
Abstract
The Csr global regulatory system coordinates gene expression in response to metabolic status. This system utilizes the RNA binding protein CsrA to regulate gene expression by binding to transcripts of structural and regulatory genes, thus affecting their structure, stability, translation, and/or transcription elongation. CsrA activity is controlled by sRNAs, CsrB and CsrC, which sequester CsrA away from other transcripts. CsrB/C levels are partly determined by their rates of turnover, which requires CsrD to render them susceptible to RNase E cleavage. Previous epistasis analysis suggested that CsrD affects gene expression through the other Csr components, CsrB/C and CsrA. However, those conclusions were based on a limited analysis of reporters. Here, we reassessed the global behavior of the Csr circuitry using epistasis analysis with RNA seq (Epi-seq). Because CsrD effects on mRNA levels were entirely lost in the csrA mutant and largely eliminated in a csrB/C mutant under our experimental conditions, while the majority of CsrA effects persisted in the absence of csrD, the original model accounts for the global behavior of the Csr system. Our present results also reflect a more nuanced role of CsrA as terminal regulator of the Csr system than has been recognized.
Similar content being viewed by others


Introduction
The Csr (Rsm) global regulatory system is conserved among Gammaproteobacteria1,2. It affects gene expression involved in major bacterial lifestyle decisions, and has been extensively studied for its roles in glycogen metabolism3,4, biofilm formation5,6,7, motility8,9, and virulence1,10. In Escherichia coli, the Csr system includes the RNA binding protein CsrA, two small regulatory RNAs (sRNAs), CsrB and CsrC (CsrB/C), which bind to and antagonize CsrA, and a protein that is specifically required for the turnover of CsrB/C, CsrD (Fig. 1a)1. Direct effects of CsrA on gene expression involve its binding to RNA sequences containing conserved GGA motifs in the 5′-untranslated or early coding region of mRNAs, leading to changes in RNA structure11, transcription elongation12, RNA stability7,9,13, and/or translation initiation3,14,15. CsrA can also regulate gene expression indirectly by controlling other regulators. For example, CsrA represses translation initiation of the transcription factor NhaR, which activates transcription of the pgaABCD operon16,17. Thus, CsrA indirectly represses transcription of pgaABCD, which is required for biofilm formation7,16,17. CsrA also directly regulates expression of FlhDC8,9, Hfq18, RelA19, DksA19, RpoE20, IraD15, PNPase21, and many other regulators that have not been investigated in detail22.

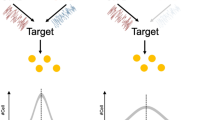
Models proposed for relationships among components of the Csr system. (a) Model 1 described in Suzuki et al.26 and others1, wherein CsrD affects gene expression through effects on CsrB/C stability, which affect the ability of CsrA to directly or indirectly control mRNA expression. (b) Model 2 proposed by Esquerré et al.33, where CsrA acts as a posttranscriptional regulator affecting RNA stability, but mediates much of its indirect effects on mRNA abundance though transcriptional effects controlled by CsrD. Bold arrows emphasize strong contributions to global gene expression in the models. Dashed lines indicate connections with limited experimental evidence. Dashed lines connecting CsrD and CsrA in Model 1 were proposed from the results in this study.
CsrA activity is controlled primarily by the steady state levels of CsrB/C, which contain many high affinity CsrA binding sites that facilitate sequestration of CsrA away from its lower affinity mRNA targets1,23,24. Transcription of CsrB/C is activated by the BarA-UvrY two-component regulatory system (TCS) in response to the accumulation of end products of metabolism, including acetate and formate2,25. Degradation of CsrB/C is initiated by the housekeeping endonuclease RNase E. CsrB/C turnover also requires the presence of CsrD and is stimulated by glucose via the interaction of unphosphorylated EIIAGlc with CsrD (Fig. 1a)26,27,28. CsrD is a membrane-bound protein that contains degenerate GGDEF and EAL domains, which do not appear to synthesize, degrade, or respond to c-di-GMP26. Its EAL domain binds specifically to EIIAGlc 28. Loss of CsrD strongly stabilizes CsrB/C but only modestly affects their steady state levels26,29, which is due to a negative feedback loop in which CsrA indirectly activates the transcription of these sRNAs via the BarA-UvrY TCS (Fig. 1a)26,29,30,31,32.
CsrD was initially described as a regulator of CsrB/C turnover and was found to regulate biofilm formation and csrB transcription in a CsrA- and CsrB/C- dependent manner26. These and other findings led to the development of a model for the workings of the Csr system, in which CsrD affects gene expression through changes in CsrB/C levels, which affect CsrA activity (Model 1, Fig. 1a). According to this model, CsrA acts as the most downstream regulator of gene expression in the Csr system. A recent transcriptomics study found that in addition to global effects of CsrA on RNA stability and steady state levels, CsrD exhibited opposing global effects on RNA levels without affecting stability33. Because the original model for the Csr system predicts that disruption of csrA and csrD should have similar effects on gene expression (Fig. 1a), an alternative model for the Csr system was formulated (Model 2, Fig. 1b). According to Model 2, CsrD is able to globally impact gene expression via transcriptional effects that are mediated downstream of CsrA, implying that CsrD can act as the terminal regulatory element of the Csr system. These two models make different predictions about the behavior of the Csr system and how it affects target gene expression on a global scale.
