc. $P < 0.05 for siProc − THR vs. siProc + THR.
Plasma analysis showed that total cholesterol levels were not significantly different between mice treated with siNEG or siProc, in both − THR or +THR mice (siNEG: 10.1 mg/mL (5.4–11.9), siProc −THR: 9.5 mg/mL (6.4–12.8), siProc + THR: 10.1 mg/mL (9.1–13.9), P = 0.43, Fig. 5B). Besides total cholesterol, fibrinogen, serum amyloid A (SAA, an acute-phase mouse protein associated with inflammation14), and interleukin 6 (IL-6, a well-established plasma marker for inflammation (see15) were determined as putative systemic markers for atherothrombosis susceptibility. In line with fibrinogen being a cardiovascular risk factor in epidemiological studies16, plasma fibrinogen levels in all groups of Apoe−/− mice were significantly higher than levels measured in normal pool plasma (NPP, pool of C57BL/6 mouse plasma). However, among siRNA treated Apoe−/− mice no differences were observed (siNEG: 363% (83–598), siProc − THR: 394% (35–562), siProc + THR: 463% (2.6–564), P = 0.74, Fig. 5C). SAA levels were slightly increased in plasma of the Apoe−/− mice compared to NPP (<0.5 ng/mL), but no differences among the Apoe−/− groups were found (siNEG: 0.9 ng/mL (0.4–3.0), siProc − THR: 1.3 ng/mL (0.4–3.1), siProc + THR: 0.8 ng/mL (0.5–3.8), P = 0.33, Fig. 5D). A similar pattern was observed for IL-6 plasma levels (siNEG: 9.4 pg/mL (0–28.4), siProc − THR: 9.7 pg/mL (0–25.9), siProc + THR: 7.7 pg/mL (0–15.8), P = 0.59, Fig. 5E). In line with previous results, PE treatment did not influence blood platelets or plasma levels of the measured parameters (Supplemental Fig. 4).
In our previous study9, we found no significant correlation between plaque size and phenotype and the susceptibility to aortic root atherothrombosis. However, with only 4 siProc + THR mice the power in that study was low. Therefore, in the current study the atherosclerotic plaques in the aortic root were also morphometrically analyzed to assess whether plaque size and characteristics could be linked to the presence of atherothrombosis (for examples of plaque stainings, see Fig. 6A,B). Plaque analysis revealed that PE treatment, similar to the atherothrombosis incidence, did not influence any of the plaque characteristics (Supplemental Fig. 5). For this reason, data of the mice with and without PE treatment were pooled to increase statistical power. In the siProc groups, the plaque area (−THR: 191 × 103 μm2 (55.0–888), +THR: 191 × 103 μm2 (112–343), P = 0.59, Fig. 6C), was not different. Interestingly, collagen and macrophage content were significantly lower in plaques with an associated thrombus (−THR: 13.4% (2.1–29.6), +THR: 8.3% (6.4–15.3), P = 0.031, Fig. 6D, and − THR: 9.1% (0.8–34.9), +THR: 6.7% (4.5–13.7), P = 0.028, Fig. 6E, respectively), unlike the necrotic core (−THR: 19.5% (4.3–80.2), +THR: 15.3% (7.8–67.0), P = 0.30, Fig. 6F). No evidence was found for the occurrence of intraplaque hemorrhages or other abnormalities in the atherosclerotic plaques (neither in − THR nor in +THR plaques).

Plaque composition of siNEG and siProc treated Apoe−/− mice. (A,B) Representative photomicrograph of a Masson’s Trichrome stained plaque in the aortic root (RCS) of mice treated with siProc, without (A) and with (B) a thrombus associated to a plaque from the aortic root. Blue areas were assessed as collagen-positive, non-stained areas as necrotic core, and pink nucleated areas as macrophage content/foam cells. The scattered black lines mark borders of the plaque. Black bars represent 200 μm. (C–F) Individual plaques from the siProc treated group are divided in plaques without a thrombus (siProc − THR) and plaques containing a thrombus (siProc + THR). (C) Total plaque area, (D) collagen, (E) cellular content, (F) necrotic core. For all panels, the indicated values represent an average measurement of three sections. Black bars indicate the median. $P < 0.05 for siProc − THR vs. siProc + THR.