The goal of this study was to examine the relationships among the Csr system components CsrA, CsrB/C and CsrD in globally regulating gene expression. Epistasis analysis was used to determine the order of their genetic effects, which were assessed by using RNA-seq, an approach that we term Epi-seq. To determine whether the effects of CsrD on gene expression require CsrA and CsrB/C, we compared the impact of a csrD null mutation on the transcriptome in isogenic wild type (WT), csrA or csrB/C mutant strains. Likewise, CsrA effects on RNA abundance were assessed in strains with or without csrD. Our results demonstrate that under our batch culture conditions, which differed from the continuous culture conditions of the previous RNA-seq study33 the Csr circuitry functions as originally proposed (Fig. 1a), wherein CsrA is required for effects of CsrD on transcript levels. Nevertheless, CsrD may act in a limited capacity outside of CsrB/C, via unresolved mechanism(s), possibly through effects on decay of other RNAs besides CsrB/C.
Results and Discussion
Characterization of the wild type and mutant strains
To minimize the effect of growth rates on gene expression and allow comparative transcriptomic analysis between strains, we used the csrA::gm gene disruption mutant as the csrA mutant in this study27. This strain expresses a functionally impaired CsrA protein with greatly reduced affinity for RNA9. Unlike a csrA deletion strain which exhibits severe growth defects and rapidly accumulates suppressor mutations, this csrA mutant grows similarly to the WT strain in rich media9,34,35. This csrA allele is similar to the partial gene deletion (csrA::51 allele) used previously33. The csrB, csrC, and csrD mutations were each complete gene deletions. As csrA/D double mutant strains showed dramatically enhanced cell aggregation and biofilm formation in liquid media (data not shown), all of the strains used in this study were constructed in a pgaC disruption mutant background36. The aggregative phenotype arises as a result of direct and indirect repression of pgaABCD expression by CsrA7,16,37. This operon encodes proteins required for biosynthesis and secretion of the polysaccharide adhesin poly-β-1,6-N-acetyl-D-glucosamine (PGA), and a pgaC mutant strain cannot synthesize PGA36,38. All strains carried an empty pBR322 plasmid or a pBR332-derivative encoding the appropriate gene for complementation analysis. Growth of all strains used in this study was essentially identical during the transition to stationary phase of growth in Kornberg (KB) medium at 37 °C, which was the condition used for the transcriptomics analyses (Fig. 2a)39.

Phenotypes of bacterial strains used in this study are consistent with previous studies. (a) Growth curves of wild type (WT) and mutant strains carrying plasmids pBR322 (not labeled), pCsrA, pCsrD or pCsrB in Kornberg (KB) medium at 37 °C. (b) Glycogen production, determined by iodine staining. (c–e) qRT-PCR analysis of the transcript levels of glgC, csrB, and csrC at the transition to stationary phase of growth in KB medium. Error bars represent s.e. of the mean of three biological replicates. Asterisks indicate level of significance of a two-tailed student’s t-test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
To confirm that the strains used in this study behave consistently with previously reported effects of the Csr system, we analyzed glycogen levels and glgC expression. CsrA represses glycogen accumulation by reducing expression of the glgCAP operon, a phenotype that has been long associated with CsrA4,13. As expected, the csrA mutant accumulated far more glycogen and glgC mRNA relative to the WT strain, whereas deletion of csrD resulted in a slight increase in glycogen and a modest but significant increase in glgC mRNA levels relative to the WT strain (Fig. 2b,c). These effects on glycogen levels were both complemented by ectopic expression of the respective genes (Fig. 2b). Ectopic expression of csrA in the csrA/D mutant strain and csrB in the csrB/C/D mutant strain significantly reduced and stimulated glycogen levels, respectively (Fig. 2b). However, ectopic expression of csrD was unable to complement effects on glycogen levels in the csrA/D background (Fig. 2b). Finally, ectopic expression of csrD had little to no effect on glycogen levels in the csrBCD strain (Fig. 2b). These results are consistent with previously reported effects of the various Csr system components on glycogen levels4,26.
Due to the complexity of the feedback loops governing CsrB/C levels (Fig. 1a), we measured these RNAs in the strains that were used in this study. As described previously29, disruption of csrA in either the WT or csrD mutant strain significantly reduced CsrB and CsrC levels, and their expression was restored by ectopic expression of csrA (Fig. 2d,e). While CsrD is essential for normal turnover of CsrB/C26, deletion of csrD only moderately increased CsrB and decreased CsrC steady state levels (Fig. 2d,e). These phenotypes were complemented by ectopic expression of csrD (Fig. 2d,e). The opposing effect of CsrD on CsrC expression is most likely a result of an increase in CsrB levels in the csrD mutant. CsrB is the principal sRNA antagonist of CsrA, and CsrA indirectly activates CsrB and CsrC transcription through the BarA-UvrY TCS1,3,24,26,29. The compensatory effects of these RNAs on each other’s expression that result from this circuitry are well documented24,33. Thus, increased CsrB levels in the csrD mutant reduce CsrA activity, thereby decreasing CsrC transcription (Fig. 1a). Overall, these observations are consistent with established regulatory circuitry (Fig. 1a), wherein negative feedback loops affect both the synthesis and turnover of CsrB/C.
CsrA retains its role as a global regulator in the absence of CsrD
Having established the strain set for the epistasis analysis, we next used RNA-seq to answer several core questions about the relationships between components of the Csr system. Differential expression analysis revealed 1,054 genes with significant changes in RNA abundance between the WT and csrA mutant strains (csrA - WT, Fig. 3a,b). CsrA repressed the expression of 828 genes and activated 226 genes (Fig. 3a). In addition, 807 out of these 1054 genes were differentially expressed between the csrA/D double mutant strain and the csrD mutant strain (csrA/D - csrD, Fig. 3b). This suggested that CsrA retains its global influence on 80% of its target genes in the absence of CsrD. We hypothesize that the remaining 20% of genes were not identified in this analysis due to the differences in CsrB/C levels resulting from the csrD mutation. These differences in CsrB/C levels would lead to changes in the amount of free CsrA, resulting in differences in the effect of CsrA on gene expression. We also compared the log2 transformed fold change in RNA abundance caused by the csrA mutation in the WT (csrA - WT) and csrD mutant strain (csrA/D - csrD) backgrounds. A relatively high Spearman’s correlation coefficient demonstrated that the absence of csrD had little impact on the overall magnitude of CsrA’s effect on gene expression (ρ = 0.78, Fig. 3c). Ectopic expression of csrA in the csrA/D double mutant resulted in vast changes in gene expression (csrA/D pCsrA - csrA/D) that were largely overlapping with those resulting from mutation of csrA in the WT strain (csrA - WT, Fig. 3b). The log2 transformed fold changes from ectopic expression of csrA in the csrA/D double mutant showed a strong negative Spearman’s correlation with those resulting from mutation of csrA in either a WT (csrA - WT, Fig. 3d) or csrD mutant strain background (csrA/D - csrD, Fig. 3e) (ρ = 0.82 and 0.87, respectively). Together these data indicate that CsrA does not require CsrD to exert global effects on transcript levels.

CsrA retains its global role in regulating mRNA levels in the absence of CsrD. (a) Volcano plot depicting the log2 transformed fold change of RNA levels between the csrA mutant and its isogenic WT strain versus log odds of significance. Genes with significant changes shown in black. The number of genes up- and down-regulated in this comparison are shown at the top of the plot. (b) Venn diagram depicting the overlap of genes differentially expressed among the following comparisons: csrA - WT, csrA/D - csrD, and csrA/D pCsrA - csrAD. (c–e) The log2 transformed fold change in RNA abundance caused by mutation or overexpression of csrA in the WT or csrD mutant backgrounds. Blue and red dots represent the genes that are only differentially expressed in one strain background. Black and grey dots represent the genes that are differentially expressed in both of the backgrounds or neither, respectively. The associated Spearman’s correlation coefficients (ρ) are shown.
CsrD effects on gene expression are lost in the csrA mutant
We identified 73 genes not including csrD that were differentially expressed between the WT and csrD mutant strains (csrD - WT, Fig. 4a), suggesting that CsrD has a limited effect on the transcriptome compared to CsrA under our experimental conditions (Figs 3a and 4a). In addition, the majority of these genes (54/73) are involved in motility and chemotaxis, suggesting that CsrD targets may exhibit limited functional roles. More critically, csrB was the only gene differentially expressed between the csrA/D double mutant and csrA mutant strains besides csrD (csrA/D - csrA, Fig. 4b), indicating that CsrD requires CsrA for essentially all of its effects on gene expression. In addition, ectopic expression of csrD was unable to complement the effects of the csrA csrD double mutant strain unlike ectopic expression of csrA in this strain (Fig. 4c). The clear inference from these findings is that csrD functions upstream of csrA to regulate gene expression.

CsrD controls gene expression through CsrA-dependent pathways. (a) Volcano plot depicting the log2 transformed fold change of RNA levels between the csrD mutant and its isogenic WT strain versus log odds of significance. The number of genes up- and down-regulated in this comparison are shown at the top of the plot. Black dots represent genes that are only differentially expressed in csrD - WT but not csrA - WT comparison. Red dots represent genes that are regulated in the same direction in the csrD and csrA mutant strains relative to the WT strain. Cyan dots represent genes that are either upregulated or downregulated in both csrD and csrA mutant strains relative to the WT strain. Grey dots represent genes that are not differentially expressed in the csrD – WT comparison. (b) Venn diagram depicting the overlap of genes differentially expressed among the following comparisons: csrD - WT, csrA/D - csrA, and csrA/D pCsrD - csrA/D. (c) Venn diagram depicting the overlap of genes differentially expressed among the following comparisons: csrA/D - WT, csrA/D pCsrA - csrA/D, and csrA/D pCsrD - csrA/D. (d–e) qRT-PCR analysis of the transcript levels of ftnB and fliA. Error bars represent the s.e. of the mean of three biological replicates. Asterisks indicate level of significance of a two-tailed student’s t-test (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
The simplest interpretation of Model 1 (Fig. 1a) predicts that mutations in csrA and csrD would result in qualitatively similar changes in gene expression in the same direction. Indeed, CsrA regulated 12 genes affected by CsrD in the same direction (cyan in Fig. 4a). We used qRT-PCR to validate these results for one such gene, ftnB (Fig. 4d). Consistent with our RNA-seq data, both csrA and csrD mutations led to an increase in ftnB mRNA levels, and these effects were complemented by ectopic expression of the respective genes (Fig. 4d). Mutation of csrA in the csrD mutant background also resulted in increased ftnB levels, but there was no significant difference in ftnB levels between the csrA and csrA/D double mutant strains. In addition, ectopic expression of csrA but not csrD complemented the change in ftnB expression in the csrA/D double mutant strain (Fig. 4d). Taken together, these qRT-PCR results are consistent with the effects of CsrD being mediated through CsrA.