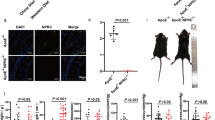
Since in our studies thrombi were exclusively found in the aortic root, we hypothesized that local hemodynamics, which can influence endothelial integrity, plays a role in atherothrombosis formation. For this reason, we re-examined the individual plaques formed in the different cusps within the aortic root (sinuses) for the presence of thrombi, by dividing the aortic root into the left coronary sinus (LCS), right coronary sinus (RCS), and non-coronary sinus (NCS, see17,18,19 and Fig. 7A). We noticed that thrombi had a preference for the RCS, over the LCS and the NCS (Fig. 7B, P = 0.018). When combining these data with the data obtained from our previous study, where we detected 4 thrombi for in total 29 siProc injected Apoe−/− mice9, the preference for the RCS was even more pronounced (P = 0.008).

siProc induced atherothrombosis in the different sinuses of the aortic root of Apoe−/− mice. (A) HE stained section of the aortic root and surrounding tissues. The non-coronary sinus (NCS) of the aortic root is covered by the two atria of the heart (atria are indicated with HA). Left and right of the NCS the left and right coronary sinuses (LCS and RCS, respectively) are present, which are both partly covered by a muscle-rich area, from which the largest part is the right ventricle (indicated with RV). *Thrombus. The scattered black lines mark borders of the plaque. Black bar represents 500 μm. (B) Overview of three independent studies, where atherothrombosis in the aortic root occurred upon siProc treatment. *P < 0.05, **P < 0.01.
To study whether the predilection for the RCS was the consequence of differences in plaque size and composition, the plaque parameters from Fig. 6 were reanalyzed for the LCS, NCS, and RCS. Interestingly, when we pooled the data from the three groups of mice (siNEG, siProc − THR, and siProc + THR) to compare the plaque area, collagen content, macrophage content, and necrotic core size, we found that plaques in the NCS were significantly smaller and contained a larger necrotic core, as compared to their counterparts in the LCS and RCS (Fig. 8). No significant differences among plaques were found when mice were separated in siNEG, siProc − THR, and siProc + THR (Supplemental Fig. 6).

Plaque composition of the LCS, NCS, and LCS. Individual plaques from all groups were pooled and analyzed for (A) total plaque area, (B) collagen, (C) cellular content, (D) and necrotic core. For all panels, the indicated values represent an average measurement of three sections. Black bars indicate the median. LCS: Left coronary sinus, NCS: Non-coronary sinus, RCS: Right coronary sinus. **P < 0.01.
Discussion
Transgenic mice with an impaired lipoprotein metabolism, such as Apoe−/− and Ldlr−/− mice, which are fed a cholesterol-rich diet rapidly develop advanced atherosclerosis1,2. However, in contrast to humans, these mice only develop atherothrombotic events upon additional (invasive) plaque damaging interventions20. The cause for the absence of atherothrombosis is unknown, but it is likely that multiple species-specific factors such as hemodynamics, plaque composition, metabolism, and life span are involved21. Recently, our group showed that transient lowering of the natural anticoagulant protein C in Apoe−/− mice resulted in spontaneous atherothrombosis at plaques formed in the sinuses of the aortic root9. This implies that potent anticoagulation (co)contributes to absence of atherothrombosis in mice. In the current study we aimed to increase the insight into the factors which associate with and possibly determine when and where a thrombus develops on top of an atherosclerotic plaque. For the first time we provide evidence that atherosclerotic plaques located in the RCS of the aortic root are more prone to the development of atherothrombosis, compared to the LCS and NCS. Interestingly, plaques with a thrombus were modestly but significantly lower in collagen and macrophage content. Therefore, we conclude that low protein C-induced atherothrombosis in mice is a low incident but reproducible phenomenon, and that it is determined by the location of the plaque, possibly in combination with differences in plaque composition.