The remaining genes regulated by CsrD were either regulated by CsrA in the opposite direction (4 genes including csrD, red in Fig. 4a) or not regulated by CsrA (58 genes, black in Fig. 4a). Interestingly, nearly all of the latter genes were regulated by CsrA under a different condition (55/58)22. These results are more complex than can be explained with a simplistic interpretation of Model 1 (Fig. 1a). According to Model 2, opposing effects of CsrA and CsrD may be mediated through CsrD effects on transcription (Fig. 1b)33. Specifically, genes whose RNA stability was not affected by CsrA were proposed as possible candidates of CsrD-dependent transcriptional regulation33. To this end, we compared genes whose expression was regulated by CsrD in this study with an analysis of CsrA-dependent regulation of RNA stability22. Out of the 73 genes regulated by CsrD (csrD-WT, not including csrD), 49 of these were not regulated at the level of their RNA stability (18 were not analyzed, 2 were destabilized by CsrA and 4 stabilized by CsrA). Thus, it appears that many of the genes regulated by CsrD in our condition are not directly regulated at the level of RNA stability by CsrA, which would make them candidates for CsrD-dependent transcriptional regulation33. However, the epistasis analysis showed that csrD exhibited no effects on gene expression (other than csrB and csrD itself) in the absence of csrA (csrA/D - csrA, Fig. 4b). Nevertheless, we wanted to validate an example of possible CsrA-independent effects of CsrD on gene expression using qRT-PCR. CsrD activated the expression of fliA, which is consistent with RNA-seq data (Fig. 4e). Although it was not significant in the RNA-seq data, CsrA repressed fliA expression (Fig. 4e), which is consistent with previous evidence22. Both of these effects were complemented by the corresponding genes (Fig. 4e). Deletion of csrD in a csrA mutant background weakly affected fliA expression (20% decrease, Fig. 4e). In stark contrast, disruption of csrA in a csrD deletion mutant resulted in a large change in fliA expression (500% increase, Fig. 4e). In addition, ectopic expression of csrA, but not csrD, was able to complement the csrA csrD double mutant (Fig. 4e). The small effect still observed for the csrD deletion in the csrA disruption mutant likely occurs because this is not a null csrA allele. Finally, deletion of csrD did not affect fliA mRNA in the csrB csrC double deletion strain and ectopic expression of csrB but not csrD was able to complement the csrB csrC csrD triple mutant (Fig. 4e). Thus, even for genes such as fliA, for which csrA and csrD single mutations have opposing effects on gene expression, the effects of CsrD are mediated via the other Csr components. Although not as simplistic as either model implies (Fig. 1), these results suggest that small and large decreases in CsrA activity (csrD mutant and csrA mutant, respectively) may lead to different and even opposing effects on gene expression. As CsrA is a global regulator (Fig. 3a), perhaps it is not surprising that it can exhibit such complex effects on gene expression, reminiscent to what has been seen for DNA binding regulators, e.g. OmpR can activate or repress ompF at low or high osmolarity, respectively40.
CsrB/C effects on gene expression are enhanced in the absence of CsrD
CsrB/C affect gene expression indirectly by sequestering CsrA from interacting with its lower affinity mRNA targets1,23,24. If CsrA acts as the terminal factor in the Csr system, then CsrB/C should still impact gene expression in the absence of CsrD. On the other hand, if CsrD were able to act downstream of CsrA to regulate transcription, then its effects on transcription should also be predicted to persist in the absence of CsrB/C. Deletion of csrB/C resulted in changes in the expression of 40 genes in the WT background (csrB/C - WT) and 218 genes in the csrD mutant strain background (csrB/C/D - csrD) (Fig. 5). This finding confirmed that CsrB/C do not require CsrD to globally affect gene expression. More importantly, the greater effects of these sRNAs on expression in the csrD mutant compared to the WT background are as would be predicted if CsrD acts by triggering CsrB/C decay. Because of increased CsrB levels, CsrA activity in the csrD mutant strain should be lower relative to the WT strain (Fig. 2D). Consequently, CsrA activity is predicted to be more greatly affected by csrB/C deletion in the csrD mutant vs the WT background. The expression of 912 genes was significantly different when CsrB was ectopically expressed in the csrB/C/D mutant strain (csrB/C/D pCsrB - csrB/C/D, Fig. 5). This further demonstrates that CsrB mediates vast effects on gene expression in the absence of CsrD. Together with the observations that CsrA globally regulates gene expression in the absence of CsrD, while CsrD requires CsrA for its effects (Fig. 3), these findings reveal that neither CsrB/C nor CsrA requires CsrD to mediate global changes in gene expression.