Since Leonardo da Vinci in the beginning of the XVI century, scientists have been intrigued by the sophisticated and sustainable tissue and milieu of the aortic valves and root22,23,24. The mouse aortic sinuses (in humans, sinuses of Valsalva) play a crucial role in regulating the closing mechanism of the valve leaflets25. The oscillating shear stress due to the opening and closing of the valves renders the cusps susceptible for the development of atherosclerotic plaques. A limited number of studies have distinguished between the three different sinuses and their plaques during their analyses. It has been reported that plaque size is reduced specifically in the RCS upon treatment with two different liver X receptor agonists18. This means that differences in local anatomy of the aortic root do not only cause the flow to be different, but also can induce different expression patterns of proteins, possibly linked to coagulation. Moreover, Bentzon et al. suggested a causal relation between lesion formation and expansive remodeling of the aortic root sinuses, which was different when comparing plaques in the LCS, NCS, and RCS26. In our study, we found that plaques formed in the RCS, the location where most thrombi are identified, and the LCS are significantly larger than their counterparts NCS. In addition, plaques in the NCS have a larger necrotic core as compared to the LCS and RCS. This observation again suggests that the plaques in the different sinuses are not similar, but since no outlying characteristics were scored for the RCS we cannot speculate on whether plaque composition contributes to atherothrombosis. The observed preference for the RCS for the development of atherothrombosis suggests that in mice local hemodynamics and wall shear stress are not only involved in atherogenesis27,28, but also in the development of atherothrombosis, as has been proposed for the human disease29,30. The identification of factors that drive the predilection for the RCS may provide novel clues on factors that drive atherothrombosis in mice and humans.
Plaques in the aortic root which were associated with a thrombus appeared to be lower in macrophage and collagen content. Although the measured values fell within the normal range, the difference was considered statistically different. Macrophages (foam cells) are crucially involved in atherogenesis and a high content of this cell population within a plaque has been associated with instability31,32. These observations make our findings in the current study, where in atherothrombotic plaques relatively fewer macrophages were found, unexpected. However, it should be noted that the plaques were advanced and that as a result the macrophage foam cell content was low. It has been described that lower collagen levels in atherosclerotic plaques are associated with instability and risk of rupture33. However, because of the current study setup, we cannot conclude whether the observed difference is a cause or consequence of the formation of a thrombus.
In the current study, siProc treatment of Apoe−/− mice and subsequent thrombus formation did not coincide with altered levels of red and white blood cells or plasma markers of inflammation (fibrinogen, SAA, and IL-6). The only blood cell population which was altered upon siProc treatment were the platelets. Most likely due to increased platelet production, atherosclerotic siProc treated mice showed significant higher levels of platelets in the circulation. To measure platelet production, a transcript analysis for genes associated with platelet production was performed in the spleen. The spleen does not contribute equally to production of platelets in mice, as compared to the bone marrow34. However, the spleen does contain megakaryocytes, which derive from hematopoietic stem cells35. In addition, mice deficient for the Apoe gene have increased extramedullary hematopoiesis36, which means that the contribution of the spleen to total platelet production is increased in these animals. Measuring spleen megakaryocytes to detect alterations of platelet production upon siProc treatment is thus a valid alternative for the bone marrow in Apoe−/− mice. Indeed we measured an upregulation of genes associated with platelet production. In addition, a follow-up study showed that in the absence of an atherosclerotic phenotype (in an independent experiment, in which wild type female C57BL/6 mice were fed a normal chow diet) platelet numbers were also increased in mice treated with an siRNA against Proc (data not shown). Both findings suggest that the anticoagulant protein C directly or indirectly influences platelet production (and possibly consumption and/or clearance), a phenomenon that, to our knowledge, has not been described before. In human studies, it has been suggested that increased platelet levels are a risk factor for atherothrombotic events, making it tempting to speculate that the higher platelet levels upon siProc-treatment contribute to the development of atherothrombosis in the aortic root37,38. For now, it is however not clear whether the increased platelets predict aortic root atherothrombosis susceptibility or whether they are a consequence of the event. For future studies, it would be of interest to dig deeper into the role of platelets and platelet activation during siProc-mediated atherothrombosis in the aortic root. For instance, modulation of platelet function (e.g. via GPVI, see39) in combination with protein C silencing may coincide with a different onset and progression of this unique atherothrombotic phenotype in mice. Moreover, the role of platelets during the local event of atherothrombosis can be studied in intravital microscopy experiments with fluorescently labeled platelets. However, because it is technically not possible to perform these type of studies in the aortic root, this is currently not feasible.