CsrB/C affect gene expression in the absence of CsrD. Venn diagram depicting the overlap of genes differentially expressed among the following comparisons: csrB/C - WT, csrB/C/D - csrD, csrB/C/D pCsrB - csrB/C/D.
CsrD regulation of target genes is largely, but not entirely CsrB/C-dependent
To further determine whether CsrD always depends on CsrB/C to mediate changes in gene expression, we compared the impact of CsrD on the transcriptome in the presence and absence of CsrB/C. Differential expression analysis revealed 60 genes that respond to csrD deletion in the WT background (csrD - WT) but not in the csrB/C mutant background (csrB/C/D - csrB/C) (Fig. 6). Thus, 82% of the genes regulated by CsrD under our conditions are controlled via a CsrB/C-dependent pathway. Using qRT-PCR, we confirmed for one of these genes, ftnB, that CsrD no longer regulated its expression in the csrB/C mutant background (Fig. 4d). In the absence of CsrB/C, 25 genes not including csrD still responded to CsrD (csrB/C/D - csrB/C, Fig. 6), suggesting that alternative regulatory pathways may allow CsrD to mediate changes in expression. Intriguingly, we observed 12 genes that were regulated by CsrD only in the csrB/C mutant background (csrB/C/D – csrB/C, Fig. 6) but not in the WT background (csrD – WT, Fig. 6). This observation suggests that a CsrB/C-independent pathway might have an opposing effect on the expression of these genes with respect to the CsrB/C-dependent pathway. However, none of these 12 changes in expression were complemented by ectopic expression of csrD (csrB/C/D pCsrD – csrB/C/D, Fig. 6), whereas overexpression of csrB complemented most of the differences identified in a csrB/C/D triple mutant (csrB/C/D pCsrB - csrB/C/D, Fig. 5). Further investigation is required to determine the relevance of these differences. A plausible explanation for CsrB/C independent effects is that CsrD may regulate the stability of other sRNAs that regulate CsrA activity. For example, the sRNAs McaS and GadY have been shown to inhibit CsrA activity when overexpressed41,42. Recently other sRNAs have been found to directly bind CsrA in vivo in both Samonella and E. coli22,43. This hypothesis will require further testing. Nevertheless, a majority of CsrD effects are mediated through CsrB/C.

CsrD regulates gene expression in CsrB/C-dependent and independent pathways. Venn diagram depicting the overlap of genes differentially expressed among the following comparisons: csrD - WT, csrB/C/D - csrB/C, and csrB/C/D pCsrD - csrB/C/D.
Indirect regulatory role of CsrA
If CsrA does not mediate vast transcriptional effects on gene expression through CsrD as proposed (Fig. 1b), this raises the question as to how CsrA affects the transcriptome on such a global scale and in the absence of effects on stability33. In addition to global post-transcriptional regulation mediated through direct binding interactions, CsrA also indirectly regulates transcript levels by controlling the expression of transcriptional and post-transcriptional regulators22. Indeed, our data revealed that CsrA affects the expression of 68 transcriptional regulators, including TCS, σ factors, and other transcription factors (Supplementary Table S1). This supports other studies, which have suggested that CsrA interacts with many mRNAs encoding transcriptional regulators in vivo19,22,43,44. CsrA also affected the abundance of 8 sRNAs in addition to CsrB/C (Supplementary Table S2). sRNAs have been shown to contribute directly and indirectly to changes in transcript levels by altering mRNA turnover and the expression of transcriptional regulators45,46. Clearly, the integration of post-transcriptional regulation into transcriptional regulatory networks is a common theme used by bacteria to control their gene expression45,46. As result, post-transcriptional regulators including CsrA and sRNAs can mediate vast effects on gene expression indirectly, which allows them to control a broad range of genes and cellular functions. Thus, CsrA no doubt mediates changes in the levels of many transcripts indirectly through its effects on transcription factors and other regulators.
Conclusions
Recent work has proposed that CsrA may exert much of its impact on the transcriptome via its effects on CsrD, which acts downstream of CsrA as a regulator of gene expression33. However, these studies rely heavily on correlational analysis of gene expression between single csrA and csrD mutant strains. On the other hand, existing models propose that CsrA is the terminal regulator in the Csr system with CsrD playing a role in gene expression solely through effects on CsrA activity26. However, the latter studies rely primarily on data from selected single gene analyses and may not capture the full effects of the Csr system on gene expression at a transcriptome-wide scale. Here we formally analyzed the relationship between different components of the Csr system on a whole genome level using epistasis analysis with RNA-seq (Epi-seq). We now provide evidence that CsrA and CsrB/C mediate vast changes in RNA abundance independently of CsrD. Moreover, CsrD affects gene expression largely through CsrB/C and seemingly entirely through CsrA. In addition, CsrD may affect expression of a limited number of genes independently of CsrB/C. The latter observation warrants further investigation, and hints that CsrD may affect the turnover of RNAs in addition to CsrB/C. Altogether, the use of Epi-seq to clarify the genetic relationships among the components of the Csr system indicates that CsrA acts as the terminal component of the Csr system to globally regulate gene expression. Because the present and previous33 studies used different experimental conditions, batch culture in Kornberg medium and ΔpgaC background vs. continuous culture in M9 glucose medium, respectively, it is conceivable that the Csr system functions differently under the two conditions. Thus, we acknowledge the formal possibility that CsrD might mediate CsrA-independent transcriptional effects under the latter growth conditions.
Methods
Media and growth conditions
Bacterial strains used in this study are listed in Supplementary Table S3. Strains were routinely grown in LB medium (1% tryptone, 1% NaCl, and 0.5% yeast extract) with antibiotics when appropriate: ampicillin (100 μg ml−1), kanamycin (50 μg ml−1), chloramphenicol (25 μg ml−1), and gentamicin (10 μg ml−1). For growth curve and RNA-seq analysis, overnight cultures grown in LB broth were inoculated into Kornberg medium (1.1% K2HPO4, 0.85% KH2PO4, 0.6% yeast extract and 0.5% glucose) at an OD600 of 0.01, and cultures were then grown at 37 °C with shaking at 250 rpm. Bacterial growth was monitored at OD600.
Construction of strains and plasmids
E. coli K-12 MG1655 pgaC880::cam was used as the WT strain, where the pgaC gene was disrupted by a mini-Tn10 transposon with a cam resistance casette36. E. coli gene deletions and disruptions were transferred by P1vir transduction using E. coli donor strains from previous studies26,27,36 and the Keio library47, as shown in Supplementary Table S3. The FRT (short flippase recognition target)-flanked antibiotic resistance cassettes introduced into mutant strains were eliminated using an flippase expression plasmid pCP20 when necessary48.
Plasmids and DNA oligonucleotides used in this study are listed in Tables S4 and S5. Plasmids p2VR112 (referred as pCsrA in this study) and pBRY4 (referred as pCsrD in this study), express csrA and csrD, respectively, under the control of their native promoters on plasmid pBR32226,27. To construct plasmid pCsrB for expression of CsrB, the csrB gene with 494 base pairs (bp) upstream and 36 bp downstream was amplified from the genomic DNA and cloned into plasmid pBR322. Strains not transformed with pCsrA, pCsrD or pCsrB were transformed with pBR322 to maintain isogenic comparisons.
Analysis of glycogen levels
Glycogen levels were analyzed by staining colonies with iodine vapor, as described previously23.
RNA extraction and purification
During transition to stationary phase of growth (OD600 of 2.0), 1 ml of cell culture was collected and immediately mixed with 0.125 mL of stop solution (10% phenol/90% ethanol) to stabilize the RNA. Total RNA was isolated using hot phenol chloroform extraction followed by ethanol precipitation. Genomic DNA was removed by treating 20 μg of nucleic acid with 4U of Turbo DNase (Ambion), and RNA was purified from these reactions with the RNeasy kit (Qiagen). The integrity of the RNA was verified using denaturing gel electrophoresis and the RNA Bioanalyzer (Agilent).
RNA-seq library preparation
For each strain, three independent biological replicates were collected. Ribosomal RNA was depleted from 5 μg of total RNA using the Ribo-Zero rRNA Removal Kit for Gram-Negative Bacteria (Illumina). The concentrations of the rRNA-depleted samples were determined with the Qubit RNA HS Assay Kit (ThermoFisher). RNA-seq libraries were then generated with the Stranded RNA-Seq Library Preparation Kit for Illumina (KAPA) and NEBNext Multiplex Oligos for Illumina adaptors (NEB) according to the manufacturer’s instructions for 100 ng of starting material and a mean insert size of 200–300 bases. Final libraries were purified with Pure Beads (KAPA). Sequencing library size and integrity were verified with DNA Bioanalyzer analysis (Agilent). Libraries were pooled and sequenced on 2 lanes of 50SE HiSeq. 2500 (Illumina) by the Genomic Services Laboratory at the HudsonAlpha Institute for Biotechnology.
RNA-seq data analysis
Raw reads were demultiplexed and mapped to the E. coli rRNA sequences with Bowtie 249. Unmapped rRNA depleted reads were then mapped to the E. coli genomic DNA sequence (NC_000913.3) with Bowtie 249. Read counts per gene were calculated with htseq-count50. Read counts were filtered to remove genes with less than an average of 10 reads per sample across all samples. Differential expression was analyzed with limma voom51; fold changes >2 and a FDR (false discovery rate) <0.05 were considered significant. The full results are presented in Supplementary Table S6.