In conclusion, atherosclerotic plaques in the RCS are a predilection site for low protein C-induced spontaneous atherothrombosis in Apoe−/− mice, a process associated with vulnerable plaques displaying a lower macrophage and collagen content and vulnerable blood with augmented platelet counts.
Materials and Methods
Animals
Female C57BL/6 Apoe−/− mice (4–7 weeks old, n = 57) were fed a Western-type diet (WTD containing 0.15% cholesterol; Special Diet Services, Sussex, UK). After 6 weeks on WTD, mice received silicone collars around both common carotid arteries, as previously described11. After 10 weeks on WTD, synthetic siRNA (5 mg/kg; Ambion, Life Technologies, Carlsbad (CA), USA) targeting protein C (siProc, cat. #S72192; n = 40), or a control siRNA without a known target in mice (siNEG, cat. #4404020; n = 17) was injected intravenously (IV). siRNA was complexed using invivofectamine 3.0, and each mouse received 2.5 nmol siRNA. Prior to siRNA injection, mouse groups were randomized based on weight and age (Table 1). siNEG- and siProc treated mice received two intravenous injections of either phenylephrine (PE, 8 μg/kg IV, Sigma-Aldrich cat. #P6126–10G, Zwijndrecht, The Netherlands; siProc: n = 20, siNEG: n = 8) or Phosphate-Buffered Saline (PBS; siProc: n = 20, siNEG: n = 9) as control. PE/PBS injections were performed 4 and 6 days after the siRNA injection. In both the siNEG and the siProc group, one mouse was removed from the experiment due to procedural errors, and thus not represented in the analyses. For an overview of the experimental procedure, see Supplementary Fig. 7. The animal experimental protocol was in agreement with the national guidelines for animal experimentation and was approved by the Ethics Committee for Animal Experiments (Leiden University, Leiden, The Netherlands).
Tissue harvesting and preparation
7 days after siRNA injection, mice were anesthetized with a subcutaneous injection of a mixture of ketamine (100 mg/kg), xylazine (12.5 mg/kg) and atropine (125 µg/kg). Citrated blood (300 μL) was drawn from the inferior vena cava, and plasma was collected as described previously40. For whole blood cell and platelet analysis, an EDTA blood sample (approximately 50 μL) was collected through retro-orbital bleeding after blood withdrawal from the vena cava. Subsequently, mice were further exsanguinated and perfused in situ with PBS, after which organs were harvested. Tissues were either snap frozen (liver and spleen, stored at −80 °C), or fixed for 24 hours in 3.7% neutral-buffered formalin (heart and carotid arteries, Formal-Fixx, Shandon Scientific Ltd, UK) and stored at room temperature for analysis.
Gene expression evaluation
Transcript of Proc was assessed by routinely quantitative PCR of hepatic tissue, with Actb as a reference housekeeping gene41.