Quantitative Reverse Transcriptase PCR (qRT-PCR)
qRT-PCR was conducted using iTaq Universal SYBR Green One-Step Kit (Bio-Rad) and an iQ5 iCycler real time PCR system (Bio-Rad) according to the manufacturer’s instructions. Reactions of 10 μl contained 200 ng of RNA or DNA standard, 300 nM of each primer, iScript reverse transcriptase, and 1× iTaq universal SYBR Green reaction mix. Reactions were incubated for 10 min of RT at 50 °C, 1 min of denaturation and RT inactivation at 95 °C, and then 45 cycles of 10 sec of denaturation at 95 °C and 20 sec of annealing, extension, and imaging at 60 °C. Melt curve analysis was used to verify the specificity of the amplicons with the parameters: 95 °C for 1 min, 55 °C for 1 min, and increasing the temperature 0.5 °C/10 sec until reaching 95 °C. RNA abundances were determined relative to a standard curve of PCR products and normalized to 16 s rRNA levels.
Statistical analysis
All statistical tests used in this paper were two-sided, and statistical significance is indicated by asterisks (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
Data availability
All datasets generated by this study are included in the Supplementary Information and/or will be uploaded to the GEO repository upon acceptance of the manuscript.
References
Vakulskas, C. A., Potts, A. H., Babitzke, P., Ahmer, B. M. M. & Romeo, T. Regulation of Bacterial Virulence by Csr (Rsm) Systems. Microbiol. Mol. Biol. Rev. 79, 193–224 (2015).
Zere, T. R. et al. Genomic Targets and Features of BarA-UvrY (-SirA) Signal Transduction Systems. PLoS One 10, e0145035 (2015).
Baker, C. S., Morozov, I., Suzuki, K., Romeo, T. & Babitzke, P. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol. Microbiol. 44, 1599–610 (2002).
Romeo, T., Gong, M., Liu, M. Y. & Brun-Zinkernagel, A. M. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J. Bacteriol. 175, 4744–55 (1993).
Jonas, K. et al. The RNA binding protein CsrA controls cyclic di-GMP metabolism by directly regulating the expression of GGDEF proteins. Mol. Microbiol. 70, 236–57 (2008).
Sterzenbach, T. et al. A novel CsrA titration mechanism regulates fimbrial gene expression in Salmonella typhimurium. EMBO J. 32, 2872–83 (2013).
Wang, X. et al. CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli. Mol. Microbiol. 56, 1648–63 (2005).
Wei, B. L. et al. Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol. Microbiol. 40, 245–56 (2001).
Yakhnin, A. V. et al. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol. Microbiol. 87, 851–66 (2013).
Bhatt, S. et al. The RNA binding protein CsrA is a pleiotropic regulator of the locus of enterocyte effacement pathogenicity island of enteropathogenic Escherichia coli. Infect. Immun. 77, 3552–68 (2009).
Patterson-Fortin, L. M., Vakulskas, C. A., Yakhnin, H., Babitzke, P. & Romeo, T. Dual posttranscriptional regulation via a cofactor-responsive mRNA leader. J. Mol. Biol. 425, 3662–77 (2013).
Figueroa-Bossi, N. et al. RNA remodeling by bacterial global regulator CsrA promotes Rho-dependent transcription termination. Genes Dev. 28, 1239–51 (2014).
Liu, M. Y., Yang, H. & Romeo, T. The product of the pleiotropic Escherichia coli gene csrA modulates glycogen biosynthesis via effects on mRNA stability. J. Bacteriol. 177, 2663–72 (1995).
Dubey, A. K. et al. CsrA regulates translation of the Escherichia coli carbon starvation gene, cstA, by blocking ribosome access to the cstA transcript. J. Bacteriol. 185, 4450–60 (2003).
Park, H. et al. Translational Repression of the RpoS Antiadapter IraD by CsrA Is Mediated via Translational Coupling to a Short Upstream Open Reading Frame Hongmarn. mBio 8, e01355–17 (2017).
Pannuri, A. et al. Translational repression of NhaR, a novel pathway for multi-tier regulation of biofilm circuitry by CsrA. J. Bacteriol. 194, 79–89 (2012).
Goller, C., Wang, X., Itoh, Y. & Romeo, T. The cation-responsive protein NhaR of Escherichia coli activates pgaABCD transcription, required for production of the biofilm adhesin poly-β-1,6-N-acetyl-D-glucosamine. J. Bacteriol. 188, 8022–8032 (2006).
Baker, C. S. et al. CsrA inhibits translation initiation of Escherichia coli hfq by binding to a single site overlapping the Shine-Dalgarno sequence. J. Bacteriol. 189, 5472–81 (2007).
Edwards, A. N. et al. Circuitry linking the Csr and stringent response global regulatory systems. Mol. Microbiol. 80, 1561–80 (2011).
Yakhnin, H., Aichele, R., Ades, S. E., Romeo, T. & Babitzke, P. Circuitry linking the global Csr and σE -dependent cell envelope stress response systems. J. Bacteriol. 199, e00484–17 (2017).
Park, H., Yakhnin, H., Connolly, M., Romeo, T. & Babitzke, P. CsrA participates in a PNPase autoregulatory mechanism by selectively repressing translation of pnp transcripts that have been previously processed by RNase III and PNPase. J. Bacteriol. 197, 3751–3759 (2015).
Potts, A. H. et al. Global role of the bacterial post-transcriptional regulator CsrA revealed by integrated transcriptomics. Nat. Commun. 8, 1596 (2017).
Liu, M. Y. et al. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J. Biol. Chem. 272, 17502–10 (1997).
Weilbacher, T. et al. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol. Microbiol. 48, 657–70 (2003).
Chavez, R. G., Alvarez, A. F., Romeo, T. & Georgellis, D. The physiological stimulus for the BarA sensor kinase. J. Bacteriol. 192, 2009–12 (2010).
Suzuki, K., Babitzke, P., Kushner, S. R. & Romeo, T. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev. 20, 2605–17 (2006).
Vakulskas, C. A. et al. Antagonistic control of the turnover pathway for the global regulatory sRNA CsrB by the CsrA and CsrD proteins. Nucleic Acids Res. 44, 7896–7910 (2016).
Leng, Y. et al. Regulation of CsrB/C sRNA decay by EIIA(Glc) of the phosphoenolpyruvate: carbohydrate phosphotransferase system. Mol. Microbiol. 99, 627–639 (2015).
Camacho, M. I. et al. Effects of the global regulator CsrA on the BarA/UvrY two-component signaling system. J. Bacteriol. 197, 983–91 (2015).
Gudapaty, S., Suzuki, K., Wang, X., Babitzke, P. & Romeo, T. Regulatory interactions of Csr components: The RNA binding protein CsrA activates csrB transcription in Escherichia coli. J. Bacteriol. 183, 6017–6027 (2001).
Adamson, D. N. & Lim, H. N. Rapid and robust signaling in the CsrA cascade via RNA-protein interactions and feedback regulation. Proc. Natl. Acad. Sci. USA 110, 13120–13125 (2013).
Suzuki, K. et al. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J. Bacteriol. 184, 5130–5140 (2002).
Esquerré, T. et al. The Csr system regulates genome-wide mRNA stability and transcription and thus gene expression in Escherichia coli. Sci. Rep. 6, 25057 (2016).
Altier, C., Suyemoto, M. & Lawhon, S. D. Regulation of Salmonella enterica serovar Typhimurium invasion genes by csrA. Infect. Immun. 68, 6790–7 (2000).
Timmermans, J. & Van Melderen, L. Conditional essentiality of the csrA gene in. Escherichia coli. J. Bacteriol. 191, 1722–1724 (2009).
Wang, X., Preston, J. F. & Romeo, T. The pgaABCD Locus of Escherichia coli Promotes the Synthesis of a Polysaccharide Adhesin Required for Biofilm Formation. J. Bacteriol. 186, 2724–2734 (2004).
Grylak-Mielnicka, A., Bidnenko, V., Bardowski, J. & Bidnenko, E. Transcription termination factor Rho: A hub linking diverse physiological processes in bacteria. Microbiology (United Kingdom) 162, 433–447 (2016).
Itoh, Y. et al. Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-β-1,6-N-acetyl-D-glucosamine. J. Bacteriol. 190, 3670–3680 (2008).
Romeo, T. & Preiss, J. Genetic regulation of glycogen biosynthesis in Escherichia coli: in vitro effects of cyclic AMP and guanosine 5′-diphosphate 3′-diphosphate and analysis of in vivo transcripts. J. Bacteriol. 171, 2773–2782 (1989).
Pratt, L. A., Hsing, W., Gibson, K. E. & Silhavy, T. J. From acids to osmZ: Multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Molecular Microbiology 20, 911–917 (1996).
Jørgensen, M. G., Thomason, M. K., Havelund, J., Valentin-Hansen, P. & Storz, G. Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev. 27, 1132–1145 (2013).
Parker, A., Cureoglu, S., De Lay, N., Majdalani, N. & Gottesman, S. Alternative pathways for Escherichia coli biofilm formation revealed by sRNA overproduction. Mol. Microbiol. 105, 309–325 (2017).
Holmqvist, E. et al. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J. 35, 991–1011 (2016).
Sowa, S. W. et al. Integrative FourD omics approach profiles the target network of the carbon storage regulatory system. Nucleic Acids Res. gkx048 (2017).
Göpel, Y. & Görke, B. Rewiring two-component signal transduction with small RNAs. Current Opinion in Microbiology 15, 132–139 (2012).
Mandin, P. & Guillier, M. Expanding control in bacteria: Interplay between small RNAs and transcriptional regulators to control gene expression. Current Opinion in Microbiology 16, 125–132 (2013).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2(2006), 0008 (2006).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–5 (2000).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–9 (2012).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Liu, R. et al. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 43, e97 (2015).
Acknowledgements
This work was supported by the National Institute of Health (R01GM059969) to T.R. and P.B. (R01AI097116) to T.R. and the National Science Foundation (DGE-1315138) to A.H.P.
Author information
Authors and Affiliations
Contributions
Y.L. and A.H.P. conceived and designed the study, conducted experiments, collected and analyzed data, and wrote the manuscript. P.B. acquired funding and contributed to writing the manuscript, T.R. acquired funding, conceived, designed and supervised the study, interpreted results, and contributed to writing of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Potts, A.H., Leng, Y., Babitzke, P. et al. Examination of Csr regulatory circuitry using epistasis analysis with RNA-seq (Epi-seq) confirms that CsrD affects gene expression via CsrA, CsrB and CsrC. Sci Rep 8, 5373 (2018). https://doi.org/10.1038/s41598-018-23713-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-23713-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