Histology
Hearts (aortic roots) and carotid arteries were formalin fixed and embedded in OCT compound (Optimum Cutting Temperature; Sakura Finetek Europe B.V., Alphen aan de Rijn, The Netherlands) before sectioning. Cryosections (10 μm, 70 μm interval) of the aortic root were prepared and mounted in a parallel series on 1% gelatin coated slides. Thrombi and clot structures in the aortic root were identified and scored blindly by two independent researchers on hematoxylin and eosin-stained sections. Plaque size per valve was determined after staining of the cyrosections for neutral lipids using Oil-Red-O and hematoxylin (Sigma-Aldrich, Zwijndrecht, The Netherlands). Corresponding sections were stained using the Masson’s Trichrome method (Sigma-Aldrich) for further morphometric analysis of the atherosclerotic plaques, including the percentage of collagen, necrotic core, and macrophage foam cells. All images were analyzed by blinded computer aided morphometric analysis using the Leica DM-RE microscope and LeicaQwin software (Leica Ltd, Cambridge, UK). Transverse serial cryosections (10 μm; 80 μm interval) of the carotid arteries (and the collars) were made. Sections were routinely stained with hematoxylin and eosin for a general assessment of histology, and were analyzed blindly by two independent researchers.
Blood and plasma analysis
Whole blood cell and platelet analysis were performed using an automated XT-2000iV veterinary hematology analyzer (Sysmex Europe GMBH, Norderstedt, Germany). Plasma cholesterol levels were measured by an enzymatic colorimetric assay (Roche diagnostics, Almere, The Netherlands). Protein C plasma levels were determined using an ELISA with a sheep anti-murine protein C polyclonal antibody (Haematologic Technologies Inc. Essex Junction (VT), USA), conjugated with Horseradish peroxidase with the EZ-Link Plus Activated Peroxidase Kit (Thermo Scientific, #31489, Waltham (MA), USA), as described previously41. Fibrinogen and Serum Amyloid A plasma levels were determined using commercial ELISA kits (Affinity Biologicals, Ancaster (ON), Canada and R&D systems, Minneapolis (MN), USA, respectively). Plasma IL-6 levels were determined using a commercially available ELISA kit (BD Biosciences, San Jose CA, USA) following the manufactures instructions.
Statistics
Statistical analyses were performed in Graphpad Instat (GraphPad Software, La Jolla (CA), USA). Data are presented with median and range. Statistical differences were assessed using the Mann-Whitney U test. A P-value < 0.05 was considered significant. Descriptive statistic and calculate proportions of the observations and the 95% confidence intervals (Wilson-score) were calculated using resources provided by the Open Source Epidemiologic Statistics for Public Health website (www.openepi.com). A Chi-Square test was performed for the valve-incidence was done in SPSS (IBM SPSS Statistics for Windows, Version 23.0, Armonk (NY), USA).
References
Zhang, S. H., Reddick, R. L., Piedrahita, J. A. & Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 258, 468–471 (1992).
Ishibashi, S. et al. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest 92, 883–893, https://doi.org/10.1172/JCI116663 (1993).
Veniant, M. M., Withycombe, S. & Young, S. G. Lipoprotein size and atherosclerosis susceptibility in Apoe(−/−) and Ldlr(−/−) mice. Arterioscler Thromb Vasc Biol 21, 1567–1570 (2001).
Veniant, M. M., Beigneux, A. P., Bensadoun, A., Fong, L. G. & Young, S. G. Lipoprotein size and susceptibility to atherosclerosis–insights from genetically modified mouse models. Curr Drug Targets 9, 174–189 (2008).
Getz, G. S. & Reardon, C. A. Do the Apoe−/− and Ldlr−/− Mice Yield the Same Insight on Atherogenesis? Arterioscler Thromb Vasc Biol 36, 1734–1741, https://doi.org/10.1161/ATVBAHA.116.306874 (2016).
Libby, P. & Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur Heart J 36, 2984–2987, https://doi.org/10.1093/eurheartj/ehv349 (2015).
Matoba, T., Sato, K. & Egashira, K. Mouse models of plaque rupture. Curr Opin Lipidol 24, 419–425, https://doi.org/10.1097/MOL.0b013e3283646e4d (2013).
Lippi, G., Franchini, M. & Targher, G. Arterial thrombus formation in cardiovascular disease. Nat Rev Cardiol 8, 502–512, https://doi.org/10.1038/nrcardio.2011.91 (2011).
Ouweneel, A. B. et al. Silencing of Anticoagulant Protein C Evokes Low-Incident but Spontaneous Atherothrombosis in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler Thromb Vasc Biol 37, 782–785, https://doi.org/10.1161/ATVBAHA.117.309188 (2017).
Selvaraj, S. et al. Pulse Pressure and Risk for Cardiovascular Events in Patients With Atherothrombosis: From the REACH Registry. J Am Coll Cardiol 67, 392–403, https://doi.org/10.1016/j.jacc.2015.10.084 (2016).
von der Thusen, J. H. et al. Induction of atherosclerotic plaque rupture in apolipoprotein E−/− mice after adenovirus-mediated transfer of p53. Circulation 105, 2064–2070 (2002).
von der Thusen, J. H., van Berkel, T. J. & Biessen, E. A. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation 103, 1164–1170 (2001).
Bot, M. et al. Atherosclerotic lesion progression changes lysophosphatidic acid homeostasis to favor its accumulation. Am J Pathol 176, 3073–3084, https://doi.org/10.2353/ajpath.2010.090009 (2010).
Uhlar, C. M. & Whitehead, A. S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 265, 501–523 (1999).
Hunter, C. A. & Jones, S. A. IL-6 as a keystone cytokine in health and disease. Nature immunology 16, 448–457, https://doi.org/10.1038/ni.3153 (2015).
Ernst, E. & Resch, K. L. Fibrinogen as a cardiovascular risk factor: a meta-analysis and review of the literature. Ann Intern Med 118, 956–963 (1993).
Icardo, J. M. & Colvee, E. Origin and course of the coronary arteries in normal mice and in iv/iv mice. J Anat 199, 473–482 (2001).
Peng, D., Hiipakka, R. A., Reardon, C. A., Getz, G. S. & Liao, S. Differential anti-atherosclerotic effects in the innominate artery and aortic sinus by the liver × receptor agonist T0901317. Atherosclerosis 203, 59–66, https://doi.org/10.1016/j.atherosclerosis.2008.05.058 (2009).
de Kerchove, L. et al. Surgical anatomy of the aortic root: implication for valve-sparing reimplantation and aortic valve annuloplasty. J Thorac Cardiovasc Surg 149, 425–433, https://doi.org/10.1016/j.jtcvs.2014.09.042 (2015).
Kuijpers, M. J. et al. Complementary roles of platelets and coagulation in thrombus formation on plaques acutely ruptured by targeted ultrasound treatment: a novel intravital model. J Thromb Haemost 7, 152–161, https://doi.org/10.1111/j.1538-7836.2008.03186.x (2009).
Tchaikovski, S. N., van Vlijmen, B. J. M., Rosing, J. & Tans, G. Development of a calibrated automated thrombography based thrombin generation test in mouse plasma. J Thromb Haemost 5, 2079–2086, https://doi.org/10.1111/j.1538-7836.2007.02719.x (2007).
Bissell, M. M., Dall’Armellina, E. & Choudhury, R. P. Flow vortices in the aortic root: in vivo 4D-MRI confirms predictions of Leonardo da Vinci. Eur Heart J 35, 1344, https://doi.org/10.1093/eurheartj/ehu011 (2014).
De Paulis, R. et al. Hemodynamics of the aortic valve and root: implications for surgery. Ann Cardiothorac Surg 2, 40–43, https://doi.org/10.3978/j.issn.2225-319X.2012.11.16 (2013).
Robicsek, F. Leonardo da Vinci and the sinuses of Valsalva. Ann Thorac Surg 52, 328–335 (1991).
Bellhouse, B. J., Bellhouse, F. H. & Reid, K. G. Fluid mechanics of the aortic root with application to coronary flow. Nature 219, 1059–1061 (1968).
Bentzon, J. F., Pasterkamp, G. & Falk, E. Expansive remodeling is a response of the plaque-related vessel wall in aortic roots of apoE-deficient mice: an experiment of nature. Arterioscler Thromb Vasc Biol 23, 257–262 (2003).
Zhu, H. et al. Differences in aortic arch geometry, hemodynamics, and plaque patterns between C57BL/6 and 129/SvEv mice. J Biomech Eng 131, 121005, https://doi.org/10.1115/1.4000168 (2009).
VanderLaan, P. A., Reardon, C. A. & Getz, G. S. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol 24, 12–22, https://doi.org/10.1161/01.ATV.0000105054.43931.f0 (2004).
Arroyo, L. H. & Lee, R. T. Mechanisms of plaque rupture: mechanical and biologic interactions. Cardiovasc Res 41, 369–375 (1999).
Chen, Y. C., Huang, A. L., Kyaw, T. S., Bobik, A. & Peter, K. Atherosclerotic Plaque Rupture: Identifying the Straw That Breaks the Camel’s Back. Arterioscler Thromb Vasc Biol 36, e63–72, https://doi.org/10.1161/ATVBAHA.116.307993 (2016).
Moore, K. J., Sheedy, F. J. & Fisher, E. A. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13, 709–721, https://doi.org/10.1038/nri3520 (2013).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355, https://doi.org/10.1016/j.cell.2011.04.005 (2011).
Hansson, G. K., Libby, P. & Tabas, I. Inflammation and plaque vulnerability. J Intern Med 278, 483–493, https://doi.org/10.1111/joim.12406 (2015).
Layendecker, S. J. & McDonald, T. P. The relative roles of the spleen and bone marrow in platelet production in mice. Experimental hematology 10, 332–342 (1982).
Morita, Y. et al. Functional characterization of hematopoietic stem cells in the spleen. Experimental hematology 39, 351–359 e353, https://doi.org/10.1016/j.exphem.2010.12.008 (2011).
Robbins, C. S. et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 125, 364–374, https://doi.org/10.1161/CIRCULATIONAHA.111.061986 (2012).
Elliott, M. A. & Tefferi, A. Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol 128, 275–290, https://doi.org/10.1111/j.1365-2141.2004.05277.x (2005).
Kumagai, N. et al. Acute coronary syndrome associated with essential thrombocythemia. J Cardiol 54, 485–489, https://doi.org/10.1016/j.jjcc.2009.03.001 (2009).
Hechler, B. & Gachet, C. Comparison of two murine models of thrombosis induced by atherosclerotic plaque injury. Thrombosis and haemostasis 105(1), S3–12, https://doi.org/10.1160/THS10-11-0730 (2011).
Safdar, H. et al. Modulation of mouse coagulation gene transcription following acute in vivo delivery of synthetic small interfering RNAs targeting HNF4alpha and C/EBPalpha. PLoS One 7, e38104, https://doi.org/10.1371/journal.pone.0038104 (2012).
Safdar, H. et al. Acute and severe coagulopathy in adult mice following silencing of hepatic antithrombin and protein C production. Blood 121, 4413–4416, https://doi.org/10.1182/blood-2012-11-465674 (2013).
Acknowledgements
The authors thank Dr. I. Bot for technical assistance with the collar operations and for critical reading of the manuscript. This work was funded by Leiden University and the Leiden University Medical Center. Furthermore, this work was supported by VICI grant 91813603 from the Netherlands Organization for Scientific Research awarded to M. van Eck. M. van Eck is an Established Investigator of the Dutch Heart Foundation (grant number 2007T056).
Author information
Authors and Affiliations
Contributions
M.v.E., B.J.M.v.V., and P.H.R. conceived the study; M.v.E., B.J.M.v.V., M.H., A.B.O., and P.H.R. designed the experiments; M.H., A.B.O., J.H., and M.K. performed the experiments; All authors analyzed the data and contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heestermans, M., Ouweneel, A.B., Hassan, J. et al. Predilection of Low Protein C-induced Spontaneous Atherothrombosis for the Right Coronary Sinus in Apolipoprotein E deficient mice. Sci Rep 8, 15106 (2018). https://doi.org/10.1038/s41598-018-32584-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32584-y
Keywords
This article is cited by
-
Atherothrombosis model by silencing of protein C in APOE*3-Leiden.CETP transgenic mice
Journal of Thrombosis and Thrombolysis (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
